Abstract
Lipid droplets are the neutral lipid storage compartments of eukaryotic cells. Mitochondria are the main source for ATP, which is generated through oxidative phosphorylation. Thus, both organelles play essential roles in fatty acid metabolism and energy homeostasis. Therefore, functional and physical interaction of lipid droplets with mitochondria is of special importance as essential processes, such as lipolysis, triacylglycerol synthesis, thermogenesis or the protection against oxidative stress, and lipotoxicity, depend on cooperation of these two organelles. Physical interaction of LDs with mitochondria is mediated by specific molecular complexes at inter-organelle membrane contact sites. Substantial progress has been achieved during the last decade in understanding the formation and the structural components of lipid droplet–mitochondria contact sites. This review gives a brief overview of the different molecular complexes that have been identified in different mammalian cell types under different conditions and their regulation.
1. Introduction
Lipid droplets (LDs) are the eukaryotic storage compartments for neutral lipids, such as triacylglycerol (TAG), cholesterol ester (CE), fat-soluble vitamins, and lipophilic toxicants [1,2,3]. LDs are, however, not “passive” containers of lipids but have important regulatory roles such as sequestering free fatty acids (to combat lipotoxicity) or the controlled release of lipids as fuel for oxidative metabolism, as substrate for membrane lipids, or as (precursors of) signaling molecules [4]. Mitochondria are the organelles that enable eukaryotes to synthesize ATP through oxidative phosphorylation (OXPHOS), which is carried out through the Krebs cycle (tricarboxylic acid cycle) and using respiratory (electron transport) chain and ATP synthase, which is the predominant source of ATP in most cells. Besides their essential role in substrate oxidation and ATP synthesis, mitochondria fulfill various other important functions, such as thermogenesis (in brown adipocytes) and regulation of the intrinsic apoptotic pathway, and they are involved in many biosynthetic pathways (for recent reviews see, e.g., [5,6,7]). The functional interaction of LDs with mitochondria is particularly important in TAG and fatty acid metabolism (Figure 1A). The preferred fuel for OXPHOS in many cells is fatty acids (especially at low glucose). They are delivered as free fatty acids bound to albumin and are released from lipoproteins by lipoprotein lipase or from cytoplasmic LDs. In the latter case, a close proximity of LDs and mitochondria is expected to enable an efficient FA transfer. In return, TAG synthesis during LD formation or LD growth requires ATP, which is usually supplied by mitochondria. This may again be improved by close contacts between LDs and mitochondria. Because of their oxidative metabolism, mitochondria are also the major site of reactive oxygen species (ROS) generation through incomplete reduction of oxygen, forming superoxide anions and peroxide [8]. LDs can protect against oxidative stress by sequestering polyunsaturated FAs (PUFAs) to prevent their peroxidation or trapping peroxidation products [9].
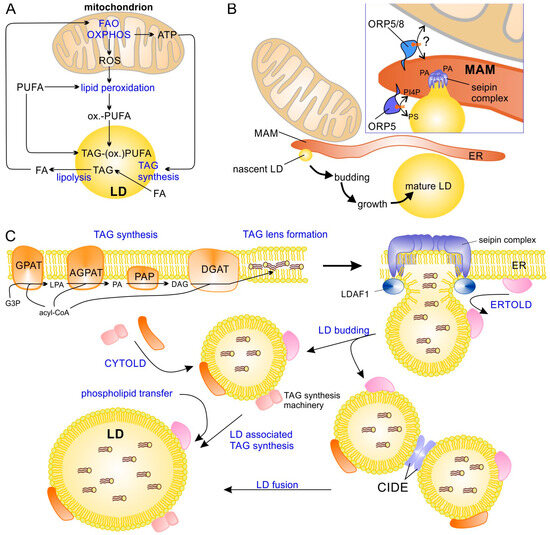
Figure 1.
LD–mitochondrial interaction and the formation of LDs. (A) Important processes that functionally link LD and mitochondria. (B) LDs are formed at mitochondrial-associated membranes (MAM) of endoplasmic reticulum (ER). The phospholipid transfer proteins ORP-5 and ORP-8 regulate LD formation by recruiting seipin oligomers to phosphatidic acid (PA)-enriched domains in MAMs. Additionally, ORP5 is required for phosphatidylinositol 4-phosphate (PI4P) and phosphatidylserine (PS) transfer/exchange between ER and the nascent LD. (C) Formation of new LDs at the ER starts with TAG synthesis, forming a TAG lens within the ER membrane and causing assembly of a seipin–LDAF1 complex that drives the formation of a new LD bud. After budding, LDs continue to grow through local TAG synthesis (at the LD surface) or through fusion. AGPAT, acyl-glycerol-3-phosphate acyltransferase; CIDE, cell death-inducing DNA fragmentation factor α-like effector; CYTOLD, cytosol-to-LD targeting (or transport pathway); DAG, diacylglycerol; DGAT, diacylglycerol acyltransferase; ER, endoplasmic reticulum; ERTOLD, ER-to-LD targeting (or transport pathway); FA, fatty acid; FAO, fatty acid β-oxidation; G3P, glycerol-3-phosphate; GPAT, glycerol-3-phosphate acyltransferase; LD, lipid droplet; LDAF1, lipid droplet assembly factor 1; LPA, lysophosphatidic acid; MAM, mitochondrial associated (ER) membrane; ORP5/8, oxysterol-binding protein-related protein family member 5/8; OXPHOS, oxidative phosphorylation; PAP, phosphatidic acid phosphatase; PUFA, polyunsaturated fatty acid; ox.-PUFA, oxidized PUFA; ROS, reactive oxygen species; TAG, triacylglycerol; TAG-(ox.)PUFA, TAG containing PUFA and/or oxidized PUFA.
Since the main topic of this article is the molecular interactions at LD–mitochondria membrane contact sites, focusing on molecular complexes that have been identified most recently, the reader is referred to several previously published reviews on LD–mitochondria interactions that discuss other aspects in more detail and in a comprehensive manner [10,11,12,13,14,15,16,17,18,19,20,21].
2. Biogenesis of Lipid Droplets
LDs differ from other organelles by being surrounded by a single phospholipid monolayer instead of a bilayer, and are extremely heterogeneous with respect to their size, ranging in diameter from about 30 nm to more than 100 µm (in univacuolar white adipose tissue) [22,23,24]. LDs not only differ with respect to their size but also their lipid composition and associated proteins; these differences can be found not only between different cell types and tissues but also within the same cell [25,26,27,28] and are strongly influenced by cellular/energy stress, metabolic demands, starvation, or infections [29,30,31]. LDs are formed at the ER in most cell types (Figure 1B), apparently in the immediate vicinity of mitochondria-associated ER membranes (MAMs) [32]. Therefore, the ER membrane protein seipin is recruited by the oxysterol binding protein (OSBP)-related proteins 5 and 8 (ORP5 and ORP8) to MAMs, and the lipid droplet assembly factor 1 (LDAF1) forms oligomers that drive the formation of LDs by stimulating TAG accumulation between the two monolayers of the ER membrane and finally budding off nascent LDs [3,32,33,34,35,36,37,38] (Figure 1C). After budding, LDs can further grow through homotypic fusion, which requires the cell death-inducing DFF45-like effector (CIDE) proteins [39], or through local TAG synthesis, as at least a subset of LDs contain enzymes of TAG biosynthesis [40,41]. Two major pathways are available to transfer new LD proteins to their destination [2,42]: (a) proteins are translated at cytosolic ribosomes and then transferred to LDs, e.g., perilipins (with the notable exception of perilipin 1 (PLIN1)) (so called CYTOLD targeting) or (b) they are translated at the rough ER and transferred to the LDs (ERTOLD targeting, seipin-dependent) at an early phase of LD budding (when a membrane bridge between ER and LDs is still present). There is also a late ERTOLD targeting that proceeds independent of seipin [2].
Degradation of lipids/TAGs stored in LDs can occur via two main routes (Figure 2): (a) lipolysis by LD-associated adipocyte triacylglycerol lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL) or (b) autophagy (lipophagy) through the formation of autophagosomes [43,44]. It is also possible that the two processes occur in succession: first partial lipolysis and then lipophagy of the residual LDs. Different pathways are involved in the regulation of LD metabolism. These include regulation of LD–mitochondrial contact formation and activity as discussed in the following. LD metabolism is also regulated by selective degradation of LD proteins (e.g., perilipins) through LAMP2A-dependent chaperone-mediated autophagy (CMA), which allows for regulation of lipolysis [45]. Starvation induces CMA-mediated degradation of PLIN2 and PLIN3, which, in concert with upregulation of ATGL and activation of macrolipophagy, stimulates cytosolic and lysosomal lipolysis [45].
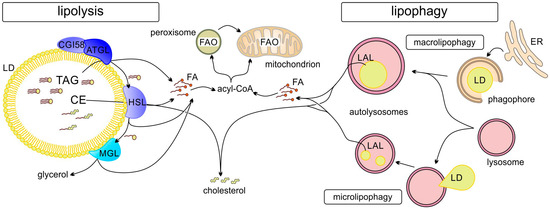
Figure 2.
Degradation of LDs can occur by lipolysis or lipophagy. Ester bonds of lipids (in TAGs or CEs) stored in LDs are hydrolyzed by lipases (ATGL, adipocyte triglyceride lipase; HSL, hormone sensitive lipase; MGL, monoacylglycerol lipase) attached to cytosolic LDs (lipolysis) or after autophagy by lysosomal acid lipase (LAL). FAs are activated by acyl-CoA synthetases and can be subjected to fatty acid oxidation (FAO) in mitochondria, or, if long chain (>C22), in peroxisomes (and after shortening are transferred to mitochondria for further β-oxidation).
3. Mitochondrial Heterogeneity and LD-Associated Mitochondria
At the morphological level, mitochondria differ in their size (regulated by fusion and fission), cristae structure, and contacts to other organelles, all of which are dynamically regulated [46,47,48]. This mitochondrial heterogeneity can be observed when different tissues or cell types are compared, though it can also be observed within a given cell [49,50]. Structural heterogeneity and differential subcellular localization of mitochondria indicate differences at the functional level. In white adipose tissue (WAT), mitochondria fuse to form thin and elongated mitochondria, while, in brown adipose tissue (BAT), mitochondria are fragmented and oval-shaped with a larger number of cristae [51]; low temperatures induce cristae formation in BAT mitochondria [52]. Mitochondria from hepatocytes are typically more rounded and short [53]. In skeletal muscle, morphologically distinct mitochondrial populations can be distinguished depending on their subcellular localization: intermyofibrillar mitochondria (IFM) are elongated (and branched), whereas subsarcolemmal mitochondria (SSM) are shorter and round-shaped [54,55]. IFM have higher electron transport chain complex I, II, and IV and ATP synthase activity when compared to SSM [54]. In addition to the variability in the overall morphology of mitochondria, their cristae structures can differ considerably. In cardiomyocytes, SSM contain mainly lamelliform cristae, whereas the majority of IFM are tubular [56].
