Using Chou’s 5-Step Rule to Evaluate the Stability of Tautomers: Susceptibility of 2-[(Phenylimino)-methyl]-cyclohexane-1,3-diones to Tautomerization Based on the Calculated Gibbs Free Energies


Abstract

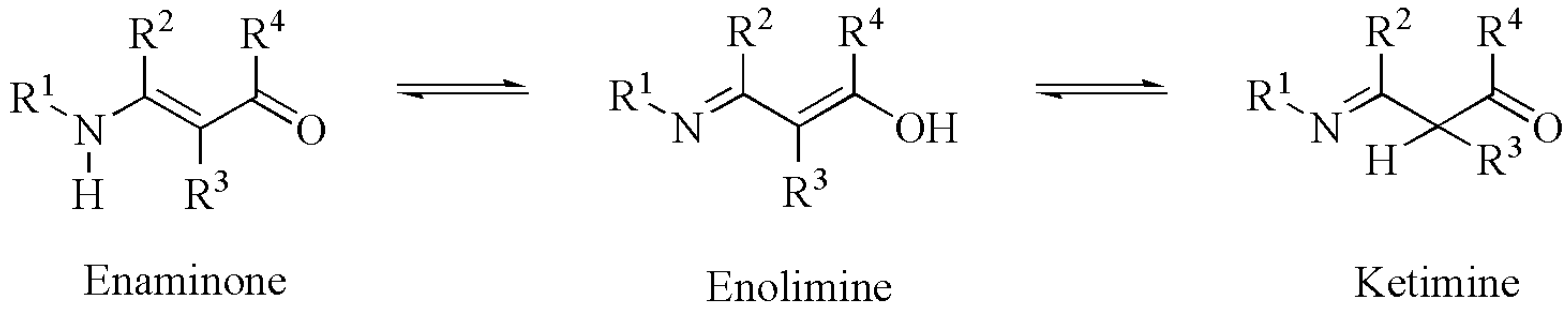
1. Introduction
2. Materials and Methods
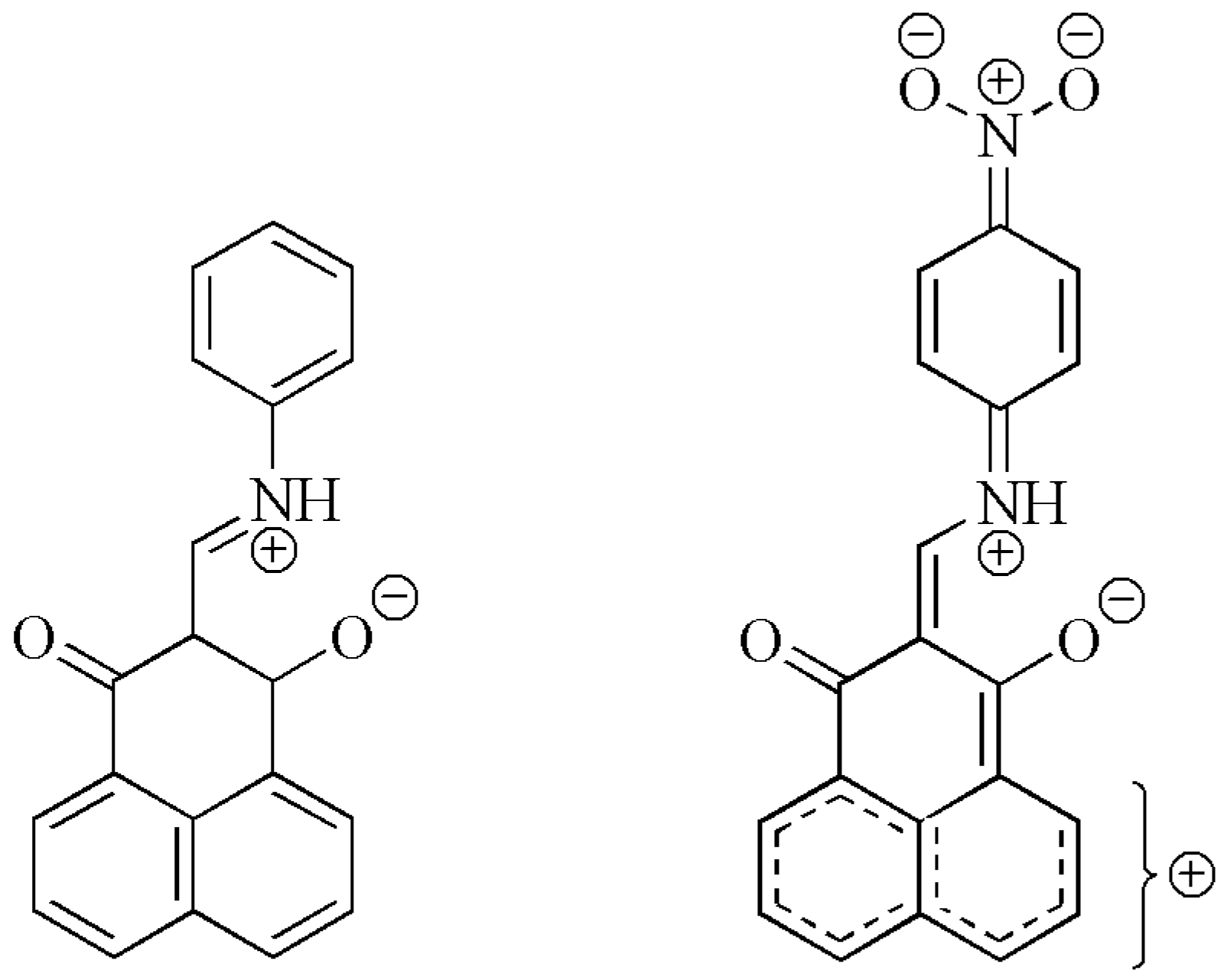
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric equilibria in relation to Pi-electron delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef]
- Dobosz, R.; Gawinecki, R. Effect of benzoannulation on tautomeric preferences of 4,6-di(pyridin-2-yl)cyclohexane-1,3-dione. J. Mol. Model. 2013, 19, 3397–3402. [Google Scholar] [CrossRef]
- Ośmiałowski, B.; Dobosz, R. The influence of secondary interactions on complex stability and double proton transfer reaction in 2-[1H]-pyridone/2-hydroxypyridine dimers. J. Mol. Model. 2011, 17, 2491–2500. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Berardi, M.J.; Shih, W.M.; Harrison, S.C.; Chou, J.J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 2011, 476, 109–113. [Google Scholar] [CrossRef] [PubMed]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.M.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the Mitochondrial Calcium Uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G.; et al. Structural Basis for Membrane Anchoring of HIV-1 Envelope Spike. Science 2016, 353, 172–175. [Google Scholar] [CrossRef]
- Chou, K.C. Review: Structural bioinformatics and its impact to biomedical science. Curr. Med. Chem. 2004, 11, 2105–2134. [Google Scholar] [CrossRef]
- Chou, K.C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015, 11, 218–234. [Google Scholar] [CrossRef]
- Kolehmainen, E.; Ośmiałowski, B.; Nissinen, M.; Kauppinen, R.; Gawinecki, R. Substituent and temperature controlled tautomerism of 2-phenacylpyridine: Hydrogen bond as a configurational lock of (Z) 2-(2-hydroxy-2-phenylvinyl)-pyridine. J. Chem. Soc. Perkin Trans. 2000, 2, 2185–2191. [Google Scholar] [CrossRef]
- Kolehmainen, E.; Ośmiałowski, B.; Krygowski, T.M.; Kauppinen, R.; Nissinen, M.; Gawinecki, R. Substituent and temperature controlled tautomerism: Multinuclear magnetic resonance, X-ray, and theoretical studies on 2-phenacylquinolines. J. Chem. Soc. Perkin Trans. 2000, 2, 1259–1266. [Google Scholar] [CrossRef]
- Gawinecki, R.; Kolehmainen, E.; Loghmani-Khouzani, H.; Ośmiałowski, B.; Lovász, T.; Rosa, P. Effect of π-electron delocalization on tautomeric equilibria. Benzoannulated 2-phenacylpyridines. Eur. J. Org. Chem. 2006, 2817–2824. [Google Scholar] [CrossRef]
- Gómez-Sánchez, A.; Paredes-León, R.; Cámpora, J. 1H and 13C NMR spectra and isomerism of 3-aminoacroleins. Magn. Reson. Chem. 1998, 36, 154–162. [Google Scholar] [CrossRef]
- Weinstein, J.; Wyman, M. A Study of β-Amino-α, β-unsaturated Ketones. J. Org. Chem. 1958, 23, 1618–1622. [Google Scholar] [CrossRef]
