
Evolutionary Conservation and Diversification of Five Pax6 Homologs in the Horseshoe Crab Species Cluster



Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Assembly Sources
2.2. Homolog Searches
2.3. Gene Model Annotation
2.4. Gene Tree Reconstruction
2.5. dN/dS Analysis
2.6. Molecular Clock Analysis
2.7. Phylogenetic Analysis of Interparalog Amino Acid Residue Differences
3. Results
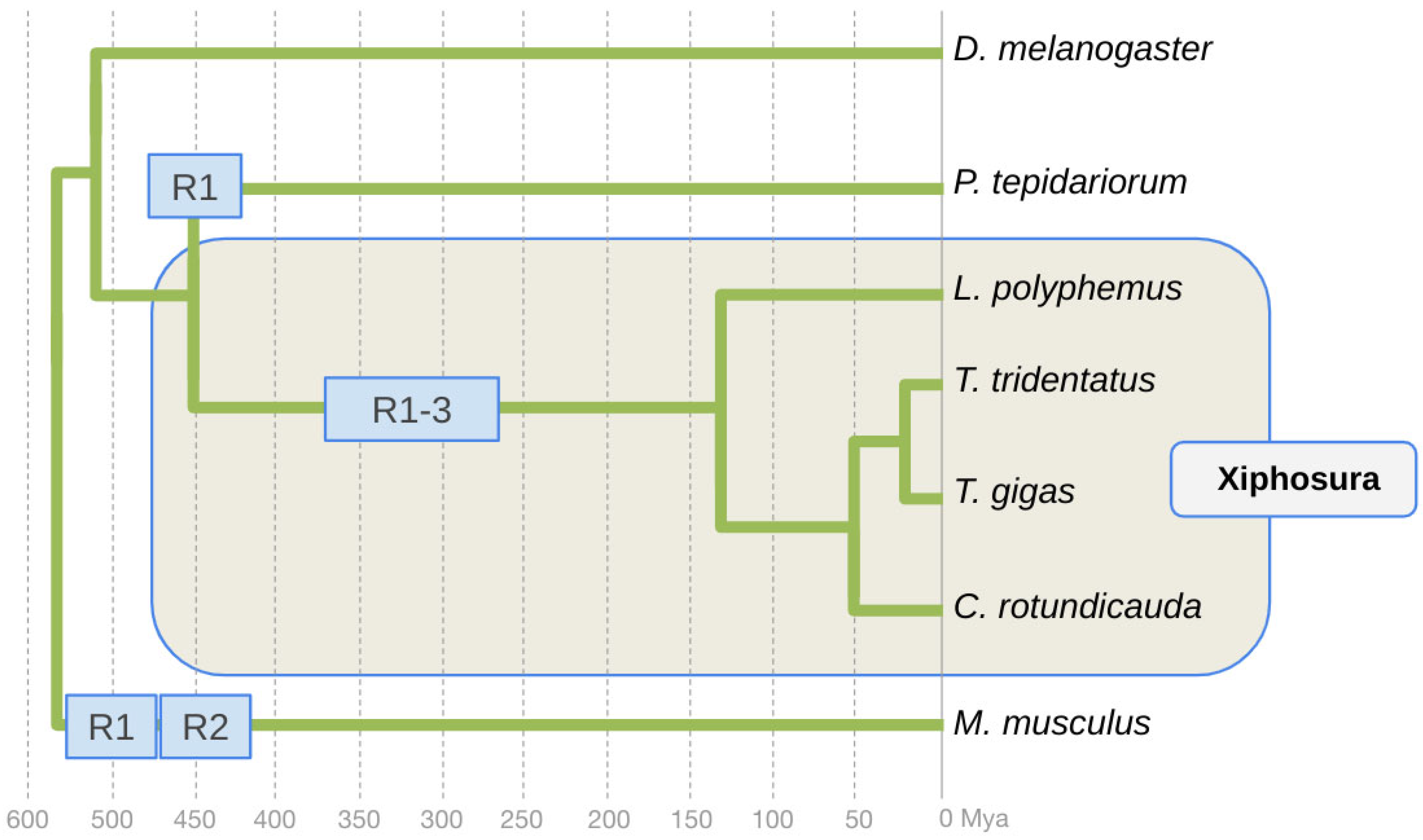
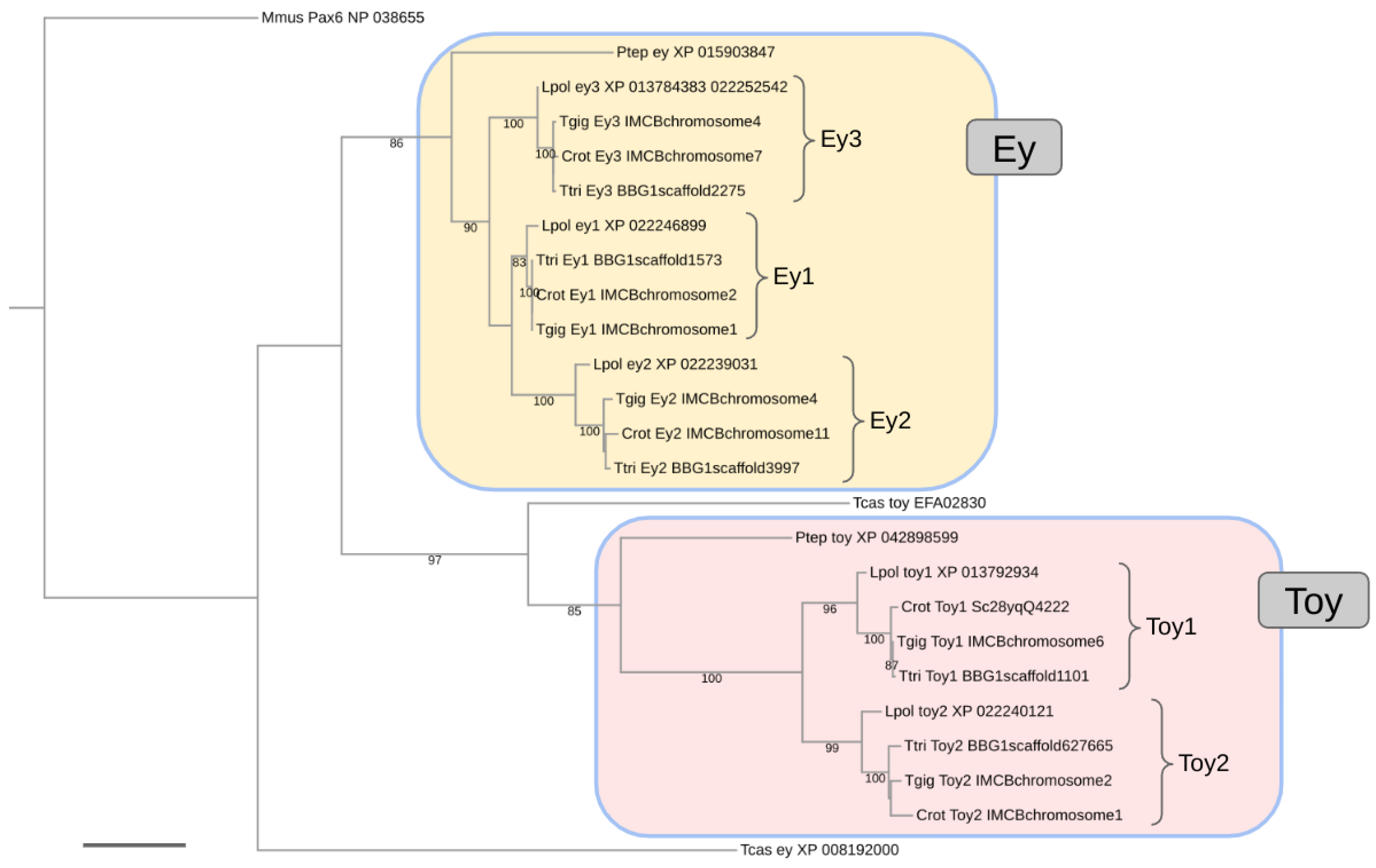
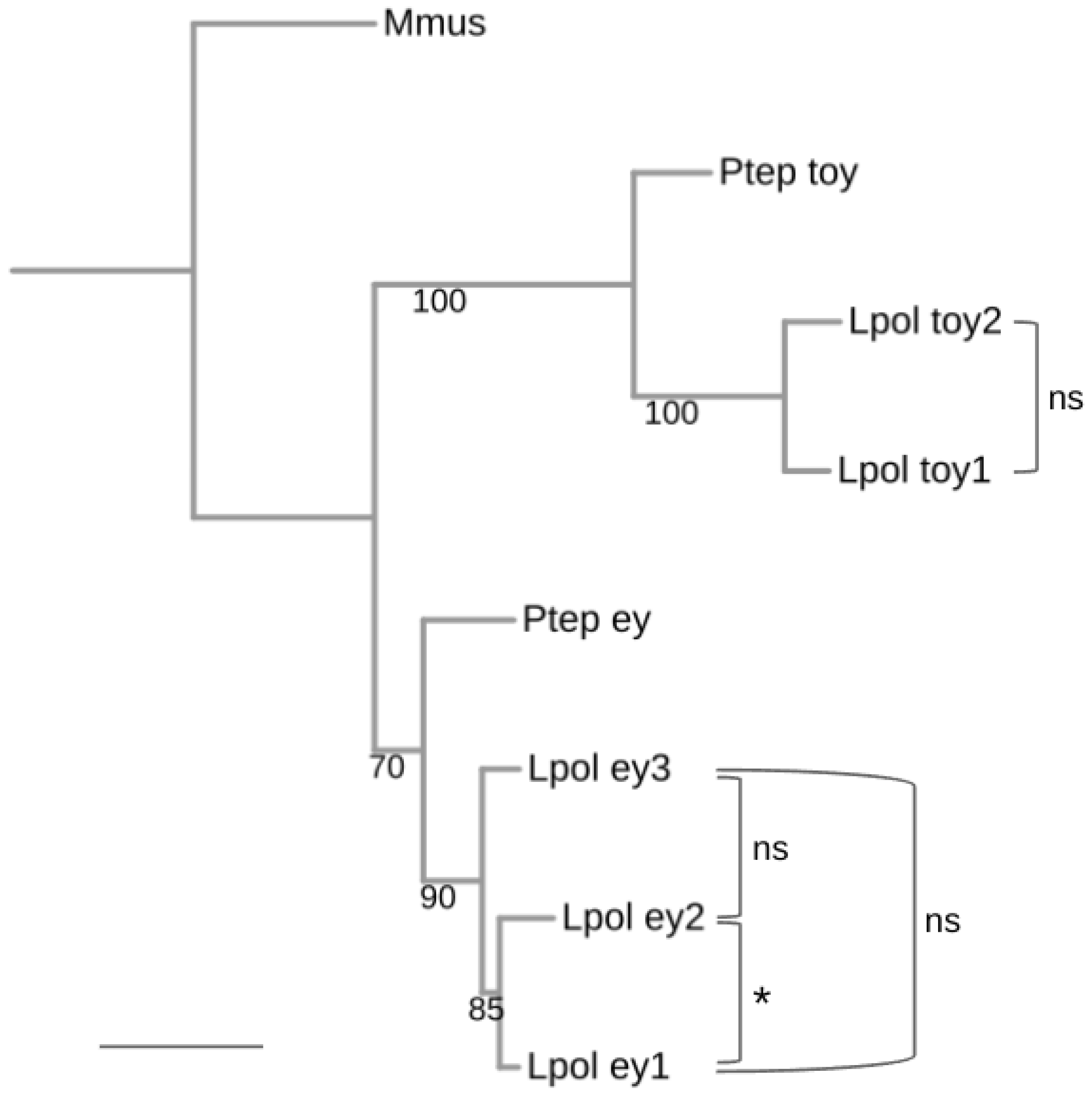
3.1. Conservation of Three Homologs of ey in Horseshoe Crabs
3.2. Conservation of Two Homologs of toy in Horseshoe Crabs
3.3. Strong Purifying Selection on All Horseshoe Crab Pax6 Homologs
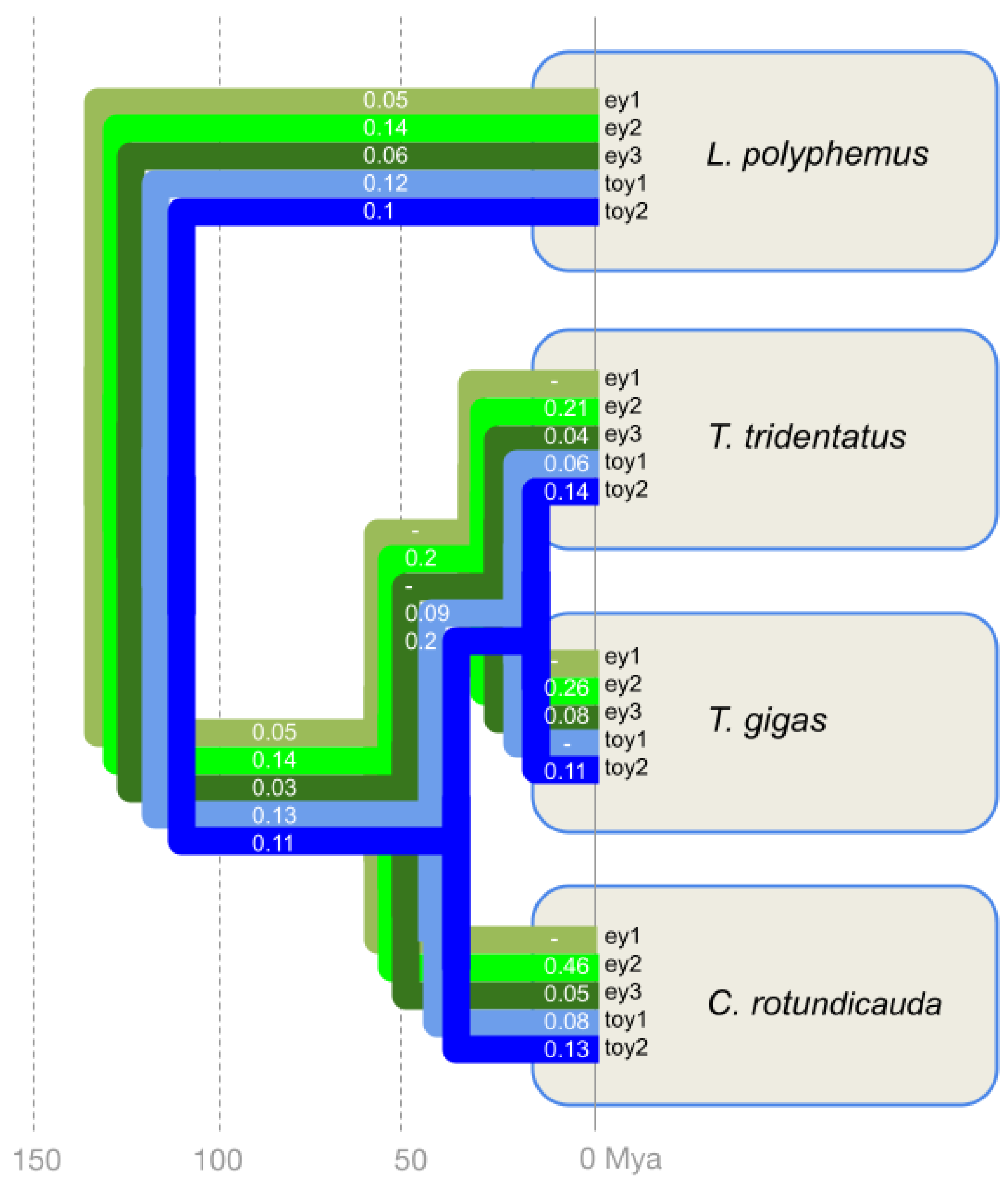
3.4. Largely Homogenous Protein Sequence Diversification Rates among the Xiphosuran ey and toy Paralogs
3.5. Evidence of Higher Paralog Protein Sequence Diversification Rates Preceding Lineage Separation in the Horseshoe Crab Species Cluster
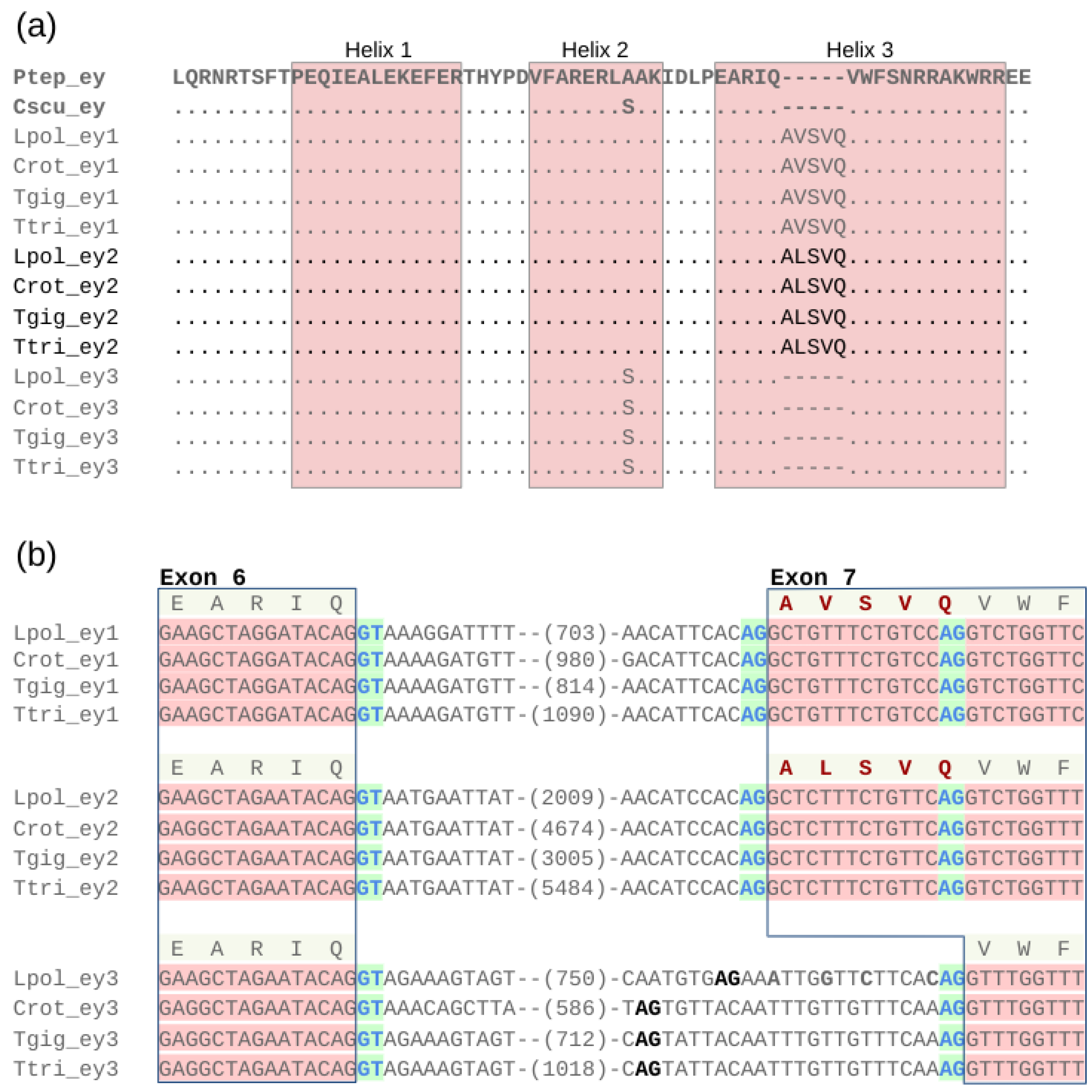
3.6. Conservation of a Homeodomain Expanding Splice Isoform in ey1 and ey2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, P.P. Chelicerates. Curr. Biol. 2018, 28, R774–R778. [Google Scholar] [CrossRef]
- Battelle, B.-A. The Eyes of Limulus polyphemus (Xiphosura, Chelicerata) and Their Afferent and Efferent Projections. Arthropod Struct. Dev. 2006, 35, 261–274. [Google Scholar] [CrossRef]
- Harzsch, S.; Hafner, G. Evolution of Eye Development in Arthropods: Phylogenetic Aspects. Arthropod Struct. Dev. 2006, 35, 319–340. [Google Scholar] [CrossRef]
