
Protein Kinases in Mediating Phage-Bacteria Interactions


Abstract
1. Introduction
2. Brief Sum of HKs and STKs in Bacterial Stress Response and Antibiotic Tolerance/Resistance
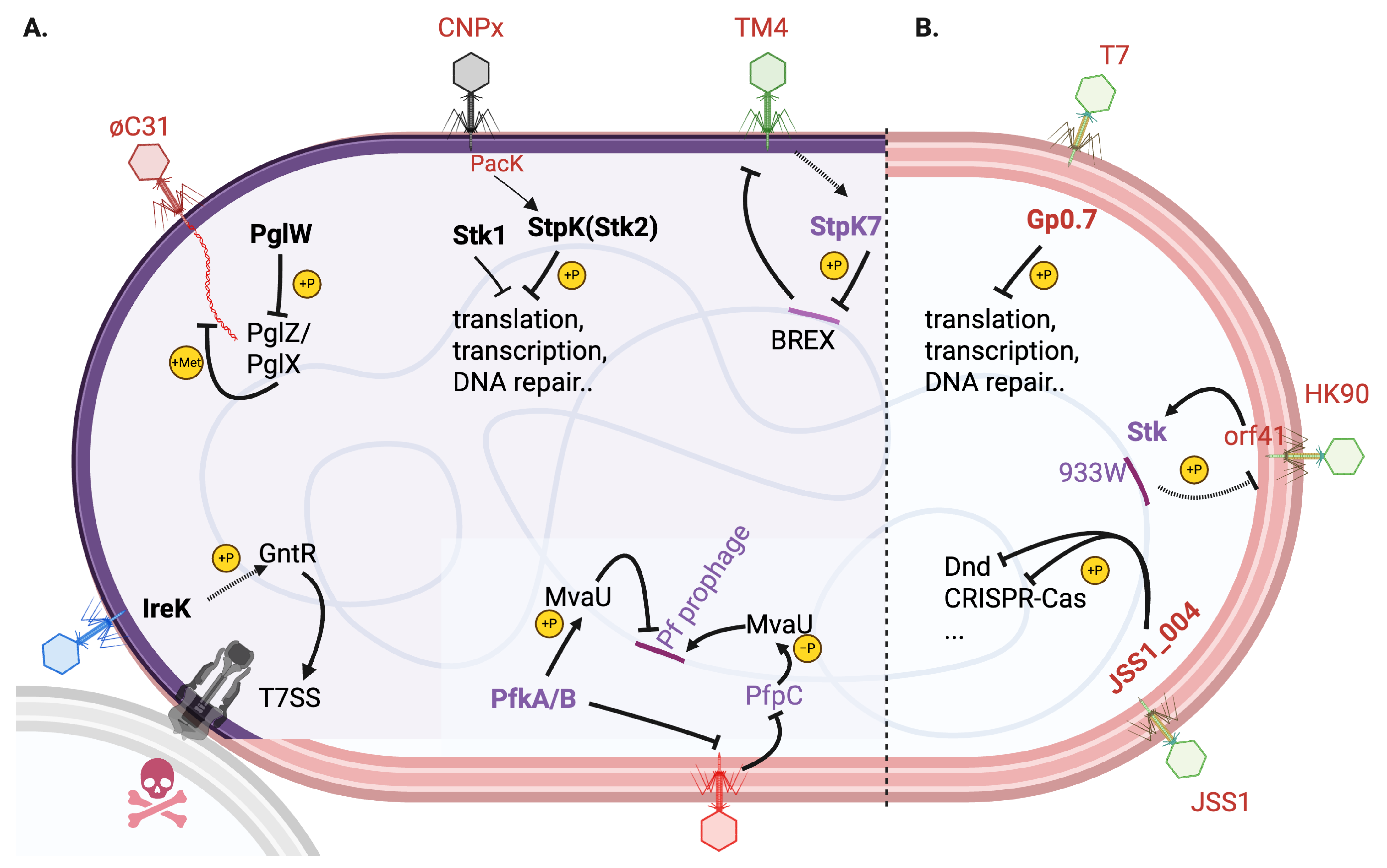
3. Bacteria-Encoded STKs in Phage-Bacteria Interactions

{kind=link}
{kind=link}
| Kinases | Origin and Types | Functions | Reference |
|---|---|---|---|
| StpK7 (MSMEG_1200) | Mycobacterium smegmatis BREX; STK | Mediates TM4 phage infection | [30] |
| StpK | Staphylococcus aureus; STK | Activated by phage protein PacK to mediate abortive infection | [31] |
| PglW | Streptomyces coelicolor; STK | Linked to phage growth limitation (Pgl) and BREX anti-phage systems | [32] |
| IreK | Enterococcus faecalis; STK | Mediates stress-responsive activation of the type VIIb secretion system (T7SS) and contact-dependent antagonism of bystander bacteria | [33] |
| PfkA/B | Pseudomonas aeruginosa; STKs (PfkA/B) and phosphatase (PfpC) | Coordinates prophage activation and antiphage defense | [34] |
| PfpC | |||
| Gp0.7 | E. coli T7 phage; STK | Phosphorylates multiple host E. coli proteins to hijack cellular processes | [35,36,37,38,39] |
| Stk933W | lambdoid phage 933W; tyrosine kinase | Exclusion of secondary phage infection | [40,41] |
| JSS1_004 | Salmonella phage JSS1; STK | Phosphorylate and dismantle multilayered host defences e.g., CRISPR-Cas | [42] |
| Rcs | Serratia sp. ATCC 39006; HK | Balancing innate and adaptive immunity | [43] |
| BarA/UvrY | Marinomonas mediterranea MMB-1; HK | Regulating the dual CRISPR-Cas systems (PAM- dependent or -independent) to confer phage resistance | [9] |
| Cpx | Klebsiella pneumoniae, etc; HK | Dual role in enhancing carbapenem resistance and conjugative plasmid transfer; potential interference with phage infection | [44,45] |
3.1. Mycobacterium smegmatis StpK7: A Pro-Phage Kinase [30]
3.2. Staphylococcus aureus StpK: Altruistic Suicide via Phosphorylation [31]
3.3. Streptomyces PglW: Component of Pgl/BREX Defense [32]
3.4. Enterococcus faecalis IreK: Sensing Phage Infection Signals [33]
3.5. Pseudomonas aeruginosa KKP: Dual-Function Module Balancing Prophage Activation and Antiphage Defense [34]
4. Phage-Encoded STKs in Phage-Bacteria Interactions [10]
5. Bacteria HKs in Phage-Bacteria Interactions
5.1. The Rcs (Regulator of Capsule Synthesis) System: A Multifaceted Envelope Stress Response Pathway [55]
5.2. BarA/UvrY (PpoS/PpoR) System in Phage Resistance via CRISPR-Cas in Marinomonas mediterranea [9]
5.3. The Cpx Envelope Stress Response System
5.3.1. Molecular Mechanisms and Physiological Relevance
5.3.2. Recent Update of Cpx in Antibiotic Hetero-Resistance and Resistance
5.3.3. Cpx in Plasmid Mobilization and Phage Interaction
5.4. Phage-Encoded Anti-HKs Proteins
6. Perspectives and Biotechnical Applications
Funding
Conflicts of Interest
References
- Janczarek, M.; Vinardell, J.M.; Lipa, P.; Karas, M. Hanks-Type Serine/Threonine Protein Kinases and Phosphatases in Bacteria: Roles in Signaling and Adaptation to Various Environments. Int. J. Mol. Sci. 2018, 19, 2872. [Google Scholar] [CrossRef] [PubMed]