- Dudek, G.O.; Volpp, G.P. Nuclear Magnetic Resonance Studies of Keto-Enol Equilibria. V. Isomerization in Aliphatic Schiff Bases. J. Am. Chem. Soc. 1963, 85, 2697–2702. [Google Scholar] [CrossRef]
- Dąbrowski, J.; Kamieńska-Trela, K. Infrared spectra and structure of substituted unsaturated carbonyl compounds—III Enamino ketones with tertiary amino group. Spectrochim. Acta 1966, 22, 211–220. [Google Scholar] [CrossRef]
- Dąbrowski, J.; Dąbrowska, U. Infrarotspektren und Struktur substituierter ungesättigter Carbonylverbindungen, VII. Enaminoketone mit starrer s-cis-und s-trans-Konformation und sekundärer Aminogruppe. Chem. Ber. 1968, 101, 2365–2374. [Google Scholar] [CrossRef]
- Kania, L.; Kamieńska-Trela, K.; Wilanowski, M. Structural increments in UV spectra of conjugated carbonyl compounds: Part, I. The α-alkyl substituted enaminones. J. Mol. Struct. 1983, 102, 1–17. [Google Scholar] [CrossRef]
- Brown, N.M.D.; Nonhebel, D.C. NMR spectra of intramolecularly hydrogen-bonded compounds—II: Schiff bases of β-diketones and o-hydroxycarbonyl compounds. Tetrahedron 1968, 24, 5655–5664. [Google Scholar] [CrossRef]
- Dąbrowski, J.; Kamieńska-Trela, K. Electronic spectra of .alpha. beta-unsaturated carbonyl compounds. I. An evaluation of increments characteristic of changes in configuration (cis/trans) and conformation (s-cis/s-trans) based on direct observation of the isomerization of enamino aldehydes and ketones. J. Am. Chem. Soc. 1976, 98, 2826–2834. [Google Scholar] [CrossRef]
- Czerwińska, E.; Kozerski, L.; Boksa, J. Structural studies by 1H and 13C d.n.m.r.: I-barrier to trans-cis isomerization in aliphatic enamino ketones of the type R-CO-CH=CH-NHR1. Org. Magn. Reson. 1976, 8, 345–349. [Google Scholar] [CrossRef]
- Kozerski, L.; Von Philipsborn, W. 15N chemical shifts as a conformational probe in enaminones A variable temperature study at natural isotope abundance. Org. Magn. Reson. 1981, 17, 306–310. [Google Scholar] [CrossRef]
- Kashima, Ch.; Yamamoto, M.; Sugiyama, N. Ultraviolet spectral study of β-amino-enones. J. Chem. Soc. C 1970, 111–114. [Google Scholar] [CrossRef]
- Kashima, C.; Aoyama, H.; Yamamoto, Y.; Nishio, T. Nuclear magnetic resonance spectral study of β-aminoenones. J. Chem. Soc. Perkin Trans. 1975, 2, 665–670. [Google Scholar] [CrossRef]
- Zhou, J.-C. NMR of enaminones. Part 8—1H, 13C and 17O NMR spectra of primary and secondary 1,2-disubstituted enaminones: Configuration, conformation and intramolecular hydrogen bonding. Magn. Reson. Chem. 1998, 36, 565–572. [Google Scholar] [CrossRef]
- Fustero, S.; De la Torre, M.G.; Jofré, V.; Carlón, R.P.; Navarro, A.; Fuentes, A.S. Synthesis and reactivity of new β-enamino acid derivatives: a simple and general approach to β-enamino esters and thioesters. J. Org. Chem. 1998, 63, 8825–8836. [Google Scholar] [CrossRef]
- Edwards, W.G.H.; Petrov, V. Some heterocyclic structures derived from acenaphthene. J. Chem. Soc. 1954, 2853–2860. [Google Scholar] [CrossRef]