- Strausfeld, N.J.; Ma, X.; Edgecombe, G.D.; Fortey, R.A.; Land, M.F.; Liu, Y.; Cong, P.; Hou, X. Arthropod Eyes: The Early Cambrian Fossil Record and Divergent Evolution of Visual Systems. Arthropod Struct. Dev. 2016, 45, 152–172. [Google Scholar] [CrossRef]
- Kin, A.; Błażejowski, B. The Horseshoe Crab of the Genus Limulus: Living Fossil or Stabilomorph? PLoS ONE 2014, 9, e108036. [Google Scholar] [CrossRef]
- Battelle, B.-A.; Ryan, J.F.; Kempler, K.E.; Saraf, S.R.; Marten, C.E.; Warren, W.C.; Minx, P.J.; Montague, M.J.; Green, P.J.; Schmidt, S.A.; et al. Opsin Repertoire and Expression Patterns in Horseshoe Crabs: Evidence from the Genome of Limulus polyphemus (Arthropoda: Chelicerata). Genome Biol. Evol. 2016, 8, 1571–1589. [Google Scholar] [CrossRef]
- Blackburn, D.C.; Conley, K.W.; Plachetzki, D.C.; Kempler, K.; Battelle, B.-A.; Brown, N.L. Isolation and Expression of Pax6 and Atonal Homologues in the American Horseshoe Crab, Limulus polyphemus. Dev. Dyn. 2008, 237, 2209–2219. [Google Scholar] [CrossRef]
- Czerny, T.; Halder, G.; Kloter, U.; Souabni, A.; Gehring, W.J.; Busslinger, M. Twin of Eyeless, a Second Pax-6 Gene of Drosophila, Acts Upstream of Eyeless in the Control of Eye Development. Mol. Cell 1999, 3, 297–307. [Google Scholar] [CrossRef]
- Friedrich, M. Ancient Genetic Redundancy of Eyeless and Twin of Eyeless in the Arthropod Ocular Segment. Dev. Biol. 2017, 432, 192–200. [Google Scholar] [CrossRef]
- Friedrich, M. Coming into Clear Sight at Last: Ancestral and Derived Events during Chelicerate Visual System Development. Bioessays 2022, 44, e2200163. [Google Scholar] [CrossRef]
