
Advantages of In Situ Mössbauer Spectroscopy in Catalyst Studies with Precaution in Interpretation of Measurements


Abstract
1. Introduction
2. Experimental Background and Selected Materials
2.1. Samples
2.2. In Situ Mössbauer Measurements
3. Mössbauer Studies on Zeolites (Example 1)
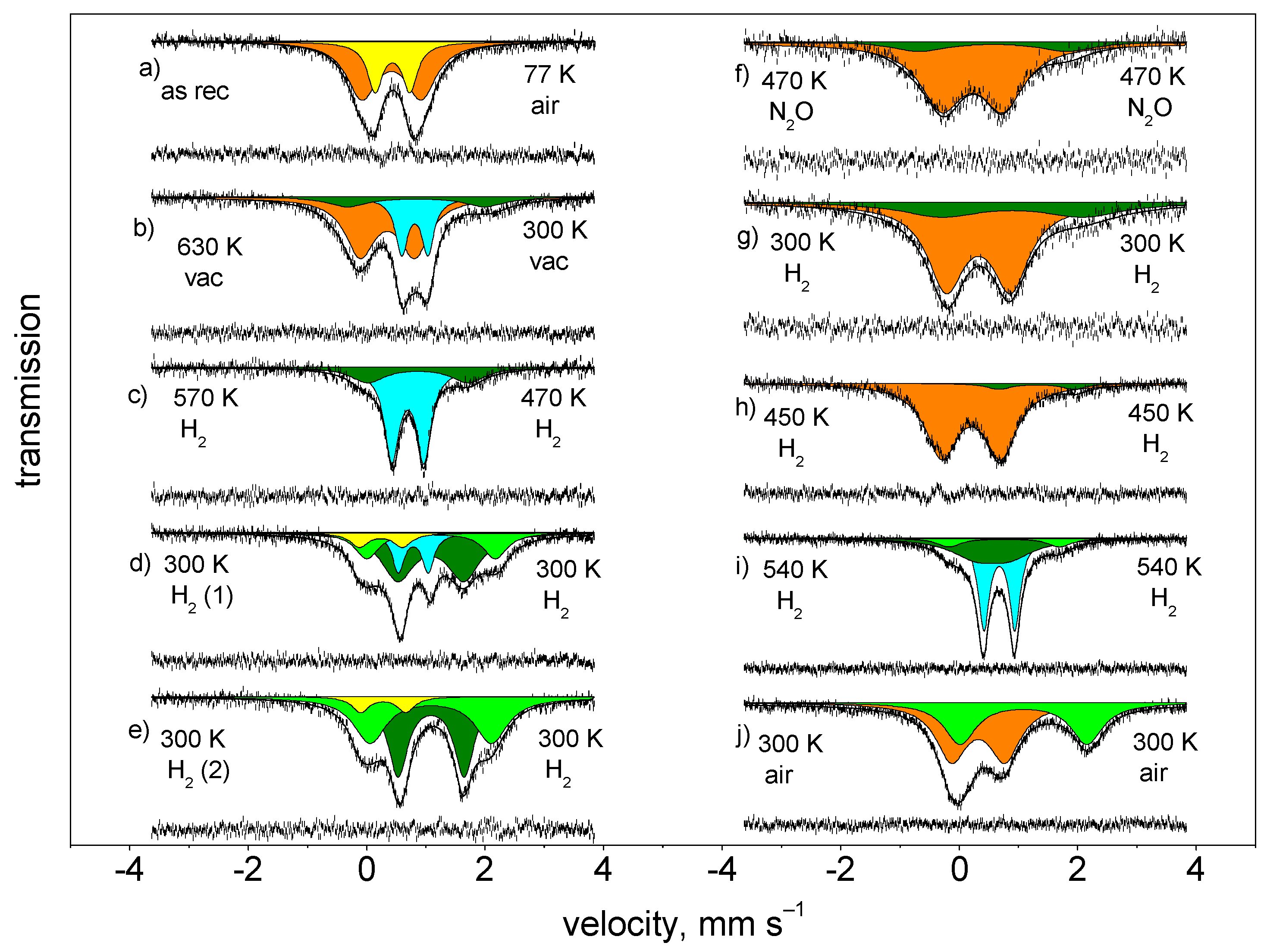
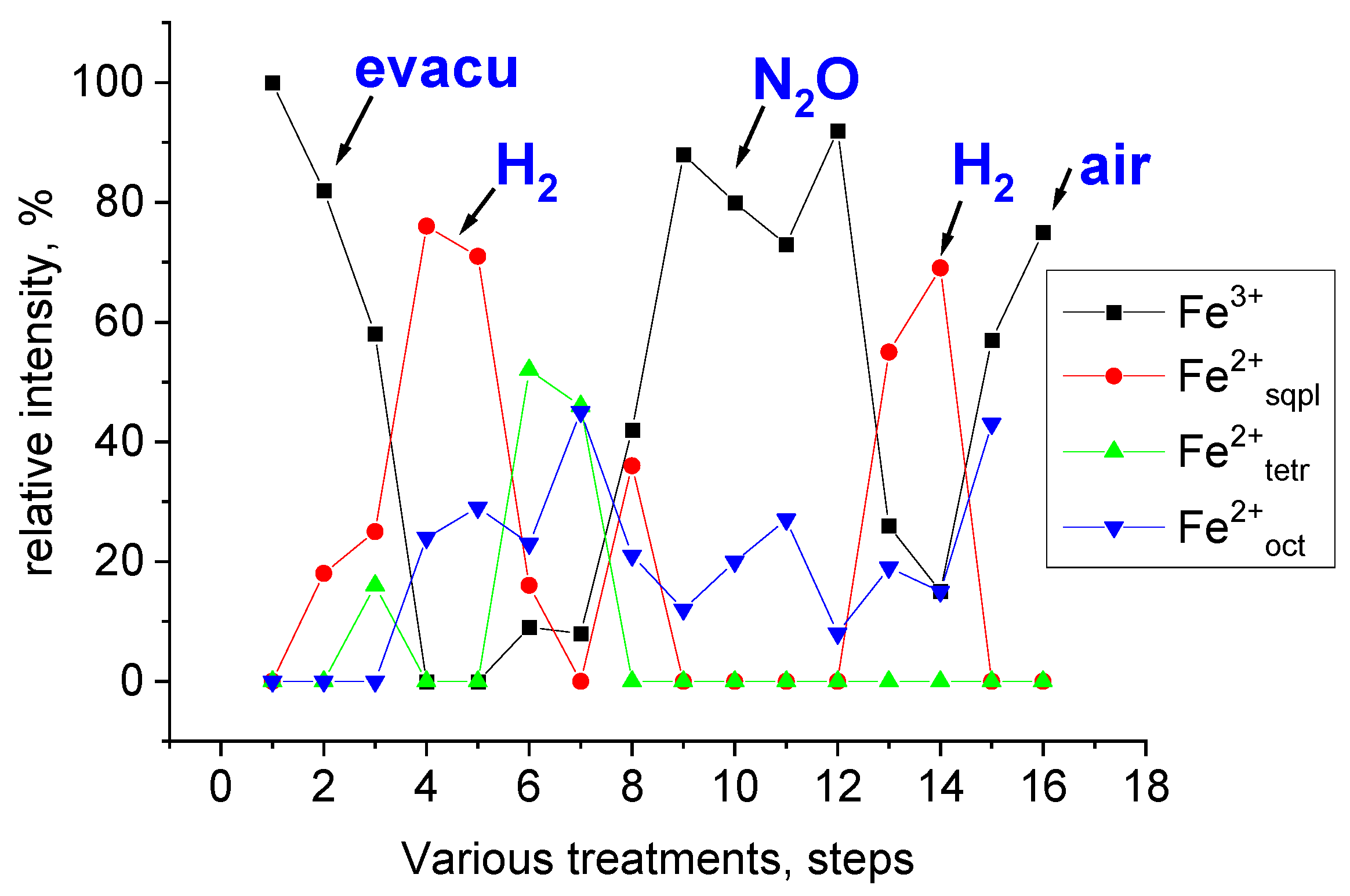
3.1. Demonstration of Extra-Framework Siting of Iron in Various Coordinations in 57Fe LTA Sample
3.2. Framework Iron Ions in Ferrierite Analog Ferrisilicate
4. Metals/Alloys (Example 2)
4.1. In Situ Dehydrochlorination of 1,2-Dichloroethane, ClCH2-CH2Cl
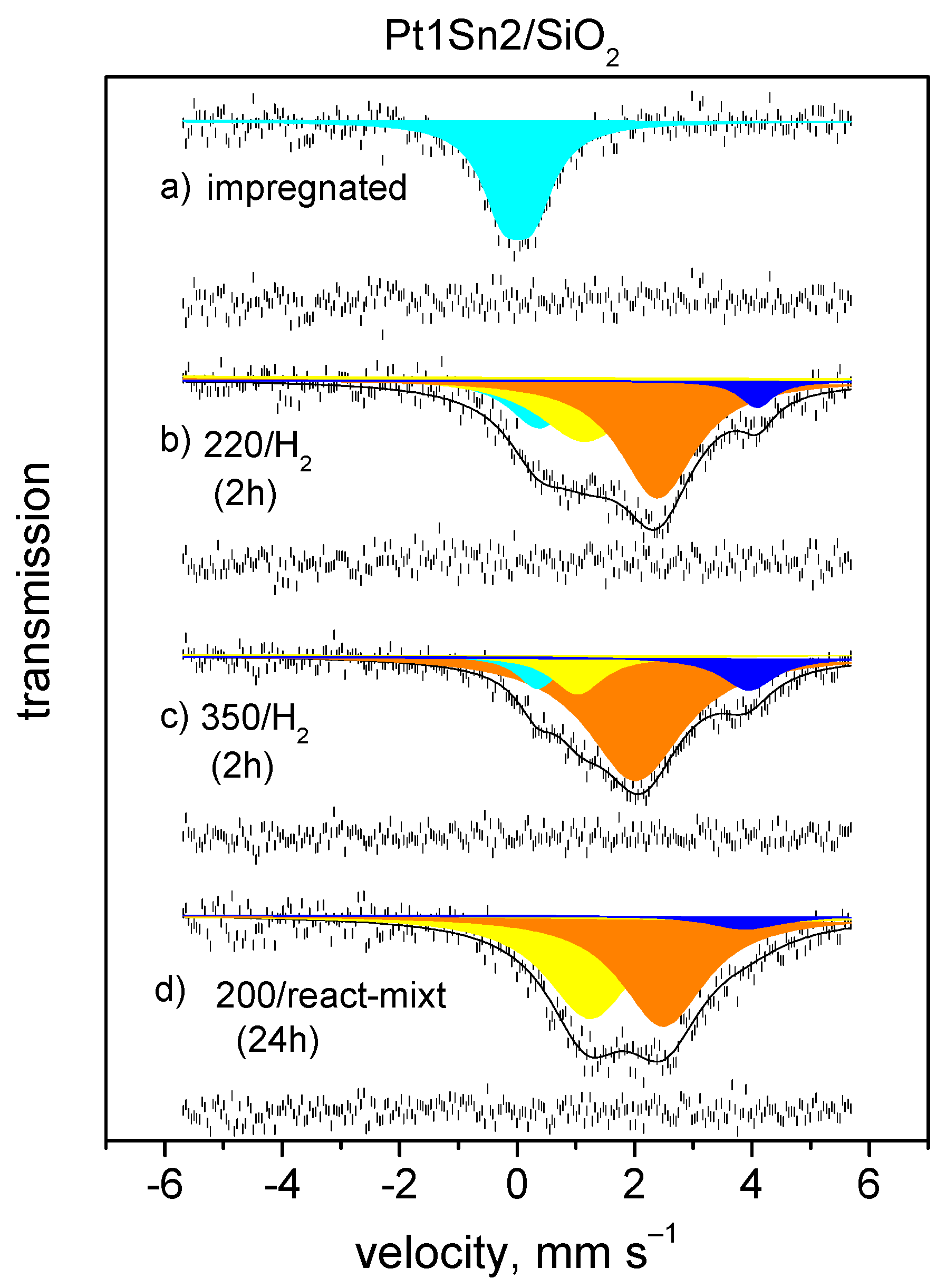
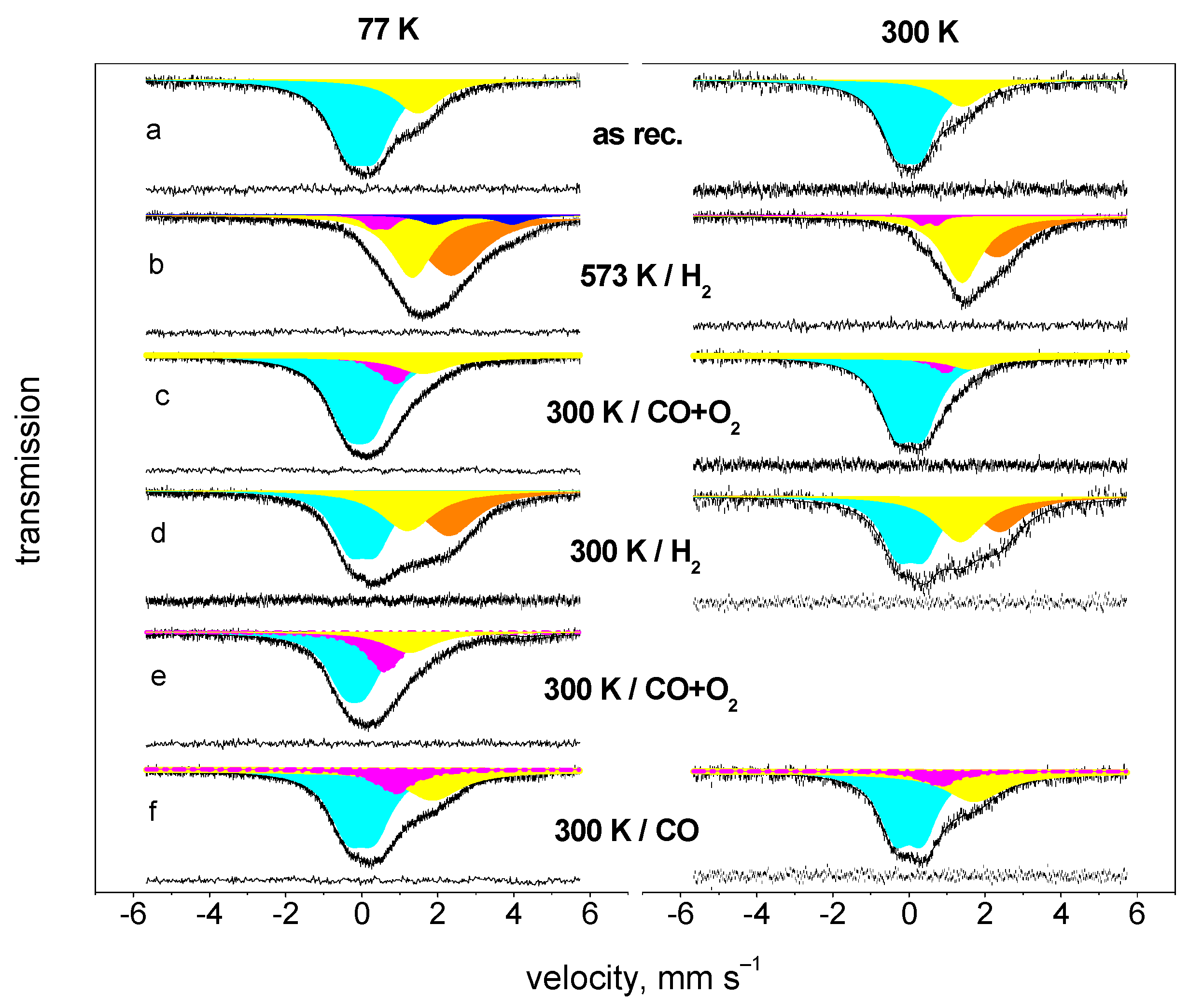
4.2. Oxidation of CO on Pt-Sn Catalyst at Room Temperature
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenwood, N.N.; Gibb, T.C. Mössbauer Spectroscopy; Chapman and Hall: London, UK, 1971. [Google Scholar]
- Cohen, R.L. Applications of Mössbauer Spectroscopy; Academic Press: Cambridge, MA, USA, 1976; Volumes 1–2. [Google Scholar]
- Long, G.J. Application of Mössbauer Spectroscopy to Inorganic Chemistry; Plenum Press: NewYork, NY, USA, 1984; Volumes 1–3. [Google Scholar]
- Gütlich, P.; Bill, E.; Trautwein, A.X. Mössbauer Spectroscopy and Transitional Metal Chemistry; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Mössbauer Spectroscopy. In Applications in Chemistry, Biology and Nanotechnology; Sharma, V.K., Klingelhöfer, G., Nishida, T., Eds.; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Modern Mössbauer Spectroscopy. In Topics in Applied Physics; Yoshida, Y., Langouche, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; Volume 137. [Google Scholar]
- Murad, E.; Cashion, J. Mössbauer Spectroscopy of Environmental Materials and Their Industrial Utilization; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Filter, W.F.; Sands, R.H.; Dunham, W.R. An ultra-high-stability Mössbauer spectrometer drive using a type-2 feedback system. Nucl. Instrum. Methods Phys. Res. B 1996, 119, 565–582. [Google Scholar] [CrossRef]
- Nagy, D.L.; Röhlich, U. An overview on model-independent data reduction methods in Mössbauer spectroscopy. Hyperfine Interact. 1991, 66, 105–126. [Google Scholar] [CrossRef]
- Alenkina, I.V.; Ushakov, M.V.; Morais, P.C.; Selvan, R.K.; Kuzmann, E.; Klencsár, Z.; Felner, I.; Homonnay, Z.; Oshtrakh, M.I. Mössbauer spectroscopy with a high velocity resolution in the studies of nanomaterials. Nanomaterials 2022, 12, 3748. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, Y.; Mehner, H.; Menzel, M.; Rogozev, B. Precision in Mössbauer spectroscopy. Hyperfine Interact. 2002, 139/140, 679–684. [Google Scholar] [CrossRef]