- Dąbrowski, J. Infra-red spectra and structure of substituted unsaturated carbonyl compounds—I: Enamino Ketones with primary amino group. Spectrochim. Acta 1963, 19, 475–496. [Google Scholar] [CrossRef]
- Ogawa, K.; Harada, J. Aggregation controlled proton tautomerization in salicylideneanilines. J. Mol. Struct. 2003, 647, 211–216. [Google Scholar] [CrossRef]
- Buemi, G.; Zuccarello, F.; Venuvanalingam, P.; Ramalingam, M. Ab initio study of tautomerism and hydrogen bonding of β-carbonylamine in the gas phase and in water solution. Theor. Chem. Acc. 2000, 104, 226–234. [Google Scholar] [CrossRef]
- Rybarczyk-Pirek, A.; Grabowski, S.J.; Małecka, M.; Nawrot-Modranka, J. Crystal and molecular structures of new chromone derivatives as empirical evidence of intramolecular proton transfer reaction; ab initio studies on intramolecular H-bonds in enaminones. J. Phys. Chem. A 2003, 106, 11956–11962. [Google Scholar] [CrossRef]
- Zabatyuk, R.I.; Volovenko, Y.M.; Shishkin, O.V.; Gorb, L.; Leszczyński, J. Aromaticity-controlled tautomerism and resonance-assisted hydrogen bonding in heterocyclic enaminone−iminoenol systems. J. Org. Chem. 2007, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Schmidt, G.M.J. Photochromy and thermochromy of anils. J. Phys. Chem. 1962, 66, 2442–2446. [Google Scholar] [CrossRef]
- Alarcón, S.H.; Olivieri, A.C.; Labadie, G.R.; Cravero, R.M.; Gonzáles-Sierra, M. Tautomerism of representative aromatic α-hydroxy carbaldehyde anils as studied by spectroscopic methods and AM1 calculations. Synthesis of 10-hydroxyphenanthrene-9-carbaldehyde. Tetrahedron 1995, 51, 4619–4626. [Google Scholar] [CrossRef]
- Vargas, V.; Amigo, L. A study of the tautomers of N-salicylidene-p-X-aniline compounds in methanol. J. Chem. Soc. Perkin Trans. 2001, 2, 1124–1129. [Google Scholar] [CrossRef]
- Dziembowska, T.; Jagodzińska, E.; Rozwadowski, Z.; Kotfica, M. Solvent effect on intramolecular proton transfer equilibrium in some N-(R-salicylidene)-alkylamines. J. Mol. Struct. 2001, 598, 229–234. [Google Scholar] [CrossRef]
- Fernández, G.J.M.; del Rio-Portilla, F.; Quiroz-García, N.; Toscano, R.A.; Salcedo, R. The structures of some ortho-hydroxy Schiff base ligands. J. Mol. Struct. 2001, 561, 197–207. [Google Scholar] [CrossRef]
- Sitkowski, J.; Stefaniak, L.; Dziembowska, T.; Grech, E.; Jagodzińska, E.; Webb, G.A. A multinuclear NMR study of proton transfer processes in Schiff bases. J. Mol. Struct. 1996, 381, 177–180. [Google Scholar] [CrossRef]
- Salman, S.R.; Lindon, J.C.; Farrant, R.D.; Carpenter, T.A. Tautomerism in 2-hydroxy-1-naphthaldehyde Schiff bases in solution and the solid state investigated using 13C NMR spectroscopy. Magn. Res. Chem. 1993, 31, 991–994. [Google Scholar] [CrossRef]
- Galić, N.; Cimerman, Z.; Tomiśić, V. Tautomeric and protonation equilibria of Schiff bases of salicylaldehyde with aminopyridines. Anal. Chim. Acta 1997, 343, 135–143. [Google Scholar] [CrossRef]