- Tierney, A.R.; Rather, P.N. Roles of two-component regulatory systems in antibiotic resistance. Future Microbiol. 2019, 14, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Francis, V.I.; Stevenson, E.C.; Porter, S.L. Two-component systems required for virulence in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2017, 364, fnx104. [Google Scholar] [CrossRef] [PubMed]
- Truong-Bolduc, Q.C.; Ding, Y.; Hooper, D.C. Posttranslational modification influences the effects of MgrA on norA expression in Staphylococcus aureus. J. Bacteriol. 2008, 190, 7375–7381. [Google Scholar] [CrossRef]
- Kaspy, I.; Rotem, E.; Weiss, N.; Ronin, I.; Balaban, N.Q.; Glaser, G. HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat. Commun. 2013, 4, 3001. [Google Scholar] [CrossRef]
- Germain, E.; Castro-Roa, D.; Zenkin, N.; Gerdes, K. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 2013, 52, 248–254. [Google Scholar] [CrossRef]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Lucas-Elio, P.; Molina-Quintero, L.R.; Xu, H.; Sanchez-Amat, A. A histidine kinase and a response regulator provide phage resistance to Marinomonas mediterranea via CRISPR-Cas regulation. Sci. Rep. 2021, 11, 20564. [Google Scholar] [CrossRef]
- Robertson, E.S. Survival of the fittest: A role for phage-encoded eukaryotic-like kinases. Mol. Microbiol. 2011, 82, 539–541. [Google Scholar] [CrossRef]
- Bonne Kohler, J.; Jers, C.; Senissar, M.; Shi, L.; Derouiche, A.; Mijakovic, I. Importance of protein Ser/Thr/Tyr phosphorylation for bacterial pathogenesis. FEBS Lett. 2020, 594, 2339–2369. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.F.; Georgellis, D. Environmental adaptation and diversification of bacterial two-component systems. Curr. Opin. Microbiol. 2023, 76, 102399. [Google Scholar] [CrossRef] [PubMed]
- Jacob-Dubuisson, F.; Mechaly, A.; Betton, J.M.; Antoine, R. Structural insights into the signalling mechanisms of two-component systems. Nat. Rev. Microbiol. 2018, 16, 585–593. [Google Scholar] [CrossRef]
- Buschiazzo, A.; Trajtenberg, F. Two-Component Sensing and Regulation: How Do Histidine Kinases Talk with Response Regulators at the Molecular Level? Annu. Rev. Microbiol. 2019, 73, 507–528. [Google Scholar] [CrossRef]
- Russo, F.D.; Silhavy, T.J. EnvZ controls the concentration of phosphorylated OmpR to mediate osmoregulation of the porin genes. J. Mol. Biol. 1991, 222, 567–580. [Google Scholar] [CrossRef]
- Hirakawa, H.; Nishino, K.; Yamada, J.; Hirata, T.; Yamaguchi, A. Beta-lactam resistance modulated by the overexpression of response regulators of two-component signal transduction systems in Escherichia coli. J. Antimicrob. Chemother. 2003, 52, 576–582. [Google Scholar] [CrossRef]
- Broder, U.N.; Jaeger, T.; Jenal, U. LadS is a calcium-responsive kinase that induces acute-to-chronic virulence switch in Pseudomonas aeruginosa. Nat. Microbiol. 2016, 2, 16184. [Google Scholar] [CrossRef]