- Kenny, N.J.; Chan, K.W.; Nong, W.; Qu, Z.; Maeso, I.; Yip, H.Y.; Chan, T.F.; Kwan, H.S.; Holland, P.W.H.; Chu, K.H.; et al. Ancestral Whole-Genome Duplication in the Marine Chelicerate Horseshoe Crabs. Heredity 2016, 116, 190–199. [Google Scholar] [CrossRef]
- Tanacredi, J.T.; Botton, M.L.; Smith, D. Biology and Conservation of Horseshoe Crabs; Springer Science & Business Media: New York, NY, USA, 2009; ISBN 9780387899596. [Google Scholar]
- Lamsdell, J.C. The Phylogeny and Systematics of Xiphosura. PeerJ 2020, 8, e10431. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Yan, Q.; Zhang, L.; Chen, D.; Ruan, L.; Kong, Y.; Shi, H.; Chen, M.; Chen, J. The Draft Genome of Horseshoe Crab Tachypleus Tridentatus Reveals Its Evolutionary Scenario and Well-Developed Innate Immunity. BMC Genom. 2020, 21, 137. [Google Scholar] [CrossRef]
- Obst, M.; Faurby, S.; Bussarawit, S.; Funch, P. Molecular Phylogeny of Extant Horseshoe Crabs (Xiphosura, Limulidae) Indicates Paleogene Diversification of Asian Species. Mol. Phylogenet. Evol. 2012, 62, 21–26. [Google Scholar] [CrossRef]
- Shingate, P.; Ravi, V.; Prasad, A.; Tay, B.-H.; Garg, K.M.; Chattopadhyay, B.; Yap, L.-M.; Rheindt, F.E.; Venkatesh, B. Chromosome-Level Assembly of the Horseshoe Crab Genome Provides Insights into Its Genome Evolution. Nat. Commun. 2020, 11, 2322. [Google Scholar] [CrossRef]
- Shingate, P.; Ravi, V.; Prasad, A.; Tay, B.-H.; Venkatesh, B. Chromosome-Level Genome Assembly of the Coastal Horseshoe Crab (Tachypleus gigas). Mol. Ecol. Resour. 2020, 20, 1748–1760. [Google Scholar] [CrossRef]