- Nazir, H.; Yildiz, M.; Yilmaz, H.; Tahir, M.N.; Ülkü, D.J. Intramolecular hydrogen bonding and tautomerism in Schiff bases. Structure of N-(2-pyridil)-2-oxo-1-naphthylidenemethylamine. J. Mol. Struct. 2000, 524, 241–250. [Google Scholar] [CrossRef]
- Becker, R.S.; Richey, W.F. Photochromic anils. Mechanisms and products of photoreactions and thermal reactions. J. Am. Chem. Soc. 1967, 89, 1298–1302. [Google Scholar] [CrossRef]
- Hansen, P.E.; Sitkowski, J.; Stefaniak, L.; Rozwadowski, Z.; Dziembowska, T. One-bond deuterium isotope effects on 15N chemical shifts in Schiff bases. Ber. Bunsenges. Phys. Chem. 1998, 102, 410–413. [Google Scholar] [CrossRef]
- Dziembowska, T. Resonance assisted intramolecular hydrogen bond in Schiff bases. Pol. J. Chem. 1998, 72, 193–202. [Google Scholar]
- Król-Starzomska, I.; Rospenk, M.; Rozwadowski, Z.; Dziembowska, T. UV-visible absorption spectroscopic studies of intramolecular proton transfer in N-(R-salicylidene)-alkylamines. Pol. J. Chem. 2000, 74, 1441–1446. [Google Scholar]
- Zhuo, J.-C. NMR of enaminones. part 6—17O and 13C NMR study of tautomerization in Schiff bases. Magn. Reson. Chem. 1999, 37, 259–268. [Google Scholar] [CrossRef]
- Antonov, L.; Fabian, W.M.F.; Nedeltcheva, D.; Kamounah, F.S. Tautomerism of 2-hydroxynaphthaldehyde Schiff bases. J. Chem. Soc. Perkin Trans. 2000, 2, 1173–1179. [Google Scholar] [CrossRef]
- Dudek, G.O.; Dudek, E.P. Spectroscopic studies of keto-enol equilibria. IX. N15-Substituted anilides. J. Am. Chem. Soc. 1966, 88, 2407–2412. [Google Scholar] [CrossRef]
- Joshi, H.; Kamounah, F.S.; van der Zwan, G.; Gooijer, C.; Antonov, L. Temperature dependent absorption spectroscopy of some tautomeric azo dyes and Schiff bases. J. Chem. Soc. Perkin Trans. 2001, 2, 2303–2308. [Google Scholar] [CrossRef]
- Gawinecki, R.; Kuczek, A.; Kolehmainen, E.; Ośmiałowski, B.; Krygowski, T.M.; Kauppinen, R. Influence of the bond fixation in benzo-annulated N-salicylideneanilines and their orto-COX derivatives (X = CH3, NH2, OCH3) on tautomeric equilibria in solution. J. Org. Chem. 2007, 72, 5598–5607. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Kasahara, Y.; Ohtani, Y.; Harada, J. Crystal structure change for the thermochromy of N-salicylideneanilines. The first observation by X-ray diffraction. J. Am. Chem. Soc. 1998, 120, 7107–7408. [Google Scholar] [CrossRef]
- Dobosz, R.; Kolehmainen, E.; Valkonen, A.; Ośmiałowski, B.; Gawinecki, R. Tautomeric preferences of phthalones and related compounds. Tetrahedron 2007, 63, 9172–9178. [Google Scholar] [CrossRef]
- Rogers, N.A.J.; Smith, H. 2-Acylcyclohexane-1,3-diones. Part II. 2-Formyl-, 2-propionyl-, 2-isobutyryl-, and 2-phenylcarbamoyl-cyclohexane1,3-dione, and their conversion into phenathridines. J. Chem. Soc. 1955, 341–346. [Google Scholar] [CrossRef]