- Pompeo, F.; Foulquier, E.; Galinier, A. Impact of Serine/Threonine Protein Kinases on the Regulation of Sporulation in Bacillus subtilis. Front. Microbiol. 2016, 7, 568. [Google Scholar] [CrossRef]
- Li, R.; Zhu, X.; Zhang, P.; Wu, X.; Jin, Q.; Pan, J. Ser/Thr protein kinase Stk1 phosphorylates the key transcriptional regulator AlgR to modulate virulence and resistance in Pseudomonas aeruginosa. Virulence 2024, 15, 2367649. [Google Scholar] [CrossRef]
- Ventura, M.; Rieck, B.; Boldrin, F.; Degiacomi, G.; Bellinzoni, M.; Barilone, N.; Alzaidi, F.; Alzari, P.M.; Manganelli, R.; O’Hare, H.M. GarA is an essential regulator of metabolism in Mycobacterium tuberculosis. Mol. Microbiol. 2013, 90, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Baerentsen, R.; Brodersen, D.E. Phylogeny Reveals Novel HipA-Homologous Kinase Families and Toxin-Antitoxin Gene Organizations. mBio 2021, 12, e0105821. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 2016, 354, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef]
- Zhang, Y.; Zborníková, E.; Rejman, D.; Gerdes, K. Novel (p)ppGpp Binding and Metabolizing Proteins of Escherichia coli. mBio 2018, 9, e02188-17. [Google Scholar] [CrossRef]
- Wang, B.; Dai, P.; Ding, D.; Del Rosario, A.; Grant, R.A.; Pentelute, B.L.; Laub, M.T. Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nat. Chem. Biol. 2019, 15, 141–150. [Google Scholar] [CrossRef]
- Zhang, Y.E.; Baerentsen, R.L.; Fuhrer, T.; Sauer, U.; Gerdes, K.; Brodersen, D.E. (p)ppGpp Regulates a Bacterial Nucleosidase by an Allosteric Two-Domain Switch. Mol. Cell 2019, 74, 1239–1249.e1234. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef]
- Georjon, H.; Bernheim, A. The highly diverse antiphage defence systems of bacteria. Nat. Rev. Microbiol. 2023, 21, 686–700. [Google Scholar] [CrossRef]
- Li, X.; Long, X.; Chen, L.; Guo, X.; Lu, L.; Hu, L.; He, Z.G. Mycobacterial phage TM4 requires a eukaryotic-like Ser/Thr protein kinase to silence and escape anti-phage immunity. Cell Host Microbe 2023, 31, 1469–1480.e1464. [Google Scholar] [CrossRef]
- Depardieu, F.; Didier, J.P.; Bernheim, A.; Sherlock, A.; Molina, H.; Duclos, B.; Bikard, D. A Eukaryotic-like Serine/Threonine Kinase Protects Staphylococci against Phages. Cell Host Microbe 2016, 20, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Hoskisson, P.A.; Sumby, P.; Smith, M.C.M. The phage growth limitation system in Streptomyces coelicolor A(3)2 is a toxin/antitoxin system, comprising enzymes with DNA methyltransferase, protein kinase and ATPase activity. Virology 2015, 477, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Willett, J.L.E.; Dunny, G.M.; Duerkop, B.A. Phage infection and sub-lethal antibiotic exposure mediate Enterococcus faecalis type VII secretion system dependent inhibition of bystander bacteria. PLoS Genet. 2021, 17, e1009204. [Google Scholar] [CrossRef]
- Guo, Y.; Tang, K.; Sit, B.; Gu, J.; Chen, R.; Shao, X.; Lin, S.; Huang, Z.; Nie, Z.; Lin, J.; et al. Control of lysogeny and antiphage defense by a prophage-encoded kinase-phosphatase module. Nat. Commun. 2024, 15, 7244. [Google Scholar] [CrossRef]