Evidence for physical association of mitochondria with LDs has been provided by electron microscopy of skeletal muscle, BAT cardiomyocytes, or tumor cells, e.g., lung adenocarcinoma [57,58,59,60,61], and by co-fractionation of mitochondria with LDs upon density gradient centrifugation [62,63,64]. Mitochondria from BAT have been separated into cytosolic mitochondria (CM) and peridroplet mitochondria (PDM), depending on their association with LDs after low-speed centrifugation [62]. The functional differences of these two types of LDs are underscored by significant differences of their proteomes [65]. Cui et al. [63] distinguished two types of LD–mitochondria associations in brown adipocytes, depending on their stable or dynamic contact with the LDs. Mitochondria in the former complex are resistant to ultracentrifugation and have been named LD-anchored mitochondria (LDAM). Although the term PDM initially referred to mitochondria isolated from the LD (fat layer) fraction after density centrifugation, it is currently also used for mitochondria in close contact with LDs as revealed by electron microscopy [66], where it may not be obvious whether the two organelles form stable of dynamic contacts. It should also be noted that mitochondria linked to single small LDs can be present in the CM fraction [67].
PDM from BAT and liver are usually larger and have higher Krebs cycle activity and oxidative phosphorylation rates (higher I+III super complex activity, cytochrome c oxidase, and ATP synthase activity) compared to CM [62,68]. In contrast, FA oxidation (FAO) is lower in PDM compared to CM in BAT [62,69]. This association between mitochondria fragmentation and a higher FAO rate is corroborated by the observation that stimulating mitochondria fusion through mitofusin 2 (MFN2) overexpression reduces FAO, whereas increased fragmentation of mitochondria (MFN2-knock out) increases FAO of long-chain acyl-CoA, i.e., acyl-CoAs that are imported and carnitine-dependent [70]. In line with this, mitochondria fragmentation results in lower sensitivity towards malonyl-CoA [70], which may be explained by the fact that increased membrane curvature reduces binding of malonyl-CoA to (and inhibition of) carnitine palmitoyltransferase 1 (CPT1) [71].
In contrast to PDM from BAT, hepatocyte LD-associated mitochondria/PDM (isolated from mouse liver) exhibit high FAO activity (two- to three-fold higher compared to CM), because of their higher level of phosphorylated (inactive) acetyl-CoA carboxylase 2 (ACC2) and higher CPT1 activity. In contrast, CM have a much higher oxidative capacity and ATP levels with increased activities of super complexes I+III and II+III [64]. Importantly, the FAO capacity was impaired in PDM isolated from a non-alcoholic fatty liver disease rat model [64]. Thus, whether LD–mitochondria contacts foster lipolysis (by facilitating FA transfer) or lipogenesis (e.g., by optimizing ATP supply) depends on the tissue/cell type and metabolic situation (Table 1). These differences in metabolic interaction depend on the specific protein interaction at the LD surface and changes in the composition and structure of the MCS with mitochondria (but also other organelles, such as ER and peroxisomes).

Table 1.
Conditions affecting LD–mitochondria contacts in different tissues and cell lines and their effect on lipolysis and FAO.
4. LD–Mitochondria MCS and Their Role in FA Metabolism
Inter-organelle MCS are structures between different (or the same type of) organelles in close proximity, i.e., at a distance below 30 nm [80], with a specific set of proteins mediating this interaction. According to a proposed scheme, these proteins can be classified into four groups [81]: (a) tethering proteins or spacers (“structural proteins”); (b) proteins mediating the physiological function of the MCS, like lipid transfer or ion flux (“functional proteins”); (c) proteins organizing the MCS by recruiting or repelling specific proteins to/from the MCS (“sorter/recruitment proteins”); and (d) proteins that regulate activity of functional proteins in the MCS, e.g., through phosphorylation (“regulator proteins”). These functional classes are not mutually exclusive and most proteins in LD–mitochondrial MCS appear to be members of more than one of these functional groups. Several protein–protein interactions between LD- and mitochondria-localized proteins at the MCS have been identified in recent years (Table 2). Different methods are used to monitor physical interactions of organelles or those in close proximity to them (the two are not always clearly distinguished), such as high-resolution fluorescence microscopy, electron microscopy (including cryo-EM), fluorescence resonance energy transfer (FRET), bimolecular fluorescence complementation, dimerization-dependent fluorescent proteins, or proximity biotinylation (for review see [82,83]). Mass spectrometric identification of co-purified proteins or proximity-dependently biotinylated proteins using BioID, split-BioID, or related approaches has been used to identify new proteins at membrane contact sites. Methods that have been used specifically to study LD–mitochondria interaction include mass spectrometry of co-precipitated proteins [74,84,85], screening of candidate genes using short hairpin RNA mediated knock-down [86], and proximity-proteomics using split BioID [87,88].
Table 2.
Protein–protein interactions identified in LD–mitochondria MCS.
Miner at al. [84,94] introduced contact-fluorescence probes (contact-FP) the exhibit dimerization-dependent fluorescence and fused them to LD and mitochondrial targeting sequences, respectively. This approach allows qualitative and quantitative examination of LD–mitochondria contact formation in living cells. At higher expression levels, these probes can also induce membrane contact formation [94], which indicates that careful adjustment of expression levels are needed; but they may also be very useful tool to study organelle interactions.
For the regulation of FA and TAG metabolism, LD–mitochondria contacts are obviously important and are implicated in both lipolysis and lipogenesis. Contacts may improve FA transfer to mitochondria but can also improve the ATP supply for LD-associated acyl-CoA synthetases to drive TAG synthesis, which is also important to reduce lipotoxicity. Starvation of cells results in a strong stimulation of FA transfer from LDs to mitochondria (as shown, e.g., using fluorescently labelled fatty acid derivatives), accompanied by an increase in the number of contacts between LDs and mitochondria [76,95], which suggests that these contacts are required for efficient FA transfer. Interaction of LDs and mitochondria is additionally influenced by motility of the organelles. During starvation, LDs and mitochondria move towards the cell periphery (along de-tyrosinylated microtubules) and the LDs become more dispersed, which improves their interaction with mitochondria and increases the FAO in mitochondria [95].
4.1. LD–Mitochondria MCS with Involvement of Perilipins
Perilipins (PLIN1 to PLIN5 in mammals) are abundant LD proteins that play essential roles in the regulation of LD metabolism and also, in part, LD formation [10,96,97,98,99] (Table 3). Therefore, perilipins are also obvious candidates to mediate tethering of other organelles to LDs. This hypothesis has been confirmed for at least some of them (PLIN1, PLIN2, PLIN5). Perilipins are not only tethering proteins but also usually also have a role as “sorter/recruitment proteins” and/or “functional proteins”. PLIN5 is highly expressed in tissues with high oxidative capacity, such as BAT, skeletal, heart muscle, and liver [100]. In skeletal muscle, there is a clear positive correlation between the PLIN5 expression level and oxidative capacity [72,101]. A specific role of PLIN5 in LD–mitochondria contacts was suggested by the observation that PLIN5-mediated LD–mitochondria contacts depend on its carboxyl-terminal domain that is missing in other perilipins [59,102]. PLIN5 interacts with fatty acid transport protein 4 (FATP4; solute carrier family 27 member 4, SLC27A4) (Figure 3A) that is localized to the outer mitochondrial membrane (OMM) [84]; this protein is an acyl-CoA synthetase (also named acyl-CoA synthetase long chain family member 4, ACSVL4). In addition, PLIN5 binds mitochondrial MFN2; however, only binding to FATP4 depends on the unique carboxyl-terminal domain of PLIN5 [84]. PLIN5 overexpression causes significant translocation of mitochondria to the LD surface [59,66] and reduces lipolysis and FAO in skeletal muscle cells [59,72,101]. This decreased FAO activity of PDM in highly oxidative tissues expressing higher levels of PLIN5 suggests that, here, PLIN5 is primarily required to reduce oxidative stress/lipotoxicity by helping to sequester FAs in LDs [96]. However, when lipolysis is activated (though β-adrenergic signaling), PLIN5 overexpression stimulates FAO [101] (Figure 3A). This switch from the “lipolytic barrier” function of PLIN5 to FAO stimulation is regulated by PKA-dependent phosphorylation of PLIN5 at serine-155 [84,102,103]. This phosphorylation of PLIN5 changes its interaction with ATGL and, as a consequence, α/β-hydrolase domain-containing protein 5 (ABHD5/CGI-58) can efficiently activate ATGL, stimulating lipolysis and FA transfer. Furthermore, PKA-phosphorylated PLIN5 binds monounsaturated FAs (MUFAs) and mediates their transfer to the nucleus, where MUFAs act as allosteric activators of sirtuin 1 (SIRT1) to promote peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α)-/peroxisome proliferator-activated receptor α (PPAR-α)-dependent expression of oxidative metabolism genes [104,105]. Expression of PLIN5 itself is stimulated by the transcription factor PPARα [100].
Table 3.
Family of mammalian perilipins (PLIN1-5). Perilipins have various functions and their activity is regulated by phosphorylation through different protein kinases.
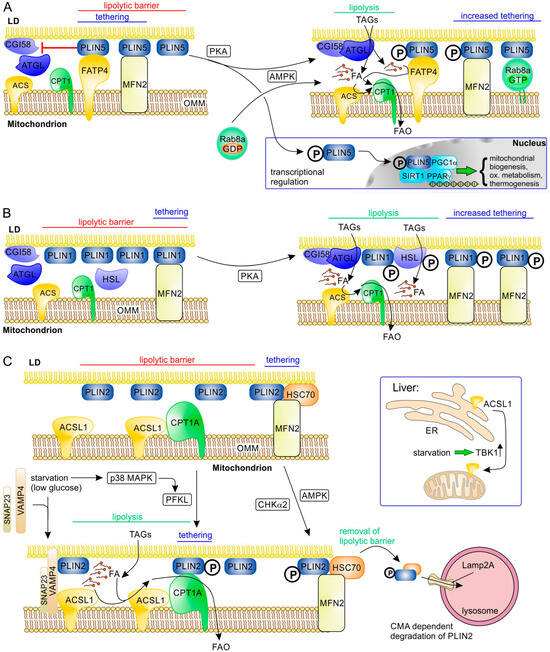
Figure 3.