- Kettmann, V.; Lokaj, J.; Milata, V.; Marko, M.; Štvrtecká, M. 2-(Phenylaminomethylidene)-cyclohexane-1,3-dione. Acta Cryst. C 2004, 60, o252–o254. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, G.; Wolfbeis, O.S.; Junek, H. Über Anilinomethylenverbindungen der Cyclohexandione. Monatshefte für Chemie 1974, 105, 1283–1291. [Google Scholar] [CrossRef]
- Donovan, P.M.; Scott, L.T. Elaboration of Diaryl Ketones into Naphthalenes Fused on Two or Four Sides: A Naphthoannulation Procedure. J. Am. Chem. Soc. 2004, 126, 3108–3112. [Google Scholar] [CrossRef]
- Zhang, M.; An, H.-Y.; Zhao, B.-G.; Xu, J.-H. Aromatic annulation strategy for naphthalenes fused at 1,2-and 3,4-positions with two heterocycles. Org. Biomol. Chem. 2006, 4, 33–35. [Google Scholar] [CrossRef]
- Panda, K.; Venkatesh, Ch.; Ila, H.; Junjappa, H. Efficient Routes to Acenaphthylene-Fused Polycyclic Arenes/Heteroarenes and Heterocyclic Fluoranthene Analogues. Eur. J. Org. Chem. 2005, 2045–2055. [Google Scholar] [CrossRef]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Chou, K.C. Progresses in predicting post-translational modification. Int. J. Pept. Res. Ther. 2019, in press. [Google Scholar] [CrossRef]
- Chou, K.C. Advance in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Curr. Med. Chem. 2019, 26, 4918–4943. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Impacts of pseudo amino acid components and 5-steps rule to proteomics and proteome analysis. Curr. Top. Med. Chem. 2019, 19, 2283–2300. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. An insightful recollection for predicting protein subcellular locations in multi-label systems. Genomics 2019, in press. [Google Scholar] [CrossRef]
- Chou, K.C. Proposing Pseudo Amino Acid Components is an Important Milestone for Proteome and Genome Analyses. Int. J. Pept. Res. Ther. 2019, in press. [Google Scholar] [CrossRef]
- Chou, C.K. Recent Progresses in Predicting Protein Subcellular Localization with Artificial Intelligence (AI) Tools Developed Via the 5-Steps Rule. Jacobs J. Gastroenterol. Hepatol. 2019, 2, 1–21. [Google Scholar]
- Chou, K.C. An insightful recollection since the distorted key theory was born about 23 years ago. Genomics 2019, in press. [Google Scholar] [CrossRef]
- Chou, K.C. Artificial intelligence (AI) tools constructed via the 5-steps rule for predicting post-translational modifications. Trends Artif. Inttell. 2019, 3, 60–74. [Google Scholar] [CrossRef]
- Chou, K.C. Distorted Key Theory and Its Implication for Drug Development. Curr. Proteom. [CrossRef]
- Chou, K.C. An insightful recollection since the birth of Gordon Life Science Institute about 17 years ago. Adv. Sci. Eng. Res. 2019, 4, 31–36. [Google Scholar] [CrossRef]
- Chou, K.C. Gordon Life Science Institute: Its philosophy, achievements, and perspective. Ann. Cancer Ther. Pharmacol. 2019, 2, 1–26. [Google Scholar]