- Robertson, E.S.; Nicholson, A.W. Protein kinase of bacteriophage T7 induces the phosphorylation of only a small number of proteins in the infected cell. Virology 1990, 175, 525–534. [Google Scholar] [CrossRef]
- Gone, S.; Nicholson, A.W. Bacteriophage T7 protein kinase: Site of inhibitory autophosphorylation, and use of dephosphorylated enzyme for efficient modification of protein in vitro. Protein Expr. Purif. 2012, 85, 218–223. [Google Scholar] [CrossRef]
- Robertson, E.S.; Aggison, L.A.; Nicholson, A.W. Phosphorylation of elongation factor G and ribosomal protein S6 in bacteriophage T7-infected Escherichia coli. Mol. Microbiol. 1994, 11, 1045–1057. [Google Scholar] [CrossRef]
- Marchand, I.; Nicholson, A.W.; Dreyfus, M. Bacteriophage T7 protein kinase phosphorylates RNase E and stabilizes mRNAs synthesized by T7 RNA polymerase. Mol. Microbiol. 2001, 42, 767–776. [Google Scholar] [CrossRef]
- Bartolec, T.; Mitosch, K.; Potel, C.; Corona, F.; Yang, A.L.J.; Burtscher, M.L.; Koumoutsi, A.; Becher, I.; Bobonis, J.; Karcher, N.; et al. Pervasive phosphorylation by phage T7 kinase disarms bacterial defenses. bioRxiv 2024. [Google Scholar] [CrossRef]
- Friedman, D.I.; Mozola, C.C.; Beeri, K.; Ko, C.C.; Reynolds, J.L. Activation of a prophage-encoded tyrosine kinase by a heterologous infecting phage results in a self-inflicted abortive infection. Mol. Microbiol. 2011, 82, 567–577. [Google Scholar] [CrossRef]
- Juhala, R.J.; Ford, M.E.; Duda, R.L.; Youlton, A.; Hatfull, G.F.; Hendrix, R.W. Genomic sequences of bacteriophages HK97 and HK022: Pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 2000, 299, 27–51. [Google Scholar] [CrossRef]
- Jiang, S.; Chen, C.; Huang, W.; He, Y.; Du, X.; Wang, Y.; Ou, H.; Deng, Z.; Xu, C.; Jiang, L.; et al. A widespread phage-encoded kinase enables evasion of multiple host antiphage defence systems. Nat. Microbiol. 2024, 9, 3226–3239. [Google Scholar] [CrossRef]
- Smith, L.M.; Jackson, S.A.; Malone, L.M.; Ussher, J.E.; Gardner, P.P.; Fineran, P.C. The Rcs stress response inversely controls surface and CRISPR-Cas adaptive immunity to discriminate plasmids and phages. Nat. Microbiol. 2021, 6, 162–172. [Google Scholar] [CrossRef]
- Liu, Z.; Guan, J.; Chen, Z.; Tai, C.; Deng, Z.; Chao, Y.; Ou, H.Y. CpxR promotes the carbapenem antibiotic resistance of Klebsiella pneumoniae by directly regulating the expression and the dissemination of bla(KPC) on the IncFII conjugative plasmid. Emerg. Microbes Infect. 2023, 12, 2256427. [Google Scholar] [CrossRef]
- Quinones-Olvera, N.; Owen, S.V.; McCully, L.M.; Marin, M.G.; Rand, E.A.; Fan, A.C.; Martins Dosumu, O.J.; Paul, K.; Sanchez Castano, C.E.; Petherbridge, R.; et al. Diverse and abundant phages exploit conjugative plasmids. Nat. Commun. 2024, 15, 3197. [Google Scholar] [CrossRef]
- Beltramini, A.M.; Mukhopadhyay, C.D.; Pancholi, V. Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect. Immun. 2009, 77, 1406–1416. [Google Scholar] [CrossRef]