- Spiering, H.; Ksenofontov, V.; Leupold, O.; Kusz, J.; Deák, L.; Németh, Z.; Bogdán, C.; Bottyán, L.; Nagy, D.L. Line shape of 57Co sources exhibiting self absorption. Hyperfine Interact. 2016, 237, 58. [Google Scholar] [CrossRef]
- Brand, R.A. Normos Mössbauer Fitting Program. The NORMOS Program is Available from Wissel GmbH. Nucl. Instrum. Methods Phys. Res. 1987, 28, 398. [Google Scholar] [CrossRef]
- Klencsár, Z.; Kuzmann, E.; Vértes, A. User-friendly software for Mössbauer spectrum analysis. J. Radioanal. Nucl. Chem. 1996, 210, 105–118. [Google Scholar] [CrossRef]
- Lagarec, K.; Rancourt, D.G. Recoil User Manual—Mössbauer Spectral Analysis Software for Windows; University of Ottawa: Ottawa, ON, Canada, 1998. [Google Scholar]
- Zak, T.; Jirásková, Y. CONFIT: Mössbauer spectra fitting program. Surf. Interface Anal. 2006, 38, 710–714. [Google Scholar] [CrossRef]
- Hjøllum, J.; Madsen, M.B. Fit;o)-A Mössbauer spectrum fitting program. arXiv 2009, arXiv:0912.0449v1. [Google Scholar]
- Kamusella, S.; Klauss, H.-H. Moessfit A free Mössbauer fitting program. Hyperfine Interact. 2016, 237, 82. [Google Scholar] [CrossRef]
- Spiering, H.; Nagy, D.L.; Németh, Z.; Bogdán, C.; Deák, L. Non-linearity correction of the velocity scale of a Mössbauer spectrum. Nucl. Instrum. Methods Phys. Res. B 2020, 480, 98–104. [Google Scholar] [CrossRef]
- Kuzmann, E.; Nagy, S.; Vértes, A. Critical review of analytical applications of Mössbauer spectroscopy illustrated by mineralogical and geological examples. Pure Appl. Chem. 2003, 75, 801–858. [Google Scholar] [CrossRef]
- Grandjean, F.; Long, G.J. Best practices and protocols in Mössbauer spectroscopy. Chem. Mater. 2021, 33, 3878–3904. [Google Scholar] [CrossRef]
- Klencsár, Z.; Wang, J.; Ge, R.; Zhou, W.; Liu, D.; Rykov, A.I.; Zhang, T. The new WEB-accessible online database of the Mössbauer Effect Data Center. Hyperfine Interact. 2021, 242, 15. [Google Scholar] [CrossRef]
- Dyar, M.D. Precision and interlaboratory reproducibility of measurements of the Mössbauer effect in minerals. Am. Mineral. 1984, 69, 1127–1144. [Google Scholar]
- Mehner, H.; Menzel, M.; Nofz, M. Laboratory intercomparison on the determination of the Fe(II)/Fe(III) ratio in glass using Mössbauer spectroscopy. Hyperfine Interact. 2004, 156/157, 347–352. [Google Scholar] [CrossRef]
- Dumesic, J.A.; Topsøe, H. Mössbauer spectroscopy applications to heterogeneous catalysis. Adv. Catal. 1977, 26, 121–246. [Google Scholar]