- Eistert, B.; Eifler, W.; Goth, H. Versuche in der Reihe des 3-Hydroxy-1-oxo-phenalens und des 1.2.3-Trioxo-2.3-dihydro-phenalens. Chem. Ber. 1968, 101, 2162–2175. [Google Scholar] [CrossRef]
- Wolfbeis, O.S.; Ziegler, E. Zur Reaktivität von C=N-Doppelbindungssystemen, X Synthesen von kondensierten Heterocyclen. Zeitschrift für Naturforschung B 1976, 31, 1519–1525. [Google Scholar] [CrossRef]
- Wolfbeis, O.S.; Ziegler, E.; Knierzinger, A.; Wipfler, H.; Trummer, I. Eine breit anwendbare Synthese fluoreszierender kondensierter α-Pyrone. Monatshefte für Chemie 1980, 111, 93–112. [Google Scholar] [CrossRef]
- Facchetti, A.; Streitwieser, A. Ion pair first and second acidities of some β-diketones and aggregation of their lithium and cesium enediolates in THF. J. Org. Chem. 2004, 69, 8345–8355. [Google Scholar] [CrossRef] [PubMed]
- Van Tinh, D.; Fischer, M.; Stadlbauer, W. Ring closure reactions of cyclic 2-arylaminomethylene-1,3-diones. J. Heterocycl. Chem. 1996, 33, 905–910. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision, B.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Woliński, K.; Hilton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Dobosz, R. Susceptibility of 2-[(Phenylimino)-methyl]-cyclohexane-1,3-diones to tautomerization. RepOD 2019. [Google Scholar] [CrossRef]
- Lloris, M.E.; Abramovitch, R.A.; Marquet, J.; Moreno-Mañas, M. Reactions of copper(II) β-diketonates under free radical conditions. II. Diazonium salts as aryl radicals source in the arylation of β-diketones. Tetrahedron 1992, 48, 6909–6916. [Google Scholar] [CrossRef]
- Olah, G.A.; Grant, J.L.; Westerman, P.W. Stable carbocations. CLXXVIII. Carbon-13 nuclear magnetic resonance spectroscopic study of protonated and diprotonated acyclic and cyclic diketones in fluorosulfuric acid-antimony pentafluoride-sulfur dioxide solution. J. Org. Chem. 1975, 40, 2102–2108. [Google Scholar] [CrossRef]
- Gawinecki, R.; Kolehmainen, E.; Dobosz, R.; Ośmiałowski, B. (1Z,3Z)-3-[Quinolin-2(1H)-ylidene]-1-(quinolin-2-yl) prop-1-en-2-ol: An unexpected most stable tautomer of 1,3-bis(quinolin-2-yl) acetone. J. Mol. Struct. 2009, 930, 78–82. [Google Scholar] [CrossRef]
- Dobosz, R.; Gawinecki, R.; Ośmiałowski, B. DFT studies on tautomeric preferences of 1-(pyridin-2-yl)-4-(quinolin-2-yl) butane-2,3-dione in the gas phase and in solution. Struct. Chem. 2010, 21, 1283–1287. [Google Scholar] [CrossRef][Green Version]
- Borowski, P.; Gawinecki, R.; Miłaczewska, A.; Skotnicka, A.; Woliński, K.; Brzyska, A. Instability of 2,2-di(pyridin-2-yl) acetic acid. Tautomerization versus decarboxylation. J. Mol. Model. 2011, 17, 857–868. [Google Scholar] [CrossRef][Green Version]
- Kurt, M.; Chinna Babu, P.; Sundaraganesan, N.; Cinar, M.; Karabacak, M. Molecular structure, vibrational, UV and NBO analysis of 4-chloro-7-nitrobenzofurazan by DFT calculations. Spectrochim. Acta Part A 2011, 79, 1162–1170. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H8 | H7 | C2 | C7 | C1 | C3 | ||