- Debarbouille, M.; Dramsi, S.; Dussurget, O.; Nahori, M.A.; Vaganay, E.; Jouvion, G.; Cozzone, A.; Msadek, T.; Duclos, B. Characterization of a serine/threonine kinase involved in virulence of Staphylococcus aureus. J. Bacteriol. 2009, 191, 4070–4081. [Google Scholar] [CrossRef]
- Chinenova, T.A.; Mkrtumian, N.M.; Lomovskaia, N.D. Genetic characteristics of a new phage resistance trait in Streptomyces coelicolor A3(2). Genetika 1982, 18, 1945–1952. [Google Scholar]
- Laity, C.; Chater, K.F.; Lewis, C.G.; Buttner, M.J. Genetic analysis of the phi C31-specific phage growth limitation (Pgl) system of Streptomyces coelicolor A3(2). Mol. Microbiol. 1993, 7, 329–336. [Google Scholar] [CrossRef]
- Sumby, P.; Smith, M.C. Phase variation in the phage growth limitation system of Streptomyces coelicolor A3(2). J. Bacteriol. 2003, 185, 4558–4563. [Google Scholar] [CrossRef]
- Bayliss, C.D.; Callaghan, M.J.; Moxon, E.R. High allelic diversity in the methyltransferase gene of a phase variable type III restriction-modification system has implications for the fitness of Haemophilus influenzae. Nucleic Acids Res. 2006, 34, 4046–4059. [Google Scholar] [CrossRef]
- Studier, F.W. The genetics and physiology of bacteriophage T7. Virology 1969, 39, 562–574. [Google Scholar] [CrossRef]
- Hochhauser, D.; Millman, A.; Sorek, R. The defense island repertoire of the Escherichia coli pan-genome. PLoS Genet. 2023, 19, e1010694. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Silhavy, T.J. Envelope stress responses: Balancing damage repair and toxicity. Nat. Rev. Microbiol. 2019, 17, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Wall, E.; Majdalani, N.; Gottesman, S. The Complex Rcs Regulatory Cascade. Annu. Rev. Microbiol. 2018, 72, 111–139. [Google Scholar] [CrossRef]
- Girgis, H.S.; Liu, Y.; Ryu, W.S.; Tavazoie, S. A comprehensive genetic characterization of bacterial motility. PLoS Genet. 2007, 3, 1644–1660. [Google Scholar] [CrossRef]
- Farris, C.; Sanowar, S.; Bader, M.W.; Pfuetzner, R.; Miller, S.I. Antimicrobial peptides activate the Rcs regulon through the outer membrane lipoprotein RcsF. J. Bacteriol. 2010, 192, 4894–4903. [Google Scholar] [CrossRef]
- Konovalova, A.; Mitchell, A.M.; Silhavy, T.J. A lipoprotein/beta-barrel complex monitors lipopolysaccharide integrity transducing information across the outer membrane. eLife 2016, 5, e15276. [Google Scholar] [CrossRef]
- Laubacher, M.E.; Ades, S.E. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J. Bacteriol. 2008, 190, 2065–2074. [Google Scholar] [CrossRef]
- Shiba, Y.; Miyagawa, H.; Nagahama, H.; Matsumoto, K.; Kondo, D.; Matsuoka, S.; Matsumoto, K.; Hara, H. Exploring the relationship between lipoprotein mislocalization and activation of the Rcs signal transduction system in Escherichia coli. Microbiology 2012, 158, 1238–1248. [Google Scholar] [CrossRef]