Molecular interactions at LD–mitochondrial contact sites involving perilipins and their regulation during stimulation of lipolysis. A general primary tethering factor in the OMM that binds all perilipins is the mitochondrial protein mitofusin-2 (MFN2). (A) The acyl-CoA synthetase ACSVL4 (FATP4) acts as an additional tethering factor, binding PLIN5, an abundant perilipin in highly oxidative tissues such as BAT and (skeletal and heart) muscle. PLIN5 forms a lipolytic barrier by inhibiting the interaction of ATGL with its activator CGI58 (ABHD5); this activity of PLIN5 is blocked upon phosphorylation by PKA or AMPK (starvation and energy stress), while its tethering function remains. Thus, lipolysis and efficient FA transfer are enabled. In addition, AMPK-dependent activation of RAB8A leads to its binding to the OMM, where it acts as a binding partner of PLIN5. Phosphorylated PLIN5 can also act as a transcriptional regulator, stimulating expression of genes involved in biogenesis of mitochondria, oxidative metabolism, and thermogenesis in BAT (e.g., UCP1). (B) PLIN1 is an important tethering factor and lipolytic barrier in adipose tissue through its binding to MFN2. PKA-dependent phosphorylation of PLIN1 blocks its lipolytic barrier function and increases its binding to MFN2. (C) The function of the ubiquitously expressed PLIN2 is regulated via different pathways. Phosphorylation by p38 MAP kinase (MAPK) under nutrient starvation causes a functional shift of liver-type phosphofructokinase (PFKL) that becomes a PLIN2 kinase; phosphorylated PLIN2 binds CPT1A in the OMM, improving tethering and FA transfer to mitochondria. In addition, low glucose induces complex formation of the SNARE proteins VAMP4 and SNAP23 with PLIN2 at LDs; this complex can associate with ACSL1 in the OMM and may therefore improve binding of the two organelles, as well as likely making activation of released FA by ACSL1 more efficient. While ACSL1 is constitutively associated with OMM in tissues such as adipose tissues, skeletal tissues, and heart muscle, it is relocalized by upregulated TBK1 from ER to mitochondria upon starvation in liver. Activation of AMPK or CHKα2 under energy stress and starvation leads to phosphorylation of PLIN2. As a consequence, the lipolytic barrier function of PLIN2 is reduced by its HSC70-dependent removal from LDs and chaperone-mediated autophagic degradation in lysosomes (this also applies to PLIN3).
The PLIN5 interaction partner MFN2 [84] is a dynamin-like GTPase (although with only low GTPase activity), localized to the OMM as well as the ER, and plays important roles in mitochondrial fusion (together with the related mitofusin 1 (MFN1)), ER-mitochondrial contacts, and mitophagy [110,111,112,113,114,115]. Lack of MFN2 in BAT results in an increased number of LDs and impaired function of mitochondria [84]. Increasing the MFN2 levels in hepatocytes increased the number of PDM (with higher CPT1 and FAO capacity compared to CM) together with stimulation of mitochondria fusion [116]. Whether the larger size of PDM, when compared to CM, is only the consequence of the dual function of MFN2 as a mediator of mitochondrial fusion and as a tethering factor for LDs is currently not clear.
Interaction of PLIN5 with MFN2 does not depend on its unique carboxyl-terminal domain; it was therefore not unexpected that (in BAT) MFN2 can also bind to PLIN1 (Figure 3B). PLIN1 is the characteristic perilipin of adipocytes (WAT and BAT) (Table 3) and is an integral membrane protein, incorporated during LD formation, in contrast to the other perilipins that follow the CYTOLD route [28]. Interaction of PLIN1 with MFN2 is strongly increased upon β-adrenergic signaling in brown adipocytes [60]. PKA-dependent phosphorylation of PLIN1 is well known to enable the release of CGI-58, the activator of ATGL, as well as docking of hormone-sensitive lipase (HSL), thereby stimulating lipolysis [117,118,119].
In addition to FATP4/ACSL4, other acyl-CoA synthetases may be involved in LD–mitochondrial contacts as well. In hepatocytes, acyl-CoA synthetase long-chain family member 1 (ACSL1) was shown to interact with synaptosome-associated protein 23 (SNAP23) and vesicle-associated membrane protein 4 (VAMP4) on LDs upon glucose deprivation [87] (Figure 3C). Localization of SNAP23 to LDs is mediated by its binding partner PLIN2 [120]. Because SNAP23 is also a component of the SNARE complex mediating GLUT glucose transporter exocytosis at the plasma membrane, increased PLIN2 levels may redistribute SNAP23 to LDs, reducing glucose uptake [121], and may thus contribute to the metabolic switch from glucose oxidation to FAO under starvation. It is, however, not clear whether PLIN2, SNAP23, VAMP4, and mitochondrial ACSL1 form a tethering complex in addition to forming a functional complex that facilitates FA import. Upon fasting, FAO in hepatocytes is stimulated by upregulation of ACSL1 expression and a shift of its subcellular localization from ER to mitochondria [122]. This is regulated by the TANK-binding kinase 1 (TBK1), which has reduced kinase activity during fasting and is responsible (in its inactive form) for moving ACSL1 to the OMM. Here, ACSL1 also interacts with CPT1 [87], possibly enabling efficient acyl-CoA synthesis and mitochondrial FA import. Obesity increases activity of TBK1 kinase through phosphorylation, which reduces its affinity for ACSL1, causing its relocalization to the ER and thus reducing mitochondrial FAO and increasing TAG synthesis [122]. In highly oxidative non-hepatic tissues (skeletal muscle, heart, BAT, and WAT), ACSL1 is mainly localized to the OMM and its localization is regulated by other mechanisms, e.g., in BAT, sortilin mediates its redistribution to lysosomal degradation, reducing FAO and thermogenesis [123].
Metabolic adaptation under starvation that directly affects LD–mitochondrial contacts can also be regulated by other mechanisms. In liver, fasting activates p38 mitogen-activated protein kinase (MAPK) that phosphorylates liver-type phosphofructokinase (PFKL) (Figure 3C). This inhibits its activity towards fructose-6-phosphate and instead favors its association with LD, where it acts as a protein kinase that phosphorylates PLIN2 (at serine 159) [90]. Phosphorylated PLIN2 binds to CPT1, and this interaction correlates with a significant increase in LD-bound mitochondria and stimulates lipolysis and FAO [90]. In human hepatocellular carcinoma cells, higher levels of phosphorylated PLIN2 correlate with increased malignancy [90].
Cardiomyocytes in mice on a high fat diet accumulate TAGs in LDs and reduce FA transfer to mitochondria, which correlates with decreased MFN2 levels and less LD–mitochondria contacts [74]. This phenotype can be rescued by MFN2 overexpression. Because PLIN1 is undetectable in heart [74], in contrast to BAT, an alternative interacting partner is required, which was identified as heat shock protein family A member 8 (HSPA8/HSC70) [90] (Figure 3C). Possibly, HSC70 is bound to LDs through its interaction with PLIN2 or PLIN3, both of which are expressed in heart muscle [78] and are known to bind HSC70 [124]. In line with its proposed role, HSPA8/HSC70 deficiency results in fewer LD–mitochondrial contacts together with lipid accumulation in LDs and less dispersed LDs [110]. PLIN2 (or PLIN3) in a complex with HSC70 can be (serine) phosphorylated by AMPK [45] and (tyrosine) phosphorylated by choline kinase α2 (CHKα2) [107], which both stimulate lipolysis by activating degradation of PLIN2 (also PLIN3) through lysosome-associated membrane protein 2A (LAMP2A)-dependent chaperone-mediated autophagy (CMA) [45,107].
4.2. LD–Mitochondria MCS Apparently Independent of Perilipins
Additional factors besides perilipins and their interaction partners may be involved in tethering and efficient FA transfer from LDs to mitochondria under nutrient deprivation. A possible candidate in this context is acyl-CoA:diacylglycerol acyltransferase 2 (DGAT2), which synthesizes TAGs at the ER. Upon induction of LD formation by oleic acid, DGAT2 partially localizes to mitochondria that are at MAMs or close to LDs [125]; however, a LD-specific binding partner has not been identified. RAB1B GTPase is able to relocalize DGAT2 from ER to LD [126]. Because DGAT2 forms homodimers [127], it was proposed that this dimerization could recruit mitochondria to LDs [62]; but whether dimerization can occur between DGAT2 enzymes in two opposing membranes is not known. However, LD-localized DGAT2 is also able to interact with FATP1/ACSVL5 (fatty acid transporter member 1, SLC27A1, acyl-CoA synthetase very long chain family member 5) in the ER [128]. Because FATP1 can also localize to mitochondria [129], it could potentially be an interaction partner of LD-associated DGAT2 at LD–mitochondria MCS.
Proteins of the vacuolar protein sorting 13 (VPS13) family are phosphoglycerolipid transfer proteins that act at MCS between different organelles [130,131]. The VPS13 homolog D (VPS13D) has been found at LD–mitochondria MCS, where it is involved in lipid transfer between the two organelles [92]. VPS13D binds via its C-terminal two amphipathic helices to LDs and an N-terminal domain (residues 404–913) is required for its interaction with mitochondria [92]. VPS13 proteins have rod-like structures that link opposing membranes and mediate lipid movement between them through a hydrophobic cavity [131]. Association of VPS13D with LD–mitochondria contact sites increased significantly upon starvation and stimulates FA transfer to mitochondria. At the LD surface, VPS13D interacts with the ESCRT-I protein TSG101 via its VPS13 adapter-binding (VAB) domain, which induces protrusions from the LDs towards mitochondria (Figure 4A). The corresponding increase in the surface/volume ratio may thereby promote lipolysis; in parallel, it may also facilitate the extraction of phospholipids from the LD monolayer. VPS13D may be recruited to the OMM by mitochondrial Rho GTPase (MIRO), which is known to recruit VPS13D to ER–mitochondria contact sites [93]. Because MIRO is also part of the MICOS complex that regulates cristae formation [132,133], it may be possible that the LD protrusions are very close to the mitochondrial cristae, which could further facilitate FA transfer and thus stimulate FAO. Similarly to this role of ESCRT-I proteins, it has been found that the ESCRT-III proteins IST1 and CHMP1B play a role in FA transfer from LDs to peroxisomes by forming protrusions from the LDs [134]. Another VPS13 family member, VPS13A, can also bind to LDs and mitochondria [133]. VPS13A deficiency results in an elevated number and reduced motility of LDs [135]. However, in contrast to VPS13D, binding sites for LDs and mitochondria are both found at the carboxyl-terminus and overlap. Thus, in contrast to VPS13D-dependent contacts, a direct linkage of LD and mitochondria by the same VPS13A may not be possible.
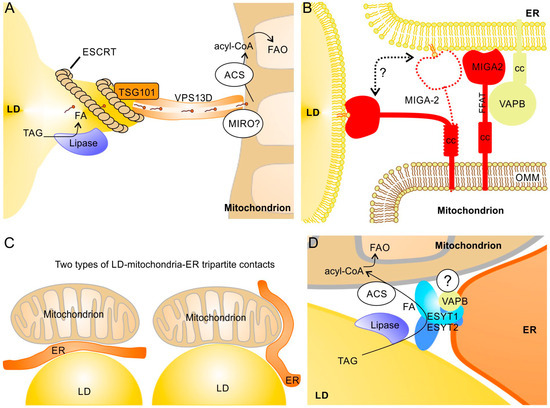
Figure 4.