- Millet, J.-M.M. Mössbauer spectroscopy in heterogeneous catalysis. Adv. Catal. 2007, 51, 309–350. [Google Scholar] [CrossRef]
- Liu, K.; Rykov, A.I.; Wang, J.; Zhang, T. Recent advances in the applications of Mössbauer spectroscopy in heterogeneous catalysis. Adv. Catal. 2015, 58, 1–142. [Google Scholar] [CrossRef]
- Lázár, K. Mössbauer spectroscopy of catalysts, Ch. 4. In Mössbauer Spectroscopy: Applications in Chemistry and Materials Science; Garcia, Y., Wang, J., Zhang, T., Eds.; Wiley: Hoboken, NJ, USA, 2024; pp. 113–143. [Google Scholar]
- Fischer, N.; Claeys, M. In situ characterization of Fischer-Tropsch catalysts: A review. J. Phys. D Appl. Phys. 2020, 53, 293001. [Google Scholar] [CrossRef]
- Bibicu, I.; Frunza, L. Characterization of oxide catalysts by in situ Mössbauer spectroscopy. Rom. Rep. Phys. 2006, 58, 427–453. [Google Scholar]
- Crajé, M.W.J.; van der Kraan, A.M.; van de Loosdrecht, J.; van Berge, P.J. The application of Mössbauer emission spectroscopy to industrial cobalt based Fischer-Tropsch catalysts. Catal. Today 2002, 71, 369–379. [Google Scholar] [CrossRef]
- Wezendonk, T.A.; Santos, V.P.; Nasalevich, M.A.; Warringa, Q.S.; Dugulan, A.I.; Chojecki, A.; Koeken, A.C.J.; Ruitenbeek, M.; Meima, G.R.; Islam, H.-U.; et al. Elucidating the nature of Fe species during pyrolysis of the Fe-BTC MOF into highly active and stable Fischer-Tropsch catalysts. ACS Catal. 2016, 6, 3236–3247. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, X.; Wang, J.; Sougrati, M.T.; Huang, Y.; Zhang, T.; Liu, B. In situ/operando Mössbauer spectroscopy for probing heterogeneous catalysis. Chem. Catal. 2021, 1, 1215–1233. [Google Scholar] [CrossRef]
- Liu, P.; Farid, S.; Liu, M.; Wang, J. In-situ/operando Mössbauer spectroscopic investigations of Fe-involved metal hydroxide-based OER electrocatalysts. Catal. Surv. Asia 2024, 28, 361–374. [Google Scholar] [CrossRef]
- Chen, R.; Zhao, J.; Li, Y.; Cui, Y.; Lu, Y.-R.; Hung, S.-F.; Wang, S.; Wang, W.; Huo, G.; Zhao, Y.; et al. Operando Mössbauer spectroscopic tracking the metastable state of atomically dispersed tin in copper oxide for selective CO2 electroreduction. J. Am. Chem. Soc. 2023, 145, 20683–20691. [Google Scholar] [CrossRef]
- Bussiere, P. Mössbauer spectroscopy, dispersion and catalysis. Hyperfine Interact. 1991, 66, 39–50. [Google Scholar] [CrossRef]
- Dubkov, K.A.; Ovanesyan, N.S.; Shteinman, A.A.; Starokon, E.V.; Panov, G.I. Evolution of iron states and formation of α-sites upon activation of FeZSM-5 zeolites. J. Catal. 2002, 207, 341–352. [Google Scholar] [CrossRef]