- Ebel, W.; Vaughn, G.J.; Peters, H.K., 3rd; Trempy, J.E. Inactivation of mdoH leads to increased expression of colanic acid capsular polysaccharide in Escherichia coli. J. Bacteriol. 1997, 179, 6858–6861. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Szewczyk, J.; Pesavento, C.; Zietek, M.; Banzhaf, M.; Roszczenko, P.; Asmar, A.; Laloux, G.; Hov, A.K.; Leverrier, P.; et al. Detecting envelope stress by monitoring beta-barrel assembly. Cell 2014, 159, 1652–1664. [Google Scholar] [CrossRef] [PubMed]
- Hussein, N.A.; Cho, S.H.; Laloux, G.; Siam, R.; Collet, J.F. Distinct domains of Escherichia coli IgaA connect envelope stress sensing and down-regulation of the Rcs phosphorelay across subcellular compartments. PLoS Genet. 2018, 14, e1007398. [Google Scholar] [CrossRef]
- Wehland, M.; Bernhard, F. The RcsAB box. Characterization of a new operator essential for the regulation of exopolysaccharide biosynthesis in enteric bacteria. J. Biol. Chem. 2000, 275, 7013–7020. [Google Scholar] [CrossRef]
- Majdalani, N.; Chen, S.; Murrow, J.; St John, K.; Gottesman, S. Regulation of RpoS by a novel small RNA: The characterization of RprA. Mol. Microbiol. 2001, 39, 1382–1394. [Google Scholar] [CrossRef]
- Francez-Charlot, A.; Laugel, B.; Van Gemert, A.; Dubarry, N.; Wiorowski, F.; Castanie-Cornet, M.P.; Gutierrez, C.; Cam, K. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 2003, 49, 823–832. [Google Scholar] [CrossRef]
- Pernestig, A.K.; Georgellis, D.; Romeo, T.; Suzuki, K.; Tomenius, H.; Normark, S.; Melefors, O. The Escherichia coli BarA-UvrY two-component system is needed for efficient switching between glycolytic and gluconeogenic carbon sources. J. Bacteriol. 2003, 185, 843–853. [Google Scholar] [CrossRef]
- Wan, J.; Gao, X.; Liu, F. Regulatory role of the Cpx ESR in bacterial behaviours. Virulence 2024, 15, 2404951. [Google Scholar] [CrossRef]
- Danese, P.N.; Silhavy, T.J. CpxP, a stress-combative member of the Cpx regulon. J. Bacteriol. 1998, 180, 831–839. [Google Scholar] [CrossRef]
- Jubelin, G.; Vianney, A.; Beloin, C.; Ghigo, J.M.; Lazzaroni, J.C.; Lejeune, P.; Dorel, C. CpxR/OmpR interplay regulates curli gene expression in response to osmolarity in Escherichia coli. J. Bacteriol. 2005, 187, 2038–2049. [Google Scholar] [CrossRef]
- Evans, K.L.; Kannan, S.; Li, G.; de Pedro, M.A.; Young, K.D. Eliminating a set of four penicillin binding proteins triggers the Rcs phosphorelay and Cpx stress responses in Escherichia coli. J. Bacteriol. 2013, 195, 4415–4424. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ishihama, A. Characterization of copper-inducible promoters regulated by CpxA/CpxR in Escherichia coli. Biosci. Biotechnol. Biochem. 2006, 70, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.L. Everything old is new again: An update on current research on the Cpx envelope stress response. Biochim. Biophys. Acta 2014, 1843, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Raivio, T.L. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J. Bacteriol. 2009, 191, 1798–1815. [Google Scholar] [CrossRef]