- Nong, W.; Qu, Z.; Li, Y.; Barton-Owen, T.; Wong, A.Y.P.; Yip, H.Y.; Lee, H.T.; Narayana, S.; Baril, T.; Swale, T.; et al. Horseshoe Crab Genomes Reveal the Evolution of Genes and microRNAs after Three Rounds of Whole Genome Duplication. Commun. Biol. 2021, 4, 83. [Google Scholar] [CrossRef]
- Simakov, O.; Marlétaz, F.; Yue, J.-X.; O’Connell, B.; Jenkins, J.; Brandt, A.; Calef, R.; Tung, C.-H.; Huang, T.-K.; Schmutz, J.; et al. Deeply Conserved Synteny Resolves Early Events in Vertebrate Evolution. Nat. Ecol. Evol. 2020, 4, 820–830. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer Science & Business Media: New York, NY, USA, 1970. [Google Scholar]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Roest Crollius, H. Chromosome Evolution at the Origin of the Ancestral Vertebrate Genome. Genome Biol. 2018, 19, 166. [Google Scholar] [CrossRef]
- Nakatani, Y.; Shingate, P.; Ravi, V.; Pillai, N.E.; Prasad, A.; McLysaght, A.; Venkatesh, B. Reconstruction of Proto-Vertebrate, Proto-Cyclostome and Proto-Gnathostome Genomes Provides New Insights into Early Vertebrate Evolution. Nat. Commun. 2021, 12, 4489. [Google Scholar] [CrossRef]
- Schwager, E.E.; Sharma, P.P.; Clarke, T.; Leite, D.J.; Wierschin, T.; Pechmann, M.; Akiyama-Oda, Y.; Esposito, L.; Bechsgaard, J.; Bilde, T.; et al. The House Spider Genome Reveals an Ancient Whole-Genome Duplication during Arachnid Evolution. BMC Biol. 2017, 15, 62. [Google Scholar] [CrossRef]
- Birchler, J.A.; Yang, H. The Multiple Fates of Gene Duplications: Deletion, Hypofunctionalization, Subfunctionalization, Neofunctionalization, Dosage Balance Constraints, and Neutral Variation. Plant Cell 2022, 34, 2466–2474. [Google Scholar] [CrossRef]
- Kuzmin, E.; Taylor, J.S.; Boone, C. Retention of Duplicated Genes in Evolution. Trends Genet. 2022, 38, 59–72. [Google Scholar] [CrossRef]