Protein complexes at LD–mitochondria contact sites that apparently do not depend on perilipins and possible LD–mitochondria–ER tripartite structures. (A) Under starvation, protrusions of the LD are formed by ESCRT-I proteins and linked via TSG101 to VPS13D. The hydrophobic cavity of VPS13D most likely enables efficient FA transfer from LDs to mitochondria. The mitochondrial GTPase MIRO may recruit VPS13D to the OMM [120,121]. (B) The mitochondrial membrane protein MIGA2 can interact with both ER and LDs. Binding to LDs is most likely mediated by direct contact of the lipid binding domain of MIGA2 with the phospholipid monolayer (whereas interaction with the ER depends on VAPB). In addition to its tethering function, MIGA2 may also transfer phospholipids between LDs and ER [35]. cc, coiled coil domain; FFAT, ER membrane targeting motif. (C) In principle, two types of tripartite structures are possible: ER sandwiched between LDs and mitochondria (left) and ER surrounding LDs and mitochondria that are in contact (right). (D) A complex of ESYT1, ESYT2, and VAPB, possibly together with additional binding partners, may exist at tripartite contacts and facilitate FA transfer. ESYT1 and ESYT2 form heterodimers attached to VAPB in the ER membrane and also bind to LDs and mitochondria; a further (currently unknown) binding partner at the OMM has been proposed [81]. Whether lipases and acyl-CoA synthetases (ACS) are also present in this complex is not known.
In white adipocytes, mitoguardin-2 (MIGA2) is another transmembrane protein of the OMM that is involved in LD–mitochondrial contacts [91] (Figure 4B). MIGA2 binds with its central phosphorylated FFAT (diphenylalanine in an acidic tract) motif to VAPA or VAPB in the ER [91] and stimulates transfer of phosphoglycerolipids, with a preference for phosphatidylserine [35]. In contrast to VAPA/B proteins in the ER, LD proteins have not yet been identified as a MIGA2 binding partner; it was proposed that MIGA2 interacts with the phospholipid monolayer of LDs via its amphiphilic C-terminal helices [91]. MIGA2 is an important factor in adipocyte differentiation and promotes de novo lipogenesis in adipocytes and TAG storage in LDs. Because MIGA2 can transfer free FAs besides phosphoglycerolipids [136], its lipogenic activity may involve transfer of FA to LDs (to serve as substrates for LD-associated acyl-CoA synthetases) as well as transfer of phospholipids to increase the surface of LDs. Accordingly, MIGA2-deficient cells have smaller and reduced numbers of LDs and MIGA2-deficient mice exhibit reduced body fat [91]. To what extent MIGA2 is required for tethering mitochondria to LDs and their functional interaction in non-adipose tissues are unclear at present.
4.3. Tripartite Contacts Between LD, Mitochondria and ER
Tripartite contacts between LDs, MT, and ER (or MAM–LD contact sites) [11,32,137] can be divided into: (a) Mito–ER–LD structures where ER is sandwiched between mitochondria and LDs and (b) ER–Mito–LD structures with a direct contact between MT and LDs with ER wrapped partially around the MT and LDs [65] (Figure 4C). The latter appears to be more common. The preferential generation of LDs at MAMs [32] suggests presence of such tripartite structures during LD formation. Tripartite contacts, however, may also be important in the context of FA transfer from LDs to mitochondria or TAG formation at LD–mitochondria contacts to promote the concurrent necessary phospholipid transfer to or from the ER.
ORP5 and ORP8 are present at ER–mitochondria contact sites and interact with the phospholipid transfer protein tyrosine phosphatase-interacting protein 51 (PTPIP51) in the OMM [138]. ORP5 regulates LD biogenesis by recruiting seipin to MAM–LD contact sites (see Figure 1B). Because ORP5 and ORP8 are known phosphoglycerolipid transfer proteins at ER–plasma membrane and ER–mitochondria contact sites [139,140,141,142], they may also enable transfer of phospholipids from the ER to LDs.
A reasonable candidate that may form tripartite contacts with mature LDs is the mitochondrial MIGA2 that can interact with both VAPA/B proteins in the ER and phospholipids of the LD membrane [93,132] (Figure 4B). In such contacts, MIGA2 could potentially transfer phospholipids between LDs and ER [35], which would be important for LD growth (and formation) as well as (in the opposite direction) during lipolysis. However, this hypothesis has not yet been tested experimentally. VPS13A can also link LDs as well as mitochondria to the ER [125] and could thus potentially also be a component of tripartite contacts. Calsyntenin-3β (CLSTN3β) links ER and LDs by simultaneously binding via a transmembrane helix to ER and via hydrophobic domains to LDs [143]. More recently, it was reported that, in the ketogenic metabolic state, CLSTN3β induced by PPARα in hepatocytes stimulates functional interaction of LDs with mitochondria (stimulation of lipolysis and FAO) [144]. However, if this is mediated through a tripartite contact is not yet clear.
Evidence for a tripartite contact where one protein complex interconnects all three organelles is rare. Such a specific structure is suggested by a recent study by Bezawork-Geleta et al. [88], which shows that, at the “triple” contact site between LDs, mitochondria, and ER, extended synaptotagmin 1 (ESYT1), ESYT2, and VAPB most likely form a heterotrimeric complex that facilitate the transfer of FAs from LDs to mitochondria (FAO was significantly reduced in ESYT1/ESYT2 and VAPB knock-out cells) (Figure 4D). This complex seems to be important to reduce lipotoxicity. However, LDs in proximity to mitochondria were not reduced in ESYT1-/ESYT2-deficient cells. The complex is thus dispensable for tethering but required for efficient FA transfer. Interestingly, in the absence of any of the three components (ESYT1, ESYT2, or VAPB) of this tripartite complex, a strong up-regulation of VPS13D, TSG101, and ESCRT-III was observed, which suggests a possible partial compensation by the above mentioned VPS13D-dependent FA transfer to mitochondria and/or the ESCRT-III-dependent FA transfer to peroxisomes [88].
5. LD–Mitochondria Interactions and Oxidative Stress
As a consequence of their metabolic function, mitochondria are the major source of reactive oxygen species (ROS) in eukaryotic cells [145]. Interaction of mitochondria with LDs plays an important role in combating ROS and oxidative stress (see Figure 1A). LDs help to mitigate ROS by sequestering excess lipids that might otherwise promote oxidative stress [15]. LDs can sequester polyunsaturated FAs (PUFA) to protect them from peroxidation reactions [146,147,148]. However, less is known about regulation of LD–mitochondria contact and specific roles of the different tethering factors in this context. The anti-oxidant role of LDs does not necessarily mean that physical contacts between LDs and mitochondria are required; functional interaction between LDs and mitochondria is even possible if they are not localized in the same cell, e.g., an increase in ROS in neurons causes transfer of peroxidized lipids to glial cells that store them in LDs and degrade these toxic lipids [149].
Oxidative stress increases the expression of PLIN5 in hepatocyte cell line HepG2 and this correlates with an increased number of LD–mitochondria contacts [150], in line with the known tethering function of PLIN5 [84]. However, through activation of the PI3K and PKB-GSK3β pathways, PLIN5 overexpression also leads to higher gene expression of anti-oxidant genes [150,151,152]. Thus, it is not clear to what extent the anti-oxidative function of PLIN5 is mediated by increasing contacts with mitochondria. In cardiomyocytes, PLIN5 overexpression increases TAG storage in LDs independent of its C-terminal domain, which is required for its interaction with mitochondria [153]. This may suggest that trapping of PUFA in LDs to reduce oxidative stress does not require PLIN5-dependent tethering of mitochondria.
6. LD–Mitochondria Contacts in Virus Replication and in the Presence of Toxins
LDs are involved in genome replication and/or virion morphogenesis of various viruses, which often requires functional interaction of LDs with the ER or other organelles [154,155,156,157,158,159]. A direct role of viral proteins in mediating LD–mitochondrial contacts was found for coronavirus SARS-CoV-2 [85]. In addition to its interaction with ER proteins BAP31 and USE1, the ORF6 protein of SARS-CoV-2 also inserts into the LD surface, where it also binds to ATGL, which abrogates PLIN2 binding to (and inhibition of) ATGL and, at the same time, promotes binding of CGI-58 to ATGL to stimulate lipolysis. In addition, ORF6 stimulates LD biogenesis (through its interaction with DGAT1 and DGAT2 at the ER) and links LDs to mitochondria, by binding to the SAMM50, MTX1, and MTX2 proteins of the SAM complex. This most likely facilitates FA transfer to mitochondria and FAO. Both LD biogenesis and ATP production through FAO appear to be important for efficient SARS-CoV-2 replication [85].
The mycotoxin aflatoxin B1 (AFB1) causes hepatic lipotoxicity and lipid accumulation in LDs [89,160,161]. AFB1 causes a relocalization of p53 to the OMM, where it mediates formation of LD–mitochondria contacts through binding to PLIN2 [161]. This reduces PLIN2-associated lipophagy.
7. Conclusions and Perspectives
Several protein complexes of LD–mitochondria MCS that are involved in the formation of contact sites and/or the functional interaction of the two organelles have been identified during the last decade. Apparently, perilipins at LDs and MFN2 in the OMM play a general role in tethering mitochondria to LDs in most (if not all) cells (though the contribution of the different perilipins depends on the specific cell type). In addition, protein complexes at LD–mitochondria contact sites that appear to be independent of perilipins have been identified more recently. It should be noted, however, that it is unclear whether the various protein complexes identified are separate structures (that may even be present within different membrane subdomains) or whether they interact and actually exist together in larger complexes (“supercomplexes”). Along this line, only few selected interacting proteins have been examined in detail, though reported mass spectrometry screenings usually list a large number of “candidate” proteins. It would be very informative to examine whether the other proteins are also present at MCS beside the “top hits” of the screenings. The complete structure of protein complexes at LD–mitochondria MCS may be examined using methods such as native gel electrophoresis, chromatography in combination with mass spectrometry (“complexome profiling” [162]), and chemical cross linking. Such “supercomplexes” could potentially enable highly efficient functional interactions between LDs and mitochondria. For example, on the one hand, the ESCRT/VPS13D complex facilitates FA-transfer from LDs to mitochondria and, on the other hand, perilipins mediate linkage of LDs to mitochondrial acyl-CoA synthetase and CPT1A (both important for the FA activation and import into mitochondria). Physical proximity of the two complexes (or the formation of a “supercomplex”) could likely enable efficient transfer of the FA into the mitochondrial matrix for FAO.
While the function and regulation of the LD–mitochondria MCS in lipolysis and FA-transfer to mitochondria have been examined in a number of studies, much less is known about the suggested and possible roles of these MCS (or tripartite contact sites that include the ER) in other processes, such as ATP-transfer from mitochondria to LDs for LD-associated TAG synthesis, the handling of oxidative stress, the exchange of phospholipid (during LD generation, lipogenesis, and lipolysis), or metabolic pathways not directly related to TAG metabolism. As an example of the latter, synthesis of steroid hormones starts in mitochondria using LD-derived cholesterol and is continued in the ER; LD–mitochondria contacts or tripartite contacts that include the ER could potentially play a functional role in this pathway as well.