|---|---|---|---|---|---|---|---|
| 1Ea | exp. | 12.97d | 8.55d | 109.61 | 151.07 | 191.81 | 196.62 |
| calc. | 12.35 | 8.61 | 113.98 | 151.98 | 195.71 | 202.93 | |
| 1Eb | exp. | 13.54d | 8.90d | 108.86 | 152.74 | 181.44 | 183.9 |
| calc. | 12.98 | 9.10 | 113.44 | 153.69 | 183.23 | 188.12 | |
| 2Ea | exp. | 12.93d | 8.61d | 109.73 | 150.88 | 196.64 | 200.25 |
| calc. | 12.30 | 8.72 | 114.12 | 151.69 | 195.88 | 203.20 | |
| 2Eb | exp. | 13.50d | 8.96d | 109.03 | 152.78 | no | no |
| calc. | 12.94 | 9.20 | 113.53 | 153.25 | 183.39 | 188.33 | |
| 3Ea | exp. | 12.76d | 8.58d | 109.78 | 150.64 | no | no |
| calc. | 12.32 | 8.76 | 114.29 | 151.46 | 196.03 | 203.42 | |
| 3Eb | exp. | 13.49d | 9.00d | 109.24 | 153.01 | no | no |
| calc. | 12.96 | 9.25 | 113.74 | 153.19 | 183.47 | 188.50 | |
| 4Ea | exp. | 12.60d | 8.47d | 109.96 | 150.46 | 195.56 | 199.67 |
| calc. | 12.30 | 8.65 | 114.45 | 151.21 | 196.03 | 203.78 | |
| 4Eb | exp. | 13.42d | 8.96d | no | 153.18 | no | no |
| calc. | 12.95 | 9.13 | 113.88 | 152.81 | 183.45 | 188.73 | |
| 5Ea | exp. | 12.98d | 8.47d | 111.17 | 149.59 | 195.82 | 200.26 |
| calc. | 12.46 | 8.80 | 115.34 | 149.93 | 196.43 | 204.79 | |
| 5Eb | exp. | 10.80bs | 8.55bs | no | 150.11 | no | no |
| calc. | 13.12 | 9.28 | 114.83 | 151.53 | 183.70 | 189.42 |
| Form | Grel | Form | Grel | ||
|---|---|---|---|---|---|
| Vacuum | DMSO | Vacuum | DMSO | ||
| 1Ea | 0.0 | 0.0 | 1Eb | 0.0 | 0.0 |
| 1Oa | 5.5 | 6.6 | 1Ob | 4.9 | 6.0 |
| 1Ka | 20.0 | 18.2 | 1Kb | 22.3 | 20.9 |
| 2Ea | 0.0 | 0.0 | 2Eb | 0.0 | 0.0 |
| 2Oa | 5.9 | 7.0 | 2Ob | 5.2 | 7.4 |
| 2Ka | 20.5 | 18.7 | 2Kb | 21.6 | 22.3 |
| 3Ea | 0.0 | 0.0 | 3Eb | 0.0 | 0.0 |
| 3Oa | 5.4 | 7.0 | 3Ob | 5.1 | 6.1 |
| 3Ka | 19.7 | 18.7 | 3Kb | 22.2 | 20.8 |
| 4Ea | 0.0 | 0.0 | 4Eb | 0.0 | 0.0 |
| 4Oa | 5.3 | 6.4 | 4Ob | 4.6 | 5.6 |
| 4Ka | 19.4 | 18.1 | 4Kb | 21.4 | 20.1 |
| 5Ea | 0.0 | 0.0 | 5Eb | 0.0 | 0.0 |
| 5Oa | 5.6 | 6.4 | 5Ob | 5.2 | 5.7 |
| 5Ka | 19.2 | 17.8 | 5Kb | 21.4 | 20.1 |
| 1Ea | 3Ea | 5Ea | 1Eb | 3Eb | 5Eb | |
|---|---|---|---|---|---|---|
| N8–C7 | 1.322 1.317 | 1.325 1.320 | 1.333 1.329 | 1.321 1.315 | 1.324 1.318 | 1.332 1.327 |
| N8–C9 | 1.409 1.413 | 1.407 1.411 | 1.396 1.397 | 1.409 1.413 | 1.406 1.411 | 1.495 1.398 |
| C7–C2 | 1.382 1.388 | 1.379 1.385 | 1.372 1.377 | 1.382 1.390 | 1.379 1.387 | 1.372 1.379 |
| C2–C3 | 1.451 1.449 | 1.453 1.451 | 1.458 1.457 | 1.447 1.444 | 1.449 1.446 | 1.454 1.452 |
| C3–O3′ | 1.229 1.234 | 1.228 1.232 | 1.226 1.230 | 1.233 1.237 | 1.232 1.236 | 1.231 1.233 |
| C2–C1 | 1.470 1.464 | 1.473 1.466 | 1.478 1.472 | 1.465 1.458 | 1.467 1.461 | 1.472 1.466 |
| C1–O1′ | 1.215 1.223 | 1.215 1.222 | 1.213 1.220 | 1.219 1.226 | 1.219 1.226 | 1.217 1.223 |
| N8–H8 | 1.021 1.020 | 1.021 1.020 | 1.021 1.020 | 1.022 1.020 | 1.021 1.020 | 1.022 1.021 |
| H8···O3′ | 1.857 1.884 | 1.861 1.887 | 1.849 1.873 | 1.849 1.875 | 1.852 1.875 | 1.839 1.862 |