- Weadick, C.J.; Chang, B.S.W. Complex Patterns of Divergence among Green-Sensitive (RH2a) African Cichlid Opsins Revealed by Clade Model Analyses. BMC Evol. Biol. 2012, 12, 206. [Google Scholar] [CrossRef]
- Des Marais, D.L.; Rausher, M.D. Escape from Adaptive Conflict after Duplication in an Anthocyanin Pathway Gene. Nature 2008, 454, 762–765. [Google Scholar] [CrossRef]
- Vavouri, T.; Semple, J.I.; Lehner, B. Widespread Conservation of Genetic Redundancy during a Billion Years of Eukaryotic Evolution. Trends Genet. 2008, 24, 485–488. [Google Scholar] [CrossRef]
- Nowak, M.A.; Boerlijst, M.C.; Cooke, J.; Smith, J.M. Evolution of Genetic Redundancy. Nature 1997, 388, 167–171. [Google Scholar] [CrossRef]
- Liberles, D.A.; Kolesov, G.; Dittmar, K. Understanding Gene Duplication through Biochemistry and Population Genetics. In Evolution after Gene Duplication; Dittmar, K., Liberles, D.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 1–21. [Google Scholar]
- Kimura, M. The Neutral Theory of Molecular Evolution. Sci. Am. 1979, 241, 98–129. [Google Scholar] [CrossRef]
- Liao, Y.Y.; Xu, P.W.; Kwan, K.Y.; Ma, Z.Y.; Fang, H.Y.; Xu, J.Y.; Wang, P.L.; Yang, S.Y.; Xie, S.B.; Xu, S.Q.; et al. Draft Genomic and Transcriptome Resources for Marine Chelicerate Tachypleus Tridentatus. Sci. Data 2019, 6, 190029. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; IEEE: Piscataway, NJ, USA, 2010; pp. 1–8. [Google Scholar]
- Roth, C.; Liberles, D.A. A Systematic Search for Positive Selection in Higher Plants (Embryophytes). BMC Plant Biol. 2006, 6, 12. [Google Scholar] [CrossRef]
- Liberles, D.A. Evaluation of Methods for Determination of a Reconstructed History of Gene Sequence Evolution. Mol. Biol. Evol. 2001, 18, 2040–2047. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A Biologist-Centric Software for Evolutionary Analysis of DNA and Protein Sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Tajima, F. Simple Methods for Testing the Molecular Evolutionary Clock Hypothesis. Genetics 1993, 135, 599–607. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Carroll, S.B.; Grenier, J.K.; Weatherbee, S.D. From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design; John Wiley & Sons: Oxford, UK, 2013. [Google Scholar]
- Chi, N.; Epstein, J.A. Getting Your Pax Straight: Pax Proteins in Development and Disease. Trends Genet. 2002, 18, 41–47. [Google Scholar] [CrossRef]
- Blanco, J.; Pauli, T.; Seimiya, M.; Udolph, G.; Gehring, W.J. Genetic Interactions of Eyes Absent, Twin of Eyeless and Orthodenticle Regulate Sine Oculis Expression during Ocellar Development in Drosophila. Dev. Biol. 2010, 344, 1088–1099. [Google Scholar] [CrossRef]
- Halder, G.; Callaerts, P.; Gehring, W.J. Induction of Ectopic Eyes by Targeting Expression of the Eyeless Gene in Drosophila. Science 1995, 267, 1788. [Google Scholar] [CrossRef]
- Baudouin-Gonzalez, L.; Harper, A.; McGregor, A.P.; Sumner-Rooney, L. Regulation of Eye Determination and Regionalization in the Spider Parasteatoda Tepidariorum. Cells 2022, 11, 631. [Google Scholar] [CrossRef]
- Schomburg, C.; Turetzek, N.; Schacht, M.I.; Schneider, J.; Kirfel, P.; Prpic, N.-M.; Posnien, N. Molecular Characterization and Embryonic Origin of the Eyes in the Common House Spider Parasteatoda Tepidariorum. Evodevo 2015, 6, 15. [Google Scholar] [CrossRef]
- Samadi, L.; Schmid, A.; Eriksson, B.J. Differential Expression of Retinal Determination Genes in the Principal and Secondary Eyes of Cupiennius Salei Keyserling (1877). Evodevo 2015, 6, 16. [Google Scholar] [CrossRef]
- Singh, P.P.; Isambert, H. OHNOLOGS v2: A Comprehensive Resource for the Genes Retained from Whole Genome Duplication in Vertebrates. Nucleic Acids Res. 2020, 48, D724–D730. [Google Scholar] [CrossRef]
- Yang, X.; Weber, M.; Zarinkamar, N.; Posnien, N.; Friedrich, F.; Wigand, B.; Beutel, R.; Damen, W.G.M.; Bucher, G.; Klingler, M.; et al. Probing the Drosophila Retinal Determination Gene Network in Tribolium (II): The Pax6 Genes Eyeless and Twin of Eyeless. Dev. Biol. 2009, 333, 215–227. [Google Scholar] [CrossRef]
- Luan, Q.; Chen, Q.; Friedrich, M. The Pax6 Genes Eyeless and Twin of Eyeless Are Required for Global Patterning of the Ocular Segment in the Tribolium Embryo. Dev. Biol. 2014, 394, 367–381. [Google Scholar] [CrossRef]
- Halder, G.; Callaerts, P.; Flister, S.; Walldorf, U.; Kloter, U.; Gehring, W.J. Eyeless Initiates the Expression of Both Sine Oculis and Eyes Absent during Drosophila Compound Eye Development. Development 1998, 125, 2181–2191. [Google Scholar] [CrossRef]
- Cvekl, A.; Callaerts, P. PAX6: 25th Anniversary and More to Learn. Exp. Eye Res. 2017, 156, 10–21. [Google Scholar] [CrossRef]
- Clements, J.; Hens, K.; Francis, C.; Schellens, A.; Callaerts, P. Conserved Role for the Drosophila Pax6 Homolog Eyeless in Differentiation and Function of Insulin-Producing Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 16183–16188. [Google Scholar] [CrossRef]
- Bürglin, T.R.; Affolter, M. Homeodomain Proteins: An Update. Chromosoma 2016, 125, 497–521. [Google Scholar] [CrossRef]
- Peterson, K.J.; Beavan, A.; Chabot, P.J.; McPeek, M.A.; Pisani, D.; Fromm, B.; Simakov, O. MicroRNAs as Indicators into the Causes and Consequences of Whole-Genome Duplication Events. Mol. Biol. Evol. 2022, 39, msab344. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homolog | Average dN/dS | Pre-Speciation Amino Acid Changes | Post-Speciation Amino Acid Changes |
|---|---|---|---|
| ey1 | 0.016 | 9 | 2 |
| ey2 | 0.219 | 24 | 9 |
| ey3 | 0.041 | 12 | 5 |
| toy1 | 0.083 | 24 | 12 |
| toy2 | 0.115 | 27 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dakarapu, T.; Friedrich, M. Evolutionary Conservation and Diversification of Five Pax6 Homologs in the Horseshoe Crab Species Cluster. Arthropoda 2024, 2, 85-98. https://doi.org/10.3390/arthropoda2010007
Dakarapu T, Friedrich M. Evolutionary Conservation and Diversification of Five Pax6 Homologs in the Horseshoe Crab Species Cluster. Arthropoda. 2024; 2(1):85-98. https://doi.org/10.3390/arthropoda2010007
Chicago/Turabian StyleDakarapu, Tanay, and Markus Friedrich. 2024. "Evolutionary Conservation and Diversification of Five Pax6 Homologs in the Horseshoe Crab Species Cluster" Arthropoda 2, no. 1: 85-98. https://doi.org/10.3390/arthropoda2010007
APA StyleDakarapu, T., & Friedrich, M. (2024). Evolutionary Conservation and Diversification of Five Pax6 Homologs in the Horseshoe Crab Species Cluster. Arthropoda, 2(1), 85-98. https://doi.org/10.3390/arthropoda2010007