Another topic to examine in the future in more detail is the molecular pathways involved in the dynamic regulation of LD–mitochondrial contacts that are currently not fully understood. Genetic approaches, e.g., genome-scale screening using Crispr/Cas9 libraries [163] in combination with “LD-mitochondrial interaction probes”, e.g., contact-FP [84], or proximity biotinylation approaches, such as contact-ID [164], are very useful to further explore this and identify regulatory factors, as well as new components of contact complexes. Such genetic screens have been used to identify genes involved in mitochondria or LD regulation (see e.g., references [165,166]), but have not yet been used for identification of genes involved in regulation of LD–mitochondria MCS. Moreover, integration of data about transcriptional regulation (transcriptomics), data about lipids in LDs (metabolomics/lipidomics), and analysis of the protein complexes (proteomics) at LD–mitochondria contact sites will be important to complete our picture of the structure, dynamics, and regulation of LD–mitochondria MCS.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ABHD5 | α/β-hydrolase domain-containing protein 5 |
| ACC2 | Acetyl-CoA carboxylase 2 |
| ACSL1 | Acyl-CoA synthetase long chain family member 1 |
| AMPK | AMP-dependent kinase |
| ATGL | Adipocyte triacylglcerol lipase |
| BAT | Brown adipose tissue |
| CE(s) | Cholesterol ester(s) |
| CGI-58 | Comparative gene identification 58 |
| CHKα2 | Choline kinase α2 |
| CM | Cytosolic mitochondria |
| CMA | Chaperone-mediated autophagy |
| CPT1/2 | Carnitine palmitoyltransferase 1/2 |
| ER | Endoplasmic reticulum |
| ESCRT-I/-III | Endosomal sorting complex required for transport-I/-III |
| ESYT1/2 | Extended synaptotagmin 1/2 |
| FA(s) | Fatty acid(s) |
| FAO | Fatty acid oxidation |
| FATP4 | Fatty acid transport protein 4 (ACSVL4; SLC27A4) |
| HSC70 | Heat shock cognate protein 70 |
| HSL | Hormone-sensitive lipase |
| HSPA8 | Heat shock protein family A member 8 |
| IFM | Intermyofibrillar mitochondria |
| IMM | Inner mitochondrial membrane |
| LAMP2A | Lysosome-associated membrane protein 2A |
| LD(s) | Lipid droplet(s) |
| MAM(s) | Mitochondria-associated ER membrane(s) |
| MCS(s) | Membrane contact site(s) |
| MIGA2 | Mitoguardin 2 |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| MGL | Monoacylglycerol lipase |
| MIGA2 | Mitogardin 2 |
| MIRO | Mitochondrial Rho GTPase |
| OMM | Outer mitochondrial membrane |
| ORF6 | Open reading frame 6 |
| ORP5/8 | OSBP-related protein 5/8 |
| OSBP | Oxysterol binding protein |
| OXPHOS | Oxidative phosphorylation |
| PDM | Peridroplet mitochondria |
| PFKL | Phosphofructokinase, liver type |
| PGC1α | Peroxisome proliferator-activated receptor γ coactivator 1α |
| PKA | Protein kinase A |
| PLIN1-5 | Perilipin 1 to 5 |
| PPARα | Peroxisome proliferator-activated receptor α |
| PUFA(s) | Polyunsaturated fatty acid(s) |
| ROS | Reactive oxygen species |
| SIRT1 | Sirtuin 1 |
| SNAP23 | Synaptosome-associated protein 23 |
| SSM | Subsarcolemmal mitochondria |
| StAR | Steroidogenic acute regulatory protein |
| TAG(s) | Triacylglycerol(s) |
| TBK1 | TANK-binding kinase 1 |
| TSG101 | Tumor susceptibility 101 |
| VAMP4 | Vesicle-associated membrane protein 4 |
| VAPA/B | VAMP-associated protein A/B |
| VDAC1/2 | Voltage-dependent anion channel 1/2 |
| VPS13 | Vacuolar protein sorting 13 |
| VPS13A/D | VPS13 homolog A/D |
| WAT | White adipose tissue |
References
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochem. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1260–1271. [Google Scholar] [CrossRef]
- Farese, R.V.; Walther, T.C. Essential biology of lipid droplets. Annu. Rev. Biochem. 2025, 94, 447–477. [Google Scholar] [CrossRef]
- Mathiowetz, A.J.; Olzmann, J.A. Lipid droplets and cellular lipid flux. Nat. Cell Biol. 2024, 26, 331–345. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. Lipid Droplets and the Management of Cellular Stress. Yale J. Biol. Med. 2019, 92, 435–452. [Google Scholar] [PubMed]
- Chen, L.; Zhou, M.; Li, H.; Liu, D.; Liao, P.; Zong, Y.; Zhang, C.; Zou, W.; Gao, J. Mitochondrial heterogeneity in diseases. Signal Transduct. Target Ther. 2023, 8, 311. [Google Scholar] [CrossRef]
- Granath-Panelo, M.; Kajimura, S. Mitochondrial heterogeneity and adaptations to cellular needs. Nat. Cell Biol. 2024, 26, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Nunnari, J. Mitochondria at the crossroads of health and disease. Cell 2024, 187, 2601–2627. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Griseti, E.; Bello, A.A.; Bieth, E.; Sabbagh, B.; Iacovoni, J.S.; Bigay, J.; Laurell, H.; Čopič, A. Molecular mechanisms of perilipin protein function in lipid droplet metabolism. FEBS Lett. 2024, 598, 1170–1198. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Giordano, F. Emerging functions of the mitochondria-ER- lipid droplet three-way junction in coordinating lipid transfer, metabolism, and storage in cells. FEBS Lett. 2024, 598, 1252–1273. [Google Scholar] [CrossRef]
- Enkler, L.; Spang, A. Functional interplay of lipid droplets and mitochondria. FEBS Lett. 2024, 598, 1235–1251. [Google Scholar] [CrossRef]
- Veliova, M.; Petcherski, A.; Liesa, M.; Shirihai, O.S. The biology of lipid droplet-bound mitochondria. Semin. Cell Dev. Biol. 2020, 108, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Renne, M.F.; Hariri, H. Lipid Droplet-Organelle Contact Sites as Hubs for Fatty Acid Metabolism, Trafficking, and Metabolic Channeling. Front. Cell Dev. Biol. 2021, 9, 726261. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W.; Xu, R.; Wang, Z.; Zhang, X.; Wang, P.; Peng, K.; Li, M.; Li, J.; Tan, Y.; et al. Plin5 Bidirectionally Regulates Lipid Metabolism in Oxidative Tissues. Oxidative Med. Cell Longev. 2022, 2022, 4594956. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.; Domingues, M.R.; Moreira, P.I.; Pereira, C.F. A Perspective on the Link between Mitochondria-Associated Membranes (MAMs) and Lipid Droplets Metabolism in Neurodegenerative Diseases. Biology 2023, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Tan, Y. Lipid Droplet-Mitochondria Contacts in Health and Disease. Int. J. Mol. Sci. 2024, 25, 6878. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, M.; Bai, R.; Chen, J.; Yang, H.; Luo, G. Molecular mechanisms of lipid droplets-mitochondria coupling in obesity and metabolic syndrome: Insights and pharmacological implications. Front. Physiol. 2024, 15, 1491815. [Google Scholar] [CrossRef]
- Litvinova, L.S.; Vulf, M.A.; Yurova, K.A.; Khaziakhmatova, O.G.; Malashchenko, V.; Bograya, M.; Kozlov, I.; Todosenko, N. Mitochondria and Lipid Droplets: Focus on the Molecular Structure of Contact Sites in the Pathogenesis of Metabolic Syndrome. Curr. Med. Chem. 2025, 32, 3006–3027. [Google Scholar] [CrossRef]
- Smolková, K.; Gotvaldová, K. Fatty Acid Trafficking Between Lipid Droplets and Mitochondria: An Emerging Perspective. Int. J. Biol. Sci. 2025, 21, 1863–1873. [Google Scholar] [CrossRef]
- Alonso-Bivou, M.; Pol, A.; Lo, H.P. Moving the fat: Emerging roles of rab GTPases in the regulation of lipid droplet contact sites. Curr. Opin. Cell Biol. 2025, 93, 102466. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cordes, K.R.; Farese, R.V., Jr.; Walther, T.C. Lipid droplets at a glance. J. Cell Sci. 2009, 122, 749–752. [Google Scholar] [CrossRef]
- Yang, H.; Galea, A.; Sytnyk, V.; Crossley, M. Controlling the size of lipid droplets: Lipid and protein factors. Curr. Opin. Cell Biol. 2012, 24, 509–516. [Google Scholar] [CrossRef]
- Kassan, A.; Herms, A.; Fernández-Vidal, A.; Bosch, M.; Schieber, N.L.; Reddy, B.J.; Fajardo, A.; Gelabert-Baldrich, M.; Tebar, F.; Enrich, C.; et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 2013, 203, 985–1001. [Google Scholar] [CrossRef]
- Brownstein, A.J.; Veliova, M.; Acin-Perez, R.; Villalobos, F.; Petcherski, A.; Tombolato, A.; Liesa, M.; Shirihai, O.S. Mitochondria isolated from lipid droplets of white adipose tissue reveal functional differences based on lipid droplet size. Life Sci. Alliance 2023, 7, e202301934. [Google Scholar] [CrossRef]
- Stiebing, C.; Matthäus, C.; Krafft, C.; Keller, A.A.; Weber, K.; Lorkowski, S.; Popp, J. Complexity of fatty acid distribution inside human macrophages on single cell level using Raman micro-spectroscopy. Anal. Bioanal. Chem. 2014, 406, 7037–7046. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Li, J.; Yu, J.; Pu, J.; Li, L.; Zhang, H.; Zhang, S.; Peng, G.; Yang, F.; et al. Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein a-I. J. Proteome Res. 2011, 10, 4757–4768. [Google Scholar] [CrossRef]
- Majchrzak, M.; Stojanović, O.; Ajjaji, D.; Ben M’barek, K.; Omrane, M.; Thiam, A.R.; Klemm, R.W. Perilipin membrane integration determines lipid droplet heterogeneity in differentiating adipocytes. Cell Rep. 2024, 43, 114093. [Google Scholar] [CrossRef] [PubMed]
- Sadh, K.; Rai, P.; Mallik, R. Feeding-fasting dependent recruitment of membrane microdomain proteins to lipid droplets purified from the liver. PLoS ONE 2017, 12, e0183022. [Google Scholar] [CrossRef] [PubMed]