| N8···O3′ | 2.656 2.669 | 2.659 2.671 | 2.653 2.665 | 2.650 2.662 | 2.652 2.662 | 2.645 2.656 |
| N8H8O3′ | 132.5 133.3 | 132.4 131.1 | 133.0 133.4 | 132.6 131.4 | 132.5 131.4 | 133.2 132.0 |
| C7N8C9C10 | 19.9 22.7 | 15.5 19.9 | −2.3 −6.8 | 20.5 23.5 | 16.2 18.7 | 3.1 9.1 |
| Tautomer | Donor (i) | Type | Acceptor (j) | Type | E(2) [kJ/mol] | |
|---|---|---|---|---|---|---|
| Vacuum | DMSO | |||||
| 1Ea | N8 | LP | C7–C2 | π* | 88.85 | 95.36 |
| C7–C2 | π | C3–O3′ | π* | 39.92 | 42.07 | |
| C7–C2 | π | C1–O1′ | π* | 33.13 | 37.43 | |
| 3Ea | N8 | LP | C7–C2 | π* | 85.37 | 91.48 |
| C7–C2 | π | C3–O3′ | π* | 38.65 | 40.77 | |
| C7–C2 | π | C1–O1′ | π* | 32.30 | 36.43 | |
| 5Ea | N8 | LP | C7–C2 | π* | 76.48 | 80.29 |
| C7–C2 | π | C3–O3′ | π* | 35.82 | 37.30 | |
| C7–C2 | π | C1–O1′ | π* | 29.77 | 33.35 | |
| 1Eb | N8 | LP | C7–C2 | π* | 88.47 | 96.54 |
| C7–C2 | π | C3–O3′ | π* | 41.96 | a | |
| C7–C2 | π | C1–O1′ | π* | 35.16 | 40.08 | |
| C7–C2 | π | C3 | LP* | a | 73.06 | |
| 3Eb | N8 | LP | C7–C2 | π* | 85.00 | 93.05 |
| C7–C2 | π | C3–O3′ | π* | 40.68 | 43.63 | |
| C7–C2 | π | C1–O1′ | π* | 34.31 | 39.03 | |
| 5Eb | N8 | LP | C7–C2 | π* | 75.86 | 81.63 |
| C7–C2 | π | C3–O3′ | π* | 37.67 | 40.11 | |
| C7–C2 | π | C1–O1′ | π* | 31.68 | 35.90 | |
| N8 | O3′ | O1′ | |
|---|---|---|---|
| 1Ea | −0.514 −0.500 | −0.628 −0.660 | −0.463 −0.640 |
| 3Ea | −0.520 −0.508 | −0.623 −0.655 | −0.583 −0.636 |
| 5Ea | −0.528 −0.517 | −0.613 −0.642 | −0.572 −0.623 |
| 1Eb | −0.510 −0.493 | −0.635 −0.660 | −0.591 −0.638 |
| 3Eb | −0.516 −0.501 | −0.630 −0.656 | −0.587 −0.634 |
| 5Eb | −0.525 −0.511 | −0.622 −0.644 | −0.578 −0.623 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobosz, R.; Mućko, J.; Gawinecki, R. Using Chou’s 5-Step Rule to Evaluate the Stability of Tautomers: Susceptibility of 2-[(Phenylimino)-methyl]-cyclohexane-1,3-diones to Tautomerization Based on the Calculated Gibbs Free Energies. Energies 2020, 13, 183. https://doi.org/10.3390/en13010183
Dobosz R, Mućko J, Gawinecki R. Using Chou’s 5-Step Rule to Evaluate the Stability of Tautomers: Susceptibility of 2-[(Phenylimino)-methyl]-cyclohexane-1,3-diones to Tautomerization Based on the Calculated Gibbs Free Energies. Energies. 2020; 13(1):183. https://doi.org/10.3390/en13010183
Chicago/Turabian StyleDobosz, Robert, Jan Mućko, and Ryszard Gawinecki. 2020. "Using Chou’s 5-Step Rule to Evaluate the Stability of Tautomers: Susceptibility of 2-[(Phenylimino)-methyl]-cyclohexane-1,3-diones to Tautomerization Based on the Calculated Gibbs Free Energies" Energies 13, no. 1: 183. https://doi.org/10.3390/en13010183
APA StyleDobosz, R., Mućko, J., & Gawinecki, R. (2020). Using Chou’s 5-Step Rule to Evaluate the Stability of Tautomers: Susceptibility of 2-[(Phenylimino)-methyl]-cyclohexane-1,3-diones to Tautomerization Based on the Calculated Gibbs Free Energies. Energies, 13(1), 183. https://doi.org/10.3390/en13010183