
Positive Inotropic Agents in Cancer Therapy: Exploring Potential Anti-Tumor Effects



Abstract
:1. Introduction
2. Positive Inotropic Agents
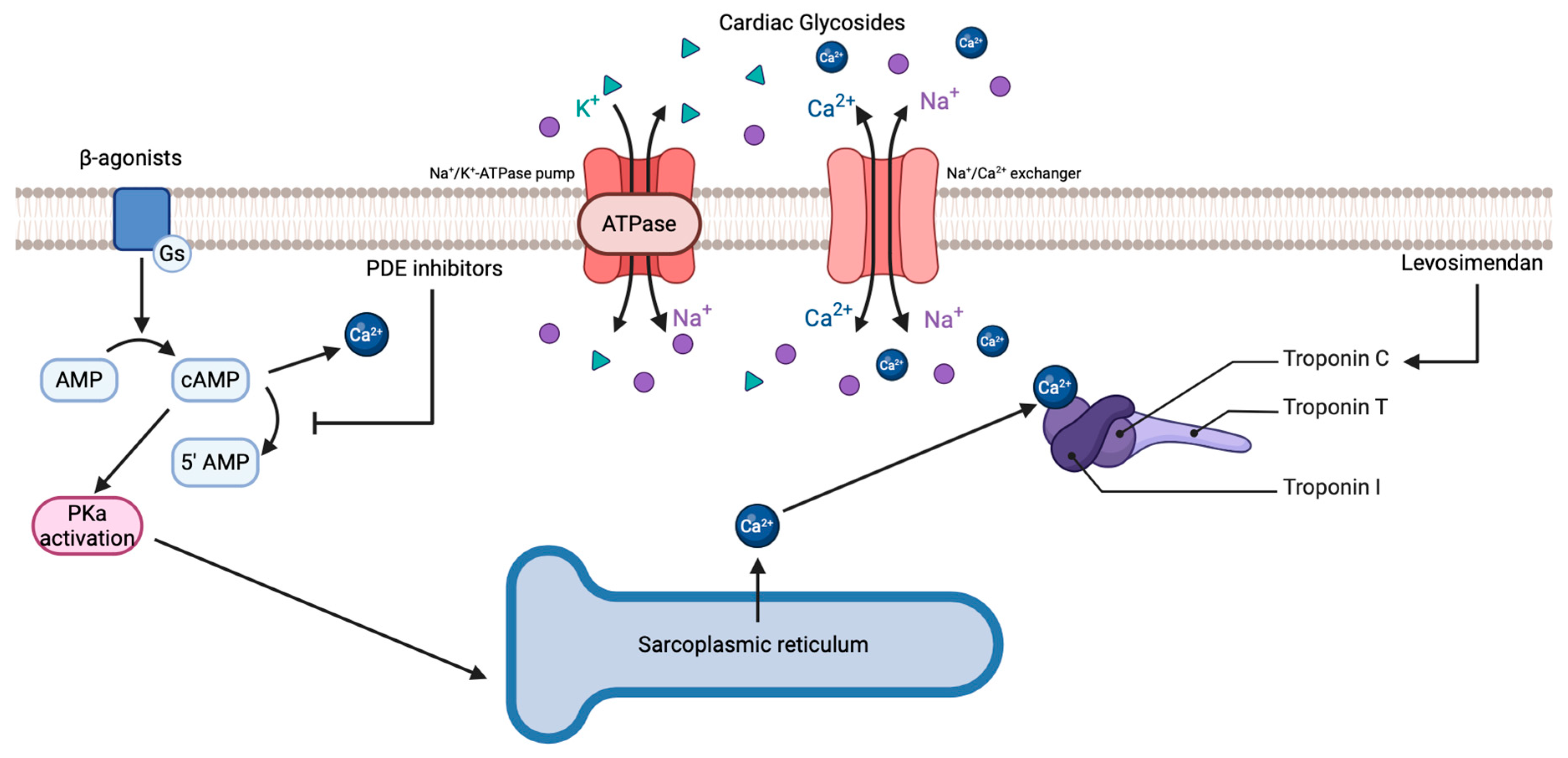
2.1. Cardiac Glycosides
2.2. β-Agonists
2.3. Phosphodiesterase (PDE) Inhibitors
2.4. Calcium Sensitizers
3. Preclinical Evidence of Anti-Tumor Activity and Current Clinical Landscape
3.1. Dopamine
3.2. Dobutamine
3.3. Digoxin
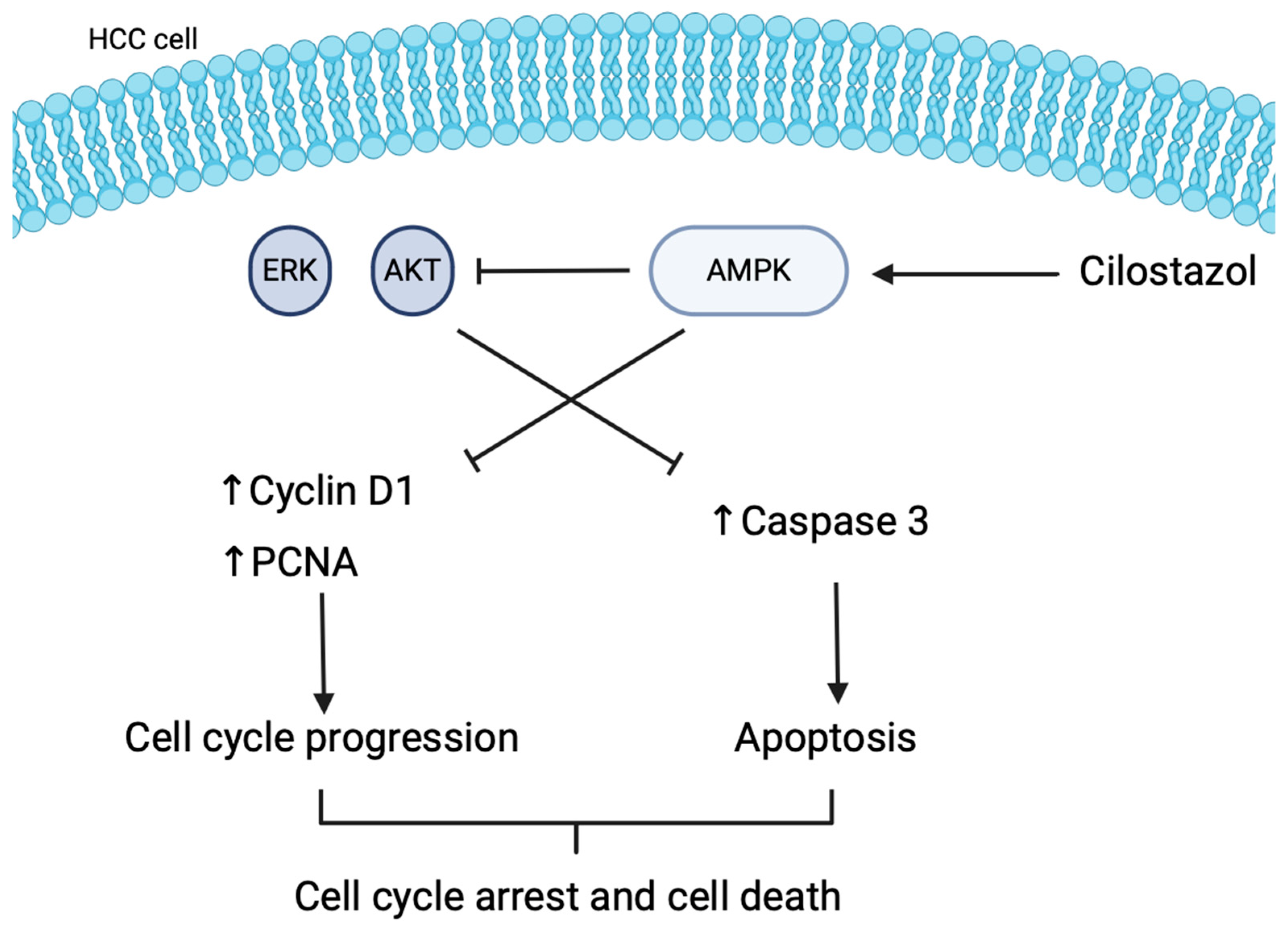
3.4. Cilostazol
3.5. Levosimendan
4. Repurposing Positive Inotropic Agents for Oncology
4.1. Inhibition of Phosphodiesterase 3
Increase in cAMP Levels
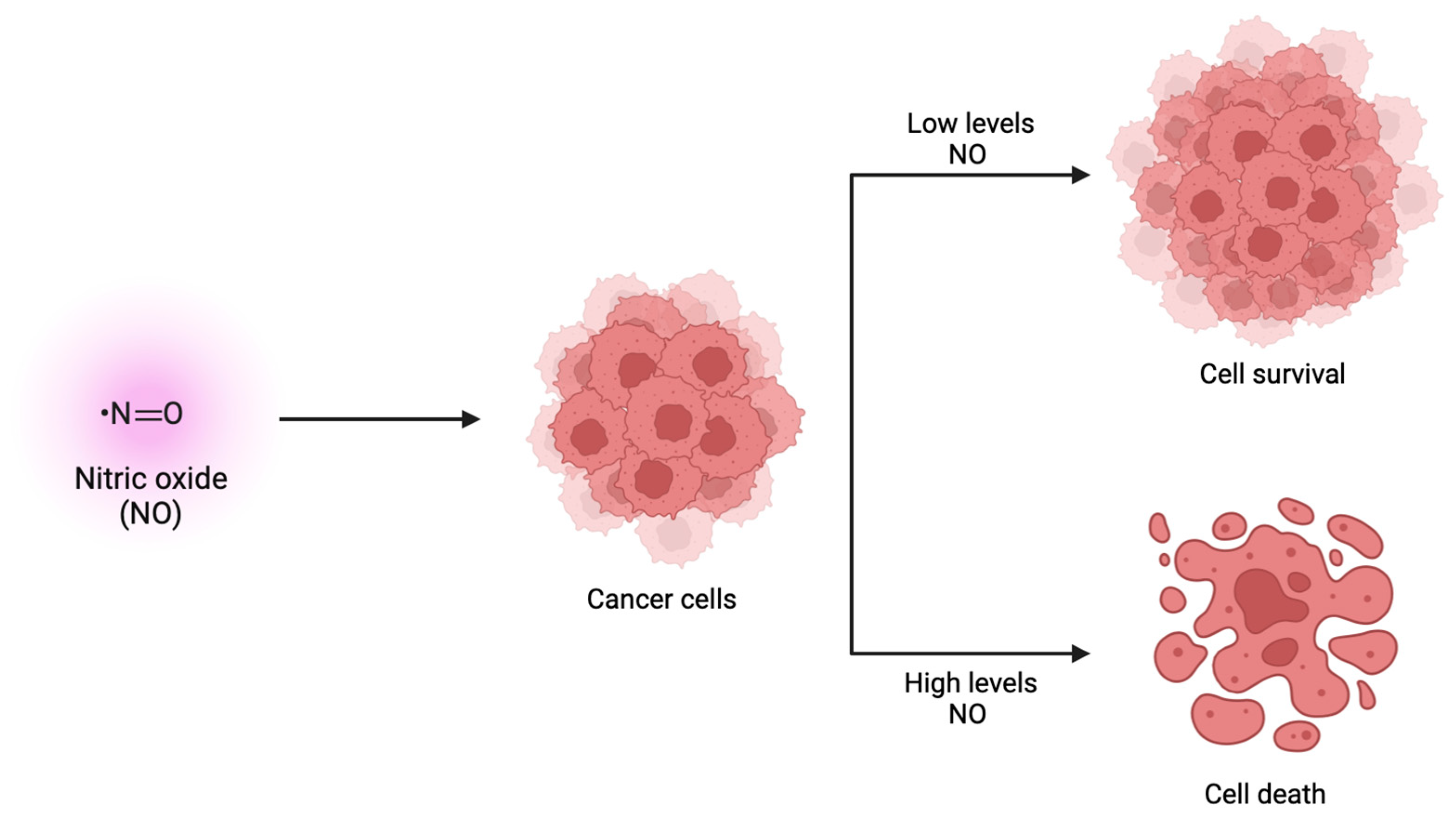
4.2. Production of Nitric Oxide
4.3. Reduction in Reactive Oxygen Species Levels
5. Challenges and Future Opportunities
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, J.S.; Amend, S.R.; Austin, R.H.; Gatenby, R.A.; Hammarlund, E.U.; Pienta, K.J. Updating the Definition of Cancer. Mol. Cancer Res. 2023, 21, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Pulumati, A.; Pulumati, A.; Dwarakanath, B.S.; Verma, A.; Papineni, R.V.L. Technological advancements in cancer diagnostics: Improvements and limitations. Cancer Rep. 2023, 6, e1764. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Derbal, Y. The Adaptive Complexity of Cancer. BioMed Res. Int. 2018, 2018, 5837235. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, M.J.; Sood, A.; Sevinsky, C.; Pris, A.D.; Zavodszky, M.I.; Ginty, F. Emerging understanding of multiscale tumor heterogeneity. Front. Oncol. 2014, 4, 366. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Frangioni, J.V.; Hoffman, R.M.; Iafrate, A.J.; Polyak, K. The challenges posed by cancer heterogeneity. Nat. Biotechnol. 2012, 30, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Charmsaz, S.; Collins, D.M.; Perry, A.S.; Prencipe, M. Novel Strategies for Cancer Treatment: Highlights from the 55th IACR Annual Conference. Cancers 2019, 11, 1125. [Google Scholar] [CrossRef] [PubMed]
- Ahire, V.; Ahmadi Bidakhvidi, N.; Boterberg, T.; Chaudhary, P.; Chevalier, F.; Daems, N.; Delbart, W.; Baatout, S.; Deroose, C.M.; Fernandez-Palomo, C.; et al. Radiobiology of Combining Radiotherapy with Other Cancer Treatment Modalities. In Radiobiology Textbook; Baatout, S., Ed.; Springer International Publishing: Cham, Switzerland, 2023; pp. 311–386. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.; Vale, N. Repurposing of the Drug Tezosentan for Cancer Therapy. Curr. Issues Mol. Biol. 2023, 45, 5118–5131. [Google Scholar] [CrossRef]