- Raivio, T.L.; Popkin, D.L.; Silhavy, T.J. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J. Bacteriol. 1999, 181, 5263–5272. [Google Scholar] [CrossRef]
- Chao, Y.; Vogel, J. A 3′ UTR-Derived Small RNA Provides the Regulatory Noncoding Arm of the Inner Membrane Stress Response. Mol. Cell 2016, 61, 352–363. [Google Scholar] [CrossRef]
- Grabowicz, M.; Koren, D.; Silhavy, T.J. The CpxQ sRNA Negatively Regulates Skp To Prevent Mistargeting of beta-Barrel Outer Membrane Proteins into the Cytoplasmic Membrane. mBio 2016, 7, e00312–e00316. [Google Scholar] [CrossRef]
- Andrieu, C.; Loiseau, L.; Vergnes, A.; Gagnot, S.; Barre, R.; Aussel, L.; Collet, J.F.; Ezraty, B. Salmonella Typhimurium uses the Cpx stress response to detect N-chlorotaurine and promote the repair of oxidized proteins. Proc. Natl. Acad. Sci. USA 2023, 120, e2215997120. [Google Scholar] [CrossRef]
- Cho, T.H.S.; Wang, J.; Raivio, T.L. NlpE Is an OmpA-Associated Outer Membrane Sensor of the Cpx Envelope Stress Response. J. Bacteriol. 2023, 205, e0040722. [Google Scholar] [CrossRef]
- Jones, C.H.; Danese, P.N.; Pinkner, J.S.; Silhavy, T.J.; Hultgren, S.J. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J. 1997, 16, 6394–6406. [Google Scholar] [CrossRef]
- Hung, D.L.; Raivio, T.L.; Jones, C.H.; Silhavy, T.J.; Hultgren, S.J. Cpx signaling pathway monitors biogenesis and affects assembly and expression of P pili. EMBO J. 2001, 20, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Debnath, I.; Norton, J.P.; Barber, A.E.; Ott, E.M.; Dhakal, B.K.; Kulesus, R.R.; Mulvey, M.A. The Cpx stress response system potentiates the fitness and virulence of uropathogenic Escherichia coli. Infect. Immun. 2013, 81, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.X.; Yi, X.X.; Cho, A.; O’Gara, F.; Wang, Y.P. CpxR Activates MexAB-OprM Efflux Pump Expression and Enhances Antibiotic Resistance in Both Laboratory and Clinical nalB-Type Isolates of Pseudomonas aeruginosa. PLoS Pathog. 2016, 12, e1005932. [Google Scholar] [CrossRef]
- Srinivasan, V.B.; Rajamohan, G. KpnEF, a new member of the Klebsiella pneumoniae cell envelope stress response regulon, is an SMR-type efflux pump involved in broad-spectrum antimicrobial resistance. Antimicrob. Agents Chemother. 2013, 57, 4449–4462. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.L.; Bina, X.R.; Slamti, L.; Waldor, M.K.; Bina, J.E. Reciprocal regulation of resistance-nodulation-division efflux systems and the Cpx two-component system in Vibrio cholerae. Infect. Immun. 2014, 82, 2980–2991. [Google Scholar] [CrossRef]
- Masi, M.; Pinet, E.; Pages, J.M. Complex Response of the CpxAR Two-Component System to beta-Lactams on Antibiotic Resistance and Envelope Homeostasis in Enterobacteriaceae. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Siryaporn, A.; Goulian, M. Cross-talk suppression between the CpxA-CpxR and EnvZ-OmpR two-component systems in E. coli. Mol. Microbiol. 2008, 70, 494–506. [Google Scholar] [CrossRef]