- Kramer, D.A.; Quiroga, A.D.; Lian, J.; Fahlman, R.P.; Lehner, R. Fasting and refeeding induces changes in the mouse hepatic lipid droplet proteome. J. Proteom. 2018, 181, 213–224. [Google Scholar] [CrossRef]
- Ventura, A.E.; Pokorna, S.; Huhn, N.; Santos, T.C.B.; Prieto, M.; Futerman, A.H.; Silva, L.C. Cell lipid droplet heterogeneity and altered biophysical properties induced by cell stress and metabolic imbalance. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2023, 1868, 159347. [Google Scholar] [CrossRef]
- Guyard, V.; Monteiro-Cardoso, V.F.; Omrane, M.; Sauvanet, C.; Houcine, A.; Boulogne, C.; Ben Mbarek, K.; Vitale, N.; Faklaris, O.; El Khallouki, N.; et al. ORP5 and ORP8 orchestrate lipid droplet biogenesis and maintenance at ER-mitochondria contact sites. J. Cell Biol. 2022, 221, e202112107. [Google Scholar] [CrossRef]
- Chung, J.; Wu, X.; Lambert, T.J.; Lai, Z.W.; Walther, T.C.; Farese, R.V., Jr. LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev. Cell 2019, 51, 551–563.E7. [Google Scholar] [CrossRef]
- Arlt, H.; Sui, X.; Folger, B.; Adams, C.; Chen, X.; Remme, R.; Hamprecht, F.A.; DiMaio, F.; Liao, M.; Goodman, J.M.; et al. Seipin forms a flexible cage at lipid droplet formation sites. Nat. Struct. Mol. Biol. 2022, 29, 194–202. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.; Jun, Y.; Lee, C. Structural basis for mitoguardin-2 mediated lipid transport at ER-mitochondrial membrane contact sites. Nat. Commun. 2022, 13, 3702. [Google Scholar] [CrossRef]
- Klug, Y.A.; Ferreira, J.V.; Carvalho, P. A unifying mechanism for seipin-mediated lipid droplet formation. FEBS Lett. 2024, 598, 1116–1126. [Google Scholar] [CrossRef]
- Schneiter, R.; Choudhary, V. Seipin collaborates with the ER membrane to control the sites of lipid droplet formation. Curr. Opin. Cell Biol. 2022, 75, 102070. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Kim, S.; Arlt, H.; Voth, G.A.; Farese, R.V., Jr. Structure and function of lipid droplet assembly complexes. Curr. Opin. Struct. Biol. 2023, 80, 102606. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, L.; Wu, L.; Li, P.; Chen, F.J. CIDE proteins and their regulatory mechanisms in lipid droplet fusion and growth. FEBS Lett. 2024, 598, 1154–1169. [Google Scholar] [CrossRef]
- Wilfling, F.; Wang, H.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.X.; Graham, M.; Christiano, R.; Fröhlich, F.; et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Airola, M.V.; Reue, K. How lipid droplets “TAG” along: Glycerolipid synthetic enzymes and lipid storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Mizrak, A.; Lee, C.W.; Cicconet, M.; Lai, Z.W.; Tang, W.C.; Lu, C.H.; Mohr, S.E.; Farese, R.V., Jr.; Walther, T.C. Identification of two pathways mediating protein targeting from ER to lipid droplets. Nat. Cell Biol. 2022, 24, 1364–1377. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Reichert, A.S. Mitochondrial Dynamics at Different Levels: From Cristae Dynamics to Interorganellar Cross Talk. Annu. Rev. Biophys. 2024, 53, 147–168. [Google Scholar] [CrossRef]
- Moura, J.P.; Oliveira, P.J.; Urbano, A.M. Mitochondria: An overview of their origin, genome, architecture, and dynamics. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167803. [Google Scholar] [CrossRef]
- Huang, C.; Deng, K.; Wu, M. Mitochondrial cristae in health and disease. Int. J. Biol. Macromol. 2023, 235, 123755. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Troppmair, J.; Sucher, R.; Hermann, M.; Saks, V.; Margreiter, R. Mitochondrial subpopulations and heterogeneity revealed by confocal imaging: Possible physiological role? Biochim. Biophys. Acta 2006, 1757, 686–691. [Google Scholar] [CrossRef]
- Michurina, S.S.; Stafeev, I.S.; Menshikov, M.Y.; Parfyonova, Y.V. Mitochondrial dynamics keep balance of nutrient combustion in thermogenic adipocytes. Mitochondrion 2021, 59, 157–168. [Google Scholar] [CrossRef]
- Latorre-Muro, P.; O’Malley, K.E.; Bennett, C.F.; Perry, E.A.; Balsa, E.; Tavares, C.D.J.; Jedrychowski, M.; Gygi, S.P.; Puigserver, P. A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab. 2021, 33, 598–614.e7. [Google Scholar] [CrossRef]
- Das, S.; Hajnóczky, N.; Antony, A.N.; Csordás, G.; Gaspers, L.D.; Clemens, D.L.; Hoek, J.B.; Hajnóczky, G. Mitochondrial morphology and dynamics in hepatocytes from normal and ethanol-fed rats. Pflug. Arch.–Eur. J. Physiol. 2012, 464, 101–109. [Google Scholar] [CrossRef]
- Ferreira, R.; Vitorino, R.; Alves, R.M.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef]
- Bosma, M. Lipid droplet dynamics in skeletal muscle. Exp. Cell Res. 2016, 340, 180–186. [Google Scholar] [CrossRef]
- Riva, A.; Tandler, B.; Loffredo, F.; Vazquez, E.; Hoppel, C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am. J. Physiol.-Heart Circ. Physiol. 2005, 289, H868–H872. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A.; Rennie, C.D.; Robertshaw, H.A.; Fedak-Tarnopolsky, S.N.; Devries, M.C.; Hamadeh, M.J. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1271–R1278. [Google Scholar] [CrossRef]
- Mirza, A.H.; Cui, L.; Zhang, S.; Liu, P. Comparative proteomics reveals that lipid droplet-anchored mitochondria are more sensitive to cold in brown adipocytes. Biochim. Biophys. Acta 2021, 1866, 158992. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Cantó, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef]
- Han, M.; Bushong, E.A.; Segawa, M.; Tiard, A.; Wong, A.; Brady, M.R.; Momcilovic, M.; Wolf, D.M.; Zhang, R.; Petcherski, A.; et al. Spatial mapping of mitochondrial networks and bioenergetics in lung cancer. Nature 2023, 615, 712–719. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef]
- Cui, L.; Mirza, A.H.; Zhang, S.; Liang, B.; Liu, P. Lipid droplets and mitochondria are anchored during brown adipocyte differentiation. Protein Cell 2019, 10, 921–926. [Google Scholar] [CrossRef]
- Talari, N.K.; Mattam, U.; Meher, N.K.; Paripati, A.K.; Mahadev, K.; Krishnamoorthy, T.; Sepuri, N.B.V. Lipid-droplet associated mitochondria promote fatty-acid oxidation through a distinct bioenergetic pattern in male Wistar rats. Nat. Commun. 2023, 14, 766. [Google Scholar] [CrossRef]
- Najt, C.P.; Adhikari, S.; Heden, T.D.; Cui, W.; Gansemer, E.R.; Rauckhorst, A.J.; Markowski, T.W.; Higgins, L.; Kerr, E.W.; Boyum, M.D.; et al. Organelle interactions compartmentalize hepatic fatty acid trafficking and metabolism. Cell Rep. 2023, 42, 112435. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.; Sun, W.; Tang, Q.; Feng, W.; Xiao, H.; Wang, J.; Yuan, X.; Xiang, M.; Gao, Y.; et al. Characteristics of different lipid droplet-mitochondrial contacts patterns during lipid droplet metabolism in T2DM-induced MASLD. Sci. Rep. 2025, 15, 3399. [Google Scholar] [CrossRef] [PubMed]
- Talari, N.K.; Mattam, U.; Rahman, A.P.; Hemmelgarn, B.K.; Wyder, M.A.; Sylvestre, P.B.; Greis, K.D.; Chella Krishnan, K. Functional compartmentalization of hepatic mitochondrial subpopulations during MASH progression. Commun. Biol. 2025, 8, 258. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, S.; Cui, L.; Wang, W.; Na, H.; Zhu, X.; Li, L.; Xu, G.; Yang, F.; Christian, M.; et al. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim. Biophys. Acta 2015, 1853, 918–928. [Google Scholar] [CrossRef]
- Ngo, J.; Choi, D.W.; Stanley, I.A.; Stiles, L.; Molina, A.J.A.; Chen, P.H.; Lako, A.; Sung, I.C.H.; Goswami, R.; Kim, M.Y.; et al. Mitochondrial morphology controls fatty acid utilization by changing CPT1 sensitivity to malonyl-CoA. EMBO J. 2023, 42, e111901. [Google Scholar] [CrossRef]
- Rao, J.N.; Warren, G.Z.L.; Estolt-Povedano, S.; Zammit, V.A.; Ulmer, T.S. An environment- dependent structural switch underlies the regulation of carnitine palmitoyltransferase 1A. J. Biol. Chem. 2011, 286, 42545–42554. [Google Scholar] [CrossRef]
- Wang, H.; Bell, M.; Sreenivasan, U.; Sreenevasan, U.; Hu, H.; Liu, J.; Dalen, K.; Londos, C.; Yamaguchi, T.; Rizzo, M.A.; et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 2011, 286, 15707–15715. [Google Scholar] [CrossRef]
- Wu, Q.; Zhao, M.; He, X.; Xue, R.; Li, D.; Yu, X.; Wang, S.; Zang, W. Acetylcholine reduces palmitate-induced cardiomyocyte apoptosis by promoting lipid droplet lipolysis and perilipin 5-mediated lipid droplet-mitochondria interaction. Cell Cycle 2021, 20, 1890–1906. [Google Scholar] [CrossRef]
- Hu, L.; Tang, D.; Qi, B.; Guo, D.; Wang, Y.; Geng, J.; Zhang, X.; Song, L.; Chang, P.; Chen, W.; et al. Mfn2/Hsc70 Complex Mediates the Formation of Mitochondria-Lipid Droplets Membrane Contact and Regulates Myocardial Lipid Metabolism. Adv. Sci. 2024, 11, e2307749. [Google Scholar] [CrossRef] [PubMed]
- He, W.; He, W.; Zeng, L.; Zhao, R.; Qiu, K.; He, P.; Sun, Z.; Tan, N. Loss of lipid droplet-mitochondria contacts confers protection against ethanol-induced cardiotoxicity. Exp. Cell Res. 2025, 447, 114517. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef]