- Candy, J.-P.; Roisin, E.; Basset, J.-M.; Uzio, D.; Morin, S.; Fischer, L.; Olivier-Fourcade, J.; Jumas, J.-C. Evidence for direct observation by Mössbauer spectroscopy of surface tin atoms in platinum-tin particles. Hyperfine Interact. 2005, 165, 55–60. [Google Scholar] [CrossRef]
- Garten, R.L.; Delgass, W.N.; Boudart, M. A Mössbauer spectroscopic study of the reversible oxidation of ferrous ions in Y zeolite. J. Catal. 1970, 18, 90–96. [Google Scholar] [CrossRef]
- Villegas, V.A.R.; Ramirez, J.I.D.L.; Pérez-Cabrera, L.; Pérez-Sicairos, S.; Chávez-Méndez, J.R.; Petranovskii, V. Analysis of catalytic sites in FeY zeolite prepared by sono-assisted exchange of Fe(II) ions. Microporous Mesoporous Mater. 2024, 380, 113306. [Google Scholar] [CrossRef]
- Fejes, P.; Kiricsi, I.; Lázár, K.; Marsi, I.; Rockenbauer, A.; Korecz, L. Attempts to produce uniform Fe(III) siting in Fe content SOD and LTA zeolites; An EPR and Mössbauer study. Appl. Catal. A Gen. 2003, 242, 63–76. [Google Scholar] [CrossRef]
- Lázár, K.; Lejeune, G.; Ahedi, R.K.; Shevade, S.; Kotasthane, A.N. Interpreting the oxidative catalytic activity in iron-substituted ferrierites using in situ Mössbauer spectroscopy. J. Chem. Phys. B 1998, 102, 4865–4870. [Google Scholar] [CrossRef]
- Rhodes, W.; Lázár, K.; Kovalchuk, V.I.; d’Itri, J.L. Hydrogen-assisted 1,2-dichloroethane dechlorination catalyzed by Pt-Sn/SiO2: Effect of the Pt/Sn atomic ratio. J. Catal. 2002, 211, 173–182. [Google Scholar] [CrossRef]
- Margitfalvi, J.L.; Borbáth, I.; Hegedűs, M.; Szegedi, Á.; Lázár, K.; Gőbölös, S.; Kristyán, S. Low-temperature oxidation of CO over tin modified Pt/SiO2 catalysts. Catal. Today 2002, 73, 343–353. [Google Scholar] [CrossRef]
- Lázár, K.; Matusek, K.; Mink, J.; Dobos, S.; Guczi, L.; Vizi-Orosz, A.; Markó, L.; Reiff, W. Spectroscopic and catalytic study on metal carbonyl clusters supported on Cab-O-Sil. J. Catal. 1984, 87, 163–178. [Google Scholar] [CrossRef]
- Kulcsár, K.; Nagy, D.L.; Pócs, L. A complete package of programs for the evaluation of Mössbauer and gamma spectra. KFKI Rep. 1971, 67. [Google Scholar]
- Zhang, J.; Tang, X.; Yi, H.; Yu, Q.; Zhang, Y.; Wei, J.; Yuan, Y. Synthesis, characterization and application of Fe zeolite: A review. Appl. Catal. A Gen. 2022, 630, 118467. [Google Scholar] [CrossRef]
- Panov, G.I.; Sobolev, V.I.; Kharitonov, A.S. The role of iron in N2O decomposition on ZSM-5 zeolite and reactivity of the surface oxygen formed. J. Mol. Catal. 1990, 61, 85–97. [Google Scholar] [CrossRef]
- Bols, M.L.; Devos, J.; Rhoda, H.M.; Plessers, D.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F.; Dusselier, M. Selective formation of α-Fe(II) sites on Fe-zeolites through one-pot synthesis. J. Am. Chem. Soc. 2021, 143, 16243–16255. [Google Scholar] [CrossRef]
- Kornas, A.; Mlekodaj, K.; Tabor, E. Nature and redox properties of iron sites in zeolites revealed by Mössbauer spectroscopy. ChemPlusChem 2024, 89, e202300543. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.G. Mineral Mössbauer spectroscopy: Correlation between chemical shift and quadrupole splitting parameters. Hyperfine Interact. 1994, 91, 739–745. [Google Scholar] [CrossRef]
- Meagher, A.; Nair, V.; Szostak, R. A Mössbauer study of ZSM-5 type ferrisilicates. Zeolites 1988, 8, 3–11. [Google Scholar] [CrossRef]
- Kumar, R.; Date, S.K.; Bill, E.; Trautwein, A. Mössbauer spectroscopic study of iron ferrisilicate analog of zeolite BETA. Zeolites 1991, 11, 211–213. [Google Scholar] [CrossRef]
- Lázár, K.; Borbély, G.; Beyer, H. In situ Mössbauer study of framework-substituted (Fe)ZSM-5 zeolites. Zeolites 1991, 11, 214–222. [Google Scholar] [CrossRef]
- Inui, T.; Tanaka, Y. Evaluation of water adsorption on different kinds of zeolite through Monte Carlo simulation. Stud. Surf. Sci. Catal. 1995, 98, 229–231. [Google Scholar]
- Lázár, K.; Vincent, C.; Fejes, P. Variety of available coordination sites for extra-framework iron in LTA and MFI zeolites. Hyperfine Interact. 2008, 187, 1–6. [Google Scholar] [CrossRef]
- Yakovlev, A.L.; Zhidomirov, G.M.; van Santen, R.A. DFT calculations on N2O decomposition by binuclear Fe complexes in Fe/ZSM-5. J. Phys. Chem. B 2001, 105, 12297–12302. [Google Scholar] [CrossRef]
- Sazama, P.; Wichterlová, B.; Tábor, E.; Šťastný, P.; Sathu, N.K.; Sobalík, Z.; Dědeček, J.; Sklenák, Š.; Klein, P.; Vondrová, A. Tailoring of the structure of Fe-cationic species in Fe-ZSM-5 by distribution of Al atoms in the framework for N2O decomposition and NH3-SCR-NOx. J. Catal. 2014, 312, 123–138. [Google Scholar] [CrossRef]
- Lázár, K.; Kotasthane, A.N.; Fejes, P. Oxygen transfer centers in Fe-FER and Fe-MFI zeolites: Redox behavior and Debye temperature derived from in situ Mössbauer spectra. Catal. Lett. 1999, 57, 171–177. [Google Scholar] [CrossRef]
- Charlton, J.S.; Cordey-Hayes, M.; Harris, I.R. A study of the 119Sn Mössbauer isomer shifts in some platinum-tin and gold-tin alloys. J. Less-Common Met. 1970, 20, 105–112. [Google Scholar] [CrossRef]
- Hu, J.; Xie, M.; Chen, Y.; Fang, J.; Zhang, Q. Thermodynamic reassessment of Au-Pt-Sn system. Mater. Res. Express 2022, 9, 016507. [Google Scholar] [CrossRef]
- Kappenstein, C.; Guerin, M.; Lázár, K.; Matusek, K.; Paál, Z. Characterization and activity in n-hexane rearrangement reactions of metallic phases on Pt-Sn/Al2O3 catalysts of different preparation. J. Chem. Soc. Faraday Trans. 1998, 94, 2463–2473. [Google Scholar] [CrossRef]
- Olivier-Fourcade, J.; Womes, M.; Jumas, J.-C.; Le Peltier, F.; Morin, S.; Didillon, B. Investigation of redox properties of different PtSn/Al2O3 catalysts. ChemPhysChem 2004, 5, 1734–1744. [Google Scholar] [CrossRef]
- Dubiel, S.M.; Cieslak, J.; Alenkina, I.V.; Oshtrakh, M.I.; Semionkin, V.A. Evaluation of the Debye temperature for iron cores in human live ferritin and its pharmaceutical analogue, Ferrum Lek, using Mössbauer spectroscopy. J. Inorg. Biochem. 2014, 140, 89–93. [Google Scholar] [CrossRef]
- Lengyel, A.; Klencsár, Z.; Homonnay, Z.; Sipos, P.; Bajnoczi, E.; Kuzmann, E. Goldanskii-Karyagin effect on hyperalkaline tin(II)-hydroxide. J. Radioanal. Nucl. Chem. 2016, 307, 1195–1201. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Saxena, A.; Shah, D.; Mahato, T.H.; Singh, B.; Shrivastava, A.; Gutch, P.; Shinde, C. Catalytic removal of carbon monoxide over carbon supported palladium catalyst. J. Hazard. Mater. 2012, 241–242, 463–471. [Google Scholar] [CrossRef]
- Parish, R.V. Structure and bonding in tin compounds, Ch. 16. In Mössbauer Spectroscopy Applied to Inorganic Chemistry; palladium catalys; Long, G.J., Ed.; Springer: Berlin/Heidelberg, Germany, 1984; Volume I, pp. 527–575. [Google Scholar]
- Sutter, E.; Ivars-Barcelo, F.; Sutter, P. Size-dependent room temperature oxidation of tin particles. Part. Part. Syst. Charact. 2014, 31, 879–885. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Atm. | Temp. of | Component | δ a | Δ b | FWHM c | Rel. Int. d |
|---|---|---|---|---|---|---|---|
| Meas. K | mm s−1 | mm s−1 | mm s−1 | % | |||
| As rec. | air | 77 | Fe3+ | 0.45 | 1.04 | 0.59 | 65.9 |
| Fe3+ | 0.46 | 0.63 | 0.35 | 34.1 | |||
| 630 K/ | vacuum | 300 | Fe3+ | 0.36 | 0.93 | 0.61 | 58.4 |
| vac | Fe2+ | 0.83 | 0.45 | 0.28 | 25.5 | ||
| Fe2+ | 0.87 | 2.44 | 0.89 | 16.1 | |||
| 570/H2 | H2 | 470 | Fe2+ | 0.72 | 0.52 | 0.29 | 71.2 |
| Fe2+ | 0.87 | 1.71 | 0.70 | 28.8 | |||
| None | H2 | 300 | Fe3+ | 0.25 | 0.75 | 0.37 | 8.7 |
| (1st part) | Fe2+ | 0.84 | 0.50 | 0.24 | 16.2 | ||
| Fe2+ | 1.08 | 1.12 | 0.68 | 52.1 | |||
| Fe2+ | 1.12 | 2.19 | 0.51 | 23.0 | |||
| None | H2 | 300 | Fe3+ | 0.28 | 0.80 | 0.35 | 8.2 |
| (2nd part) | Fe2+ | 1.10 | 1.08 | 0.39 | 46.2 | ||
| Fe2+ | 1.08 | 2.04 | 0.65 | 45.6 | |||
| 470/N20 | N2O | 470 | Fe3+ | 0.25 | 1.00 | 0.83 | 85.2 |
| Fe2+ | 0.57 | 2.60 | 0.90 | 14.7 | |||
| RT/H2 | H2 | 300 | Fe3+ | 0.34 | 1.06 | 0.74 | 73.0 |
| Fe2+ | 0.88 | 2.46 | 1.62 | 27.0 | |||
| 450/H2 | H2 | 450 | Fe3+ | 0.21 | 0.99 | 0.67 | 94.1 |
| Fe2+ | 1.37 | 1.25 | 0.52 | 6.9 | |||
| 540/H2 | H2 | 540 | Fe2+ | 0.57 | 0.60 | 0.95 | 39.2 |
| Fe2+ | 0.69 | 0.53 | 0.23 | 50.5 | |||
| Fe2+ | 0.78 | 1.89 | 0.48 | 10.2 | |||
| 300/air | air | 300 | Fe3+ | 0.32 | 0.85 | 0.61 | 56.7 |
| Fe2+ | 1.15 | 2.09 | 0.63 | 43.2 |
| Treatment | Temp. | Atm. | Comp. | δ a | Δ b | FWHM c | Rel. Int. d |
|---|---|---|---|---|---|---|---|
| Meas. (K) | mm s−1 | mm s−1 | mm s−1 | % | |||
| Calcined | 300 | air | Fe3+ | 0.28 | 0.60 | 0.43 | 13.4 |
| /air | Fe3+ | 0.35 | 1.03 | 0.74 | 86.6 | ||
| 620 K/vac | 300 | vacuum | Fe3+ | 0.24 | 1.98 | 0.61 | 70.9 |
| Fe3+ | 0.37 | 1.17 | 1.08 | 29.1 | |||
| 620 K/H2 | 77 | hydrogen | Fe3+ | 0.33 | 1.83 | 0.38 | 13.6 |
| Fe3+ | 0.54 | 1.16 | 0.82 | 37.6 | |||
| Fe2+ | 1.17 | 1.98 | 0.77 | 36.4 | |||
| Fe2+ | 1.15 | 3.03 | 0.56 | 12.3 | |||
| None | 300 | hydrogen | Fe3+ | 0.30 | 1.62 | 0.59 | 35.6 |
| Fe3+ | 0.31 | 0.97 | 0.61 | 56.1 | |||
| Fe2+ | 0.78 | 2.46 | 0.44 | 3.3 | |||
| Fe2+ | 1.26 | 2.49 | 0.49 | 5.0 | |||
| None | 77 | hydrogen | Fe3+ | 0.40 | 1.48 | 0.69 | 52.5 |
| (repeat) | Fe3+ | 0.40 | 0.88 | 0.54 | 35.9 | ||
| Fe2+ | 1.25 | 2.26 | 0.25 | 1.9 | |||
| Fe2+ | 1.40 | 2.96 | 0.61 | 9.7 | |||
| 620 K/vac | 300 | vacuum | Fe3+ | 0.22 | 1.98 | 0.56 | 60.8 |
| Fe3+ | 0.46 | 1.52 | 1.25 | 39.2 | |||
| 620 K/H2 | 77 | hydrogen | Fe3+ | 0.37 | 1.66 | 0.45 | 13.5 |
| Fe3+ | 0.42 | 1.13 | 0.79 | 28.6 | |||
| Fe2+ | 1.09 | 2.21 | 0.76 | 36.9 | |||
| Fe2+ | 1.42 | 2.53 | 0.59 | 21.0 | |||
| 140 K/vac | 140 K | vacuum | Fe3+ | 0.31 | 1.73 | 0.53 | 27.9 |
| Fe3+ | 0.32 | 1.14 | 0.61 | 38.6 | |||
| Fe2+ | 0.99 | 2.02 | 0.58 | 12.4 | |||
| Fe2+ | 1.31 | 2.30 | 0.77 | 21.1 | |||
| 300 K/vac | 300 K | vacuum | Fe3+ | 0.25 | 1.63 | 0.52 | 47.5 |
| Fe3+ | 0.32 | 0.98 | 0.59 | 26.9 | |||
| Fe2+ | 1.04 | 2.14 | 0.96 | 25.6 | |||
| 620 K/vac | 300 | vacuum | Fe3+ | 0.24 | 1.96 | 0.50 | 58.6 |
| Fe3+ | 0.40 | 1.27 | 0.95 | 27.6 | |||
| Fe2+ | 1.22 | 1.50 | 0.91 | 13.7 |
| Treatment | Comp. | δ a | Δ b | FWHM c | Rel. Int. d | Srel e |
|---|---|---|---|---|---|---|
| mm s−1 | mm s−1 | mm s−1 | % | |||
| As prepared | Sn4+ | 0.07 | 0.50 | 0.89 | 100 | 1.00 |
| H2/493 K | Sn4+ | 0.54 | - | 1.31 | 24 | 1.54 |
| Pt-Sn(a) | 1.39 | - | 1.64 | 21 | ||
| Pt-Sn(b) | 2.45 | - | 1.48 | 50 | ||
| SnCl2 | 4.15 | - | 0.74 | 5 | ||
| H2/623 K | Sn4+ | 0.38 | - | 0.68 | 6 | 1.74 |
| Pt-Sn(a) | 1.11 | - | 1.29 | 19 | ||
| Pt-Sn(b) | 2.16 | - | 1.69 | 64 | ||
| SnCl2 | 3.95 | - | 1.14 | 12 | ||
| React. mixt. f | Pt-Sn(a) | 1.22 | - | 1.59 | 43 | 1.42 |
| 473 K | Pt-Sn(b) ≈ β-Sn | 2.53 | - | 1.76 | 52 | |
| SnCl2 | 3.96 | - | 1.58 | 5 |
| Treatment | Comp. | 77 K | 300 K | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| δ a | Δ b | FWHM c | Rel. Int. d | δ a | Δ b | FWHM c | Rel. Int. d | f(rel) e | ||
| mm s−1 | mm s−1 | mm s−1 | % | mm s−1 | mm s−1 | mm s−1 | % | |||
| As received | Sn4+ | 0.00 | 0.72 | 1.17 | 74 | 0.01 | 0.64 | 1.07 | 77 | 4.14 |
| PtSn(a) | 1.49 | - | 1.43 | 26 | 1.42 | - | 1.16 | 23 | 4.87 | |
| H2/573 K | Sn4+(surf) | 0.49 | 0.42 | 0.59 | 6 | 0.53 | 0.43 | 0.39 | 4 | 7.43 |
| Sn2+ | 2.88 | 2.11 | 1.06 | 9 | ||||||
| PtSn(a) | 1.31 | - | 1.27 | 37 | 1.41 | - | 1.25 | 55 | 3.78 | |
| PtSn(b) | 2.32 | - | 2.00 | 48 | 2.38 | - | 1.60 | 40 | 6.50 | |
| CO + O2 | Sn4+ | 0.00 | 0.68 | 1.25 | 72 | 0.01 | 0.72 | 1.06 | 83 | 2.88 |
| 300 K | Sn4+(surf) | 0.86 | - | 1.10 | 16 | 0.93 | - | 0.88 | 8 | 6.28 |
| PtSn | 1.77 | - | 1.41 | 12 | 1.60 | - | 1.32 | 9 | 4.06 | |
| H2/300 K | Sn4+ | 0.04 | 0.70 | 1.09 | 42 | 0.05 | 0.71 | 1.03 | 45 | 5.49 |
| PtSn(a) | 1.22 | - | 1.58 | 24 | 1.36 | - | 1.54 | 32 | 4.53 | |
| PtSn(b) | 2.31 | - | 1.79 | 33 | 2.39 | - | 1.48 | 23 | 7.35 | |
| CO + O2 | Sn4+ | −0.19 | 0.57 | 1.10 | 53 | |||||
| 300 K | Sn4+(surf) | 0.66 | - | 1.24 | 29 | |||||
| Sn2+ | 3.23 | 2.30 | 1.30 | 7 | ||||||
| PtSn(a) | 1.40 | - | 1.41 | 12 | ||||||
| CO/300 K | Sn4+ | −0.02 | 0.71 | 1.13 | 62 | 0.00 | 0.73 | 0.99 | 64 | 3.54 |
| Sn4+(surf) | 0.88 | - | 1.17 | 14 | 0.80 | - | 1.10 | 8 | 5.79 | |
| PtSn | 1.86 | - | 1.69 | 24 | 1.72 | - | 1.74 | 27 | 3.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lázár, K. Advantages of In Situ Mössbauer Spectroscopy in Catalyst Studies with Precaution in Interpretation of Measurements. Spectrosc. J. 2025, 3, 10. https://doi.org/10.3390/spectroscj3010010
Lázár K. Advantages of In Situ Mössbauer Spectroscopy in Catalyst Studies with Precaution in Interpretation of Measurements. Spectroscopy Journal. 2025; 3(1):10. https://doi.org/10.3390/spectroscj3010010
Chicago/Turabian StyleLázár, Károly. 2025. "Advantages of In Situ Mössbauer Spectroscopy in Catalyst Studies with Precaution in Interpretation of Measurements" Spectroscopy Journal 3, no. 1: 10. https://doi.org/10.3390/spectroscj3010010
APA StyleLázár, K. (2025). Advantages of In Situ Mössbauer Spectroscopy in Catalyst Studies with Precaution in Interpretation of Measurements. Spectroscopy Journal, 3(1), 10. https://doi.org/10.3390/spectroscj3010010