- Lopez, C.; Checa, S.K.; Soncini, F.C. CpxR/CpxA Controls scsABCD Transcription To Counteract Copper and Oxidative Stress in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2018, 200, e00126-18. [Google Scholar] [CrossRef]
- Yan, K.; Liu, T.; Duan, B.Z.; Liu, F.; Cao, M.M.; Peng, W.; Dai, Q.; Chen, H.C.; Yuan, F.Y.; Bei, W.C. The CpxAR Two-Component System Contributes to Growth, Stress Resistance, and Virulence of by Upregulating Transcription. Front. Microbiol. 2020, 11, 1026. [Google Scholar] [CrossRef]
- Jaswal, K.; Shrivastava, M.; Roy, D.; Agrawal, S.; Chaba, R. Metabolism of long-chain fatty acids affects disulfide bond formation in Escherichia coli and activates envelope stress response pathways as a combat strategy. PLoS Genet. 2020, 16, e1009081. [Google Scholar] [CrossRef]
- Kumar, A.; Sperandio, V. Indole Signaling at the Host-Microbiota-Pathogen Interface. mBio 2019, 10, e01031-19. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Russell, R.M.; Pifer, R.; Menezes-Garcia, Z.; Cuesta, S.; Narayanan, S.; MacMillan, J.B.; Sperandio, V. The Serotonin Neurotransmitter Modulates Virulence of Enteric Pathogens. Cell Host Microbe 2020, 28, 41–53.e48. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.J.; Bennison, D.J.; Kulkarni, E.; Azar, H.; Sun, H.; Li, H.; Bradshaw, J.; Yeap, H.W.; Lim, N.; Mishra, V.; et al. Aminoglycoside heteroresistance in Enterobacter cloacae is driven by the cell envelope stress response. mBio 2024, 15, e0169924. [Google Scholar] [CrossRef] [PubMed]
- McEwen, J.; Silverman, P. Chromosomal mutations of Escherichia coli that alter expression of conjugative plasmid functions. Proc. Natl. Acad. Sci. USA 1980, 77, 513–517. [Google Scholar] [CrossRef]
- McEwen, J.; Silverman, P. Genetic analysis of Escherichia coli K-12 chromosomal mutants defective in expression of F-plasmid functions: Identification of genes cpxA and cpxB. J. Bacteriol. 1980, 144, 60–67. [Google Scholar] [CrossRef]
- Schroven, K.; Putzeys, L.; Kerremans, A.; Ceyssens, P.J.; Vallino, M.; Paeshuyse, J.; Haque, F.; Yusuf, A.; Koch, M.D.; Lavigne, R. The phage-encoded PIT4 protein affects multiple two-component systems of Pseudomonas aeruginosa. Microbiol. Spectr. 2023, 11, e0237223. [Google Scholar] [CrossRef]
- Leprince, A.; Mahillon, J. Phage Adsorption to Gram-Positive Bacteria. Viruses 2023, 15, 196. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.E. Protein Kinases in Mediating Phage-Bacteria Interactions. Kinases Phosphatases 2025, 3, 14. https://doi.org/10.3390/kinasesphosphatases3030014
Zhang YE. Protein Kinases in Mediating Phage-Bacteria Interactions. Kinases and Phosphatases. 2025; 3(3):14. https://doi.org/10.3390/kinasesphosphatases3030014
Chicago/Turabian StyleZhang, Yong Everett. 2025. "Protein Kinases in Mediating Phage-Bacteria Interactions" Kinases and Phosphatases 3, no. 3: 14. https://doi.org/10.3390/kinasesphosphatases3030014
APA StyleZhang, Y. E. (2025). Protein Kinases in Mediating Phage-Bacteria Interactions. Kinases and Phosphatases, 3(3), 14. https://doi.org/10.3390/kinasesphosphatases3030014