- Bórquez, J.C.; Díaz-Castro, F.; La Fuente, F.P.; Espinoza, K.; Figueroa, A.M.; Martínez-Ruíz, I.; Hernández, V.; López-Soldado, I.; Ventura, R.; Domingo, J.C.; et al. Mitofusin-2 induced by exercise modifies lipid droplet-mitochondria communication, promoting fatty acid oxidation in male mice with NAFLD. Metabolism 2024, 152, 155765. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Yang, E.J.; Molina David, J.; Cho, S.; Ficarella, M.; Pape, N.; Schiffer, J.E.; Njeim, R.; Kim, S.S.; Lo Re, C.; et al. Ezetimibe Enhances Lipid Droplet and Mitochondria Contact Formation, Improving Fatty Acid Transfer and Reducing Lipotoxicity in Alport Syndrome Podocytes. Int. J. Mol. Sci. 2024, 25, 13134. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Sarhadi, T.R.; Panse, J.S.; Nagotu, S. Mind the gap: Methods to study membrane contact sites. Exp. Cell Res. 2023, 431, 113756. [Google Scholar] [CrossRef]
- Gamuyao, R.; Chang, C.L. Imaging and proteomics toolkits for studying organelle contact sites. Front. Cell Dev. Biol. 2024, 12, 1466915. [Google Scholar] [CrossRef] [PubMed]
- Miner, G.E.; So, C.M.; Edwards, W.; Ragusa, J.V.; Wine, J.T.; Wong Gutierrez, D.; Airola, M.V.; Herring, L.E.; Coleman, R.A.; Klett, E.L.; et al. PLIN5 interacts with FATP4 at membrane contact sites to promote lipid droplet-to-mitochondria fatty acid transport. Dev. Cell 2023, 58, 1250–1265.e6. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Hu, B.; Li, J.; Chen, R.; Yuan, Z.; Xiao, H.; Chang, H.; Jiu, Y.; Cai, K.; Ding, B. Coronaviral ORF6 protein mediates inter-organelle contacts and modulates host cell lipid flux for virus production. EMBO J. 2023, 42, e112542. [Google Scholar] [CrossRef]
- Ouyang, Q.; Chen, Q.; Ke, S.; Ding, L.; Yang, X.; Rong, P.; Feng, W.; Cao, Y.; Wang, Q.; Li, M.; et al. Rab8a as a mitochondrial receptor for lipid droplets in skeletal muscle. Dev. Cell 2023, 58, 289–305.e6. [Google Scholar] [CrossRef]
- Young, P.A.; Senkal, C.E.; Suchanek, A.L.; Grevengoed, T.J.; Lin, D.D.; Zhao, L.; Crunk, A.E.; Klett, E.L.; Füllekrug, J.; Obeid, L.M.; et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem. 2018, 293, 16724–16740. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Devereux, C.J.; Keenan, S.N.; Lou, J.; Cho, E.; Nie, S.; De Souza, D.P.; Narayana, V.K.; Siddall, N.A.; Rodrigues, C.H.M.; et al. Proximity proteomics reveals a mechanism of fatty acid transfer at lipid droplet- mitochondria- endoplasmic reticulum contact sites. Nat. Commun. 2025, 16, 2135. [Google Scholar] [CrossRef]
- Che, L.; Huang, J.; Lin, J.X.; Xu, C.Y.; Wu, X.M.; Du, Z.B.; Wu, J.S.; Lin, Z.N.; Lin, Y.C. Aflatoxin B1 exposure triggers hepatic lipotoxicity via p53 and perilipin 2 interaction-mediated mitochondria-lipid droplet contacts: An in vitro and in vivo assessment. J. Hazard. Mater. 2023, 445, 130584. [Google Scholar] [CrossRef]
- Meng, Y.; Guo, D.; Lin, L.; Zhao, H.; Xu, W.; Luo, S.; Jiang, X.; Li, S.; He, X.; Zhu, R.; et al. Glycolytic enzyme PFKL governs lipolysis by promoting lipid droplet-mitochondria tethering to enhance β-oxidation and tumor cell proliferation. Nat. Metab. 2024, 6, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Freyre, C.A.C.; Rauher, P.C.; Ejsing, C.S.; Klemm, R.W. MIGA2 Links Mitochondria, the ER, and Lipid Droplets and Promotes De Novo Lipogenesis in Adipocytes. Mol. Cell 2019, 76, 811–825.e14. [Google Scholar] [CrossRef]
- Wang, J.; Fang, N.; Xiong, J.; Du, Y.; Cao, Y.; Ji, W.K. An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D-TSG101 interactions. Nat. Commun. 2021, 12, 1252. [Google Scholar] [CrossRef]
- Guillén-Samander, A.; Leonzino, M.; Hanna, M.G.; Tang, N.; Shen, H.; De Camilli, P. VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004. [Google Scholar] [CrossRef]
- Miner, G.E.; Smith, S.Y.; Showalter, W.K.; So, C.M.; Ragusa, J.V.; Powers, A.E.; Zanellati, M.C.; Hsu, C.H.; Marchan, M.F.; Cohen, S. Contact-FP: A Dimerization-Dependent Fluorescent Protein Toolkit for Visualizing Membrane Contact Site Dynamics. Contact 2024, 7, 25152564241228911. [Google Scholar] [CrossRef]
- Herms, A.; Bosch, M.; Reddy, B.J.; Schieber, N.L.; Fajardo, A.; Rupérez, C.; Fernández- Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef]
- Barneda, D.; Frontini, A.; Cinti, S.; Christian, M. Dynamic changes in lipid droplet-associated proteins in the “browning” of white adipose tissues. Biochim. Biophys. Acta 2013, 1831, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Najt, C.P.; Devarajan, M.; Mashek, D.G. Perilipins at a glance. J. Cell Sci. 2022, 135, jcs259501. [Google Scholar] [CrossRef]
- Gianazza, E.; Papaianni, G.G.; Brocca, L.; Banfi, C.; Mallia, A. Omics Approaches to Study Perilipins and Their Significant Biological Role in Cardiometabolic Disorders. Int. J. Mol. Sci. 2025, 26, 557. [Google Scholar] [CrossRef]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Croce, M.A.; Gropler, M.C.; Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Kern, P.A.; et al. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006, 55, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Laurens, C.; Bourlier, V.; Mairal, A.; Louche, K.; Badin, P.M.; Mouisel, E.; Montagner, A.; Marette, A.; Tremblay, A.; Weisnagel, J.S.; et al. Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 2016, 6, 38310. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Jaeger, D.; Kolleritsch, S.; Zimmermann, R.; Zechner, R.; Lass, A.; Haemmerle, G. The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J. Biol. Chem. 2015, 290, 1295–1306. [Google Scholar] [CrossRef]
- Keenan, S.N.; De Nardo, W.; Lou, J.; Schittenhelm, R.B.; Montgomery, M.K.; Granneman, J.G.; Hinde, E.; Watt, M.J. Perilipin 5 S155 phosphorylation by PKA is required for the control of hepatic lipid metabolism and glycemic control. J. Lipid Res. 2021, 62, 100016. [Google Scholar] [CrossRef]
- Najt, C.P.; Khan, S.A.; Heden, T.D.; Witthuhn, B.A.; Perez, M.; Heier, J.L.; Mead, L.E.; Franklin, M.P.; Karanja, K.K.; Graham, M.J.; et al. Lipid Droplet-Derived Monounsaturated Fatty Acids Traffic via PLIN5 to Allosterically Activate SIRT1. Mol. Cell 2020, 77, 810–824.e8. [Google Scholar] [CrossRef]
- Gallardo-Montejano, V.I.; Saxena, G.; Kusminski, C.M.; Yang, C.; McAfee, J.L.; Hahner, L.; Hoch, K.; Dubinsky, W.; Narkar, V.A.; Bickel, P.E. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1α/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat Commun. 2016, 7, 12723. [Google Scholar] [CrossRef]
- Zhang, H.H.; Souza, S.C.; Muliro, K.V.; Kraemer, F.B.; Obin, M.S.; Greenberg, A.S. Lipase-selective functional domains of perilipin A differentially regulate constitutive and protein kinase A-stimulated lipolysis. J. Biol. Chem. 2003, 278, 51535–51542. [Google Scholar] [CrossRef]
- Liu, R.; Lee, J.H.; Li, J.; Yu, R.; Tan, L.; Xia, Y.; Zheng, Y.; Bian, X.L.; Lorenzi, P.L.; Chen, Q.; et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol. Cell 2021, 81, 2722–2735.e9. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef]
- Kleinert, M.; Parker, B.L.; Chaudhuri, R.; Fazakerley, D.J.; Serup, A.; Thomas, K.C.; Krycer, J.R.; Sylow, L.; Fritzen, A.M.; Hoffman, N.J.; et al. mTORC2 and AMPK differentially regulate muscle triglyceride content via Perilipin 3. Mol. Metab. 2016, 5, 646–655. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Kulkarni, P.G.; Mohire, V.M.; Bhaisa, P.K.; Joshi, M.M.; Puranik, C.M.; Waghmare, P.P.; Banerjee, T. Mitofusin-2: Functional switch between mitochondrial function and neurodegeneration. Mitochondrion 2023, 69, 116–129. [Google Scholar] [CrossRef]
- Tábara, L.C.; Segawa, M.; Prudent, J. Molecular mechanisms of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2025, 26, 123–146. [Google Scholar] [CrossRef]
- Wang, L.J.; Lai, X.H.; Luo, Z.; Feng, G.L.; Song, Y.F. Diallyl disulfide alleviates hepatic steatosis by the conservative mechanism from fish to tetrapod: Augment Mfn2/Atgl-Mediated lipid droplet-mitochondria coupling. Redox Biol. 2024, 77, 103395. [Google Scholar] [CrossRef]
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2018, 19, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Rothenberg, A.; Gomez, C.; Cohen, A.W.; Garcia, A.; Bhattacharyya, S.; Shapiro, L.; Dolios, G.; Wang, R.; Lisanti, M.P.; et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 2004, 279, 42062–42071. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Xu, G.; Dorward, H.; Tansey, J.T.; Contreras, J.A.; Kimmel, A.R.; Londos, C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 2003, 161, 1093–1103. [Google Scholar] [CrossRef]
- Senthivinayagam, S.; McIntosh, A.L.; Moon, K.C.; Atshaves, B.P. Plin2 inhibits cellular glucose uptake through interactions with SNAP23, a SNARE complex protein. PLoS ONE 2013, 8, e73696. [Google Scholar] [CrossRef]
- Foster, L.J.; Yaworsky, K.; Trimble, W.S.; Klip, A. SNAP23 promotes insulin-dependent glucose uptake in 3T3-L1 adipocytes: Possible interaction with cytoskeleton. Am. J. Physiol. 1999, 276, C1108–C1114. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Reilly, S.M.; Abu-Odeh, M.; Murphy, A.N.; Mahata, S.K.; Zhang, J.; Cho, Y.; Seo, J.B.; Hung, C.W.; Green, C.R.; et al. TANK-Binding Kinase 1 Regulates the Localization of Acyl-CoA Synthetase ACSL1 to Control Hepatic Fatty Acid Oxidation. Cell Metab. 2020, 32, 1012–1027.e7. [Google Scholar] [CrossRef]
- Yang, M.; Ge, J.; Liu, Y.L.; Wang, H.Y.; Wang, Z.H.; Li, D.P.; He, R.; Xie, Y.Y.; Deng, H.Y.; Peng, X.M.; et al. Sortilin-mediated translocation of mitochondrial ACSL1 impairs adipocyte thermogenesis and energy expenditure in male mice. Nat. Commun. 2024, 15, 7746. [Google Scholar] [CrossRef]