- Okuyama, R. Advancements in Drug Repurposing: Examples in Psychiatric Medications. Int. J. Mol. Sci. 2023, 24, 11000. [Google Scholar] [CrossRef]
- Ribeiro, E.; Vale, N. Understanding the Clinical Use of Levosimendan and Perspectives on its Future in Oncology. Biomolecules 2023, 13, 1296. [Google Scholar] [CrossRef]
- Khataniar, A.; Pathak, U.; Rajkhowa, S.; Jha, A.N. A Comprehensive Review of Drug Repurposing Strategies against Known Drug Targets of COVID-19. COVID 2022, 2, 148–167. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- de Mello, T.P.; Silva, L.N.; de Souza, R.; Frota, H.F.; Branquinha, M.H.; dos Santos, A.L.S. Drug repurposing strategy against fungal biofilms. Curr. Top. Med. Chem. 2020, 20, 509–516. [Google Scholar] [CrossRef]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef]
- Schein, C.H. Repurposing approved drugs on the pathway to novel therapies. Med. Res. Rev. 2020, 40, 586–605. [Google Scholar] [CrossRef]
- Suchonwanit, P.; Thammarucha, S.; Leerunyakul, K. Minoxidil and its use in hair disorders: A review. Drug Des. Devel Ther. 2019, 13, 2777–2786. [Google Scholar] [CrossRef]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef]
- Latif, T.; Chauhan, N.; Khan, R.; Moran, A.; Usmani, S.Z. Thalidomide and its analogues in the treatment of Multiple Myeloma. Exp. Hematol. Oncol. 2012, 1, 27. [Google Scholar] [CrossRef]
- Sperling, R. Zidovudine. Infect. Dis. Obstet. Gynecol. 1998, 6, 197–203. [Google Scholar] [CrossRef]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Devel Ther. 2013, 8, 87–100. [Google Scholar] [CrossRef]
- Cummings, S.R.; Eckert, S.; Krueger, K.A.; Grady, D.; Powles, T.J.; Cauley, J.A.; Norton, L.; Nickelsen, T.; Bjarnason, N.H.; Morrow, M.; et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 1999, 281, 2189–2197. [Google Scholar] [CrossRef]
- Victoir, B.; Croix, C.; Gouilleux, F.; Prié, G. Targeted Therapeutic Strategies for the Treatment of Cancer. Cancers 2024, 16, 461. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. [Google Scholar] [CrossRef]
- Weth, F.R.; Hoggarth, G.B.; Weth, A.F.; Paterson, E.; White, M.P.J.; Tan, S.T.; Peng, L.; Gray, C. Unlocking hidden potential: Advancements, approaches, and obstacles in repurposing drugs for cancer therapy. Br. J. Cancer 2024, 130, 703–715. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, K.; Ren, Z.; Yin, D.; Zhou, Y