- Liu, S.; Geng, B.; Zou, L.; Wei, S.; Wang, W.; Deng, J.; Xu, C.; Zhao, X.; Lyu, Y.; Su, X.; et al. Development of hypertrophic cardiomyopathy in perilipin-1 null mice with adipose tissue dysfunction. Cardiovasc. Res. 2015, 105, 20–30. [Google Scholar] [CrossRef]
- Stone, S.J.; Levin, M.C.; Zhou, P.; Han, J.; Walther, T.C.; Farese, R.V., Jr. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 2009, 284, 5352–5361. [Google Scholar] [CrossRef] [PubMed]
- Malis, Y.; Armoza-Eilat, S.; Nevo-Yassaf, I.; Dukhovny, A.; Sklan, E.H.; Hirschberg, K. Rab1b facilitates lipid droplet growth by ER-to-lipid droplet targeting of DGAT2. Sci. Adv. 2024, 10, eade7753. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; McFie, P.J.; Banman, S.L.; Brandt, C.; Stone, S.J. Diacylglycerol acyltransferase-2 (DGAT2) and monoacylglycerol acyltransferase-2 (MGAT2) interact to promote triacylglycerol synthesis. J. Biol. Chem. 2014, 289, 28237–28248. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, S.O.; Cole, R.A.; McKinney, S.A.; Guo, F.; Haas, J.T.; Bobba, S.; Farese, R.V., Jr.; Mak, H.Y. The FATP1-DGAT2 complex facilitates lipid droplet expansion at the ER- lipid droplet interface. J. Cell Biol. 2012, 198, 895–911. [Google Scholar] [CrossRef]
- Guitart, M.; Osorio-Conles, O.; Pentinat, T.; Cebrià, J.; García-Villoria, J.; Sala, D.; Sebastián, D.; Zorzano, A.; Ribes, A.; Jiménez-Chillarón, J.C.; et al. Fatty acid transport protein 1 (FATP1) localizes in mitochondria in mouse skeletal muscle and regulates lipid and ketone body disposal. PLoS ONE 2014, 9, e98109. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef]
- Dziurdzik, S.K.; Conibear, E. The Vps13 Family of Lipid Transporters and Its Role at Membrane Contact Sites. Int. J. Mol. Sci. 2021, 22, 2905. [Google Scholar] [CrossRef]
- Modi, S.; López-Doménech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia-Cárcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 2019, 10, 4399. [Google Scholar] [CrossRef]
- Anand, R.; Reichert, A.S.; Kondadi, A.K. Emerging Roles of the MICOS Complex in Cristae Dynamics and Biogenesis. Biology 2021, 10, 600. [Google Scholar] [CrossRef]
- Chang, C.L.; Weigel, A.V.; Ioannou, M.S.; Pasolli, H.A.; Xu, C.S.; Peale, D.R.; Shtengel, G.; Freeman, M.; Hess, H.F.; Blackstone, C.; et al. Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J. Cell Biol. 2019, 218, 2583–2599. [Google Scholar] [CrossRef]
- Yeshaw, W.M.; van der Zwaag, M.; Pinto, F.; Lahaye, L.L.; Faber, A.I.; Gómez-Sánchez, R.; Dolga, A.M.; Poland, C.; Monaco, A.P.; van IJzendoorn, S.C.; et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. eLife 2019, 8, e43561. [Google Scholar] [CrossRef]
- Hong, Z.; Adlakha, J.; Wan, N.; Guinn, E.; Giska, F.; Gupta, K.; Melia, T.J.; Reinisch, K.M. Mitoguardin-2-mediated lipid transfer preserves mitochondrial morphology and lipid droplet formation. J. Cell Biol. 2022, 221, e202207022. [Google Scholar] [CrossRef]
- Boone, C.; Lewis, S.C. Bridging lipid metabolism and mitochondrial genome maintenance. J. Biol. Chem. 2024, 300, 107498. [Google Scholar] [CrossRef]
- Galmes, R.; Houcine, A.; van Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Du, X.; Wang, H.; Dong, J.; Ferguson, C.; Brown, A.J.; Parton, R.G.; Wu, J.W.; Yang, H. ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P2) and regulate its level at the plasma membrane. Nat. Commun. 2017, 8, 757. [Google Scholar] [CrossRef] [PubMed]
- Sohn, M.; Korzeniowski, M.; Zewe, J.P.; Wills, R.C.; Hammond, G.R.V.; Humpolickova, J.; Vrzal, L.; Chalupska, D.; Veverka, V.; Fairn, G.D.; et al. PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER-PM contact sites. J. Cell Biol. 2018, 217, 1797–1813. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Rochin, L.; Arora, A.; Houcine, A.; Jääskeläinen, E.; Kivelä, A.M.; Sauvanet, C.; Le Bars, R.; Marien, E.; Dehairs, J.; et al. ORP5/8 and MIB/MICOS link ER- mitochondria and intra-mitochondrial contacts for non-vesicular transport of phosphatidylserine. Cell Rep. 2022, 40, 111364. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Tol, M.J.; Wu, J.; Uchiyama, L.F.; Xiao, X.; Cui, L.; Bedard, A.H.; Weston, T.A.; Rajendran, P.S.; Vergnes, L.; et al. CLSTN3β enforces adipocyte multilocularity to facilitate lipid utilization. Nature 2023, 613, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, L.F.; Nguyen, A.; Qian, K.; Cui, L.; Pham, K.T.; Xiao, X.; Gao, Y.; Shimanaka, Y.; Tol, M.J.; Vergnes, L.; et al. PPARα regulates ER-lipid droplet protein Calsyntenin-3β to promote ketogenesis in hepatocytes. Proc. Natl. Acad. Sci. USA 2025, 122, e2426338122. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Cortini, M.; Ilieva, E.; Massari, S.; Bettini, G.; Avnet, S.; Baldini, N. Uncovering the protective role of lipid droplet accumulation against acid-induced oxidative stress and cell death in osteosarcoma. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167576. [Google Scholar] [CrossRef]
- Danielli, M.; Perne, L.; Jarc Jovičić, E.; Petan, T. Lipid droplets and polyunsaturated fatty acid trafficking: Balancing life and death. Front. Cell Dev. Biol. 2023, 11, 1104725. [Google Scholar] [CrossRef]
- Goodman, L.D.; Ralhan, I.; Li, X.; Lu, S.; Moulton, M.J.; Park, Y.J.; Zhao, P.; Kanca, O.; Ghaderpour Taleghani, Z.S.; Jacquemyn, J.; et al. Tau is required for glial lipid droplet formation and resistance to neuronal oxidative stress. Nat. Neurosci. 2024, 27, 1918–1933. [Google Scholar] [CrossRef]
- Tan, Y.; Jin, Y.; Wang, Q.; Huang, J.; Wu, X.; Ren, Z. Perilipin 5 Protects against Cellular Oxidative Stress by Enhancing Mitochondrial Function in HepG2 Cells. Cells 2019, 8, 1241. [Google Scholar] [CrossRef]
- Zhu, Y.; Ren, C.; Zhang, M.; Zhong, Y. Perilipin 5 Reduces Oxidative Damage Associated With Lipotoxicity by Activating the PI3K/ERK-Mediated Nrf2-ARE Signaling Pathway in INS-1 Pancreatic β-Cells. Front. Endocrinol. 2020, 11, 166. [Google Scholar] [CrossRef] [PubMed]
- Huo, K.; Ma, K.G.; Guo, Q.Y.; Duan, P.; Xu, J. Perilipin 5 protects against oxygen- glucose deprivation/reoxygenation-elicited neuronal damage by inhibiting oxidative stress and inflammatory injury via the Akt-GSK-3β-Nrf2 pathway. Int. Immunopharmacol. 2022, 108, 108718. [Google Scholar] [CrossRef] [PubMed]
- Kien, B.; Kolleritsch, S.; Kunowska, N.; Heier, C.; Chalhoub, G.; Tilp, A.; Wolinski, H.; Stelzl, U.; Haemmerle, G. Lipid droplet-mitochondria coupling via perilipin 5 augments respiratory capacity but is dispensable for FA oxidation. J. Lipid Res. 2022, 63, 100172. [Google Scholar] [CrossRef] [PubMed]
- Awadh, A.A. The Role of Cytosolic Lipid Droplets in Hepatitis C Virus Replication, Assembly, and Release. Biomed. Res. Int. 2023, 2023, 5156601. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, W.; Xiao, M.Z.X.; Zheng, Y.; Liang, Q. The interplay between lipid droplets and virus infection. J. Med. Virol. 2023, 95, e28967. [Google Scholar] [CrossRef]
- Cesar-Silva, D.; Pereira-Dutra, F.S.; Giannini, A.L.M.; Maya-Monteiro, C.M.; de Almeida, C.J.G. Lipid compartments and lipid metabolism as therapeutic targets against coronavirus. Front. Immunol. 2023, 14, 1268854. [Google Scholar] [CrossRef]
- Hsia, J.Z.; Liu, D.; Haynes, L.; Cruz-Cosme, R.; Tang, Q. Lipid Droplets: Formation, Degradation, and Their Role in Cellular Responses to Flavivirus Infections. Microorganisms 2024, 12, 647. [Google Scholar] [CrossRef]
- Herker, E. Lipid Droplets in Virus Replication. FEBS Lett. 2024, 598, 1299–1300. [Google Scholar] [CrossRef]
- Farías, M.A.; Diethelm-Varela, B.; Kalergis, A.M.; González, P.A. Interplay between lipid metabolism, lipid droplets and RNA virus replication. Crit. Rev. Microbiol. 2024, 50, 515–539. [Google Scholar] [CrossRef]
- Ren, X.L.; Han, P.; Meng, Y. Aflatoxin B1-Induced COX-2 Expression Promotes Mitophagy and Contributes to Lipid Accumulation in Hepatocytes In Vitro and In Vivo. Int. J. Toxicol. 2020, 39, 594–604. [Google Scholar] [CrossRef]
- Rotimi, O.A.; Rotimi, S.O.; Duru, C.U.; Ebebeinwe, O.J.; Abiodun, A.O.; Oyeniyi, B.O.; Faduyile, F.A. Acute aflatoxin B1-Induced hepatotoxicity alters gene expression and disrupts lipid and lipoprotein metabolism in rats. Toxicol. Rep. 2017, 4, 408–414. [Google Scholar] [CrossRef]
- Cabrera-Orefice, A.; Potter, A.; Evers, F.; Hevler, J.F.; Guerrero-Castillo, S. Complexome Profiling-Exploring Mitochondrial Protein Complexes in Health and Disease. Front. Cell Dev. Biol. 2022, 9, 796128. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Kwak, C.; Shin, S.; Park, J.S.; Jung, M.; Nhung, T.T.M.; Kang, M.G.; Lee, C.; Kwon, T.H.; Park, S.K.; Mun, J.Y.; et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. USA 2020, 117, 12109–12120. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.A.; Deol, K.K.; Mathiowetz, A.J.; Lange, M.; Leto, D.E.; Stevenson, J.; Hashemi, S.H.; Morgens, D.W.; Easter, E.; Heydari, K.; et al. Parallel CRISPR-Cas9 screens identify mechanisms of PLIN2 and lipid droplet regulation. Dev. Cell 2023, 58, 1782–1800.e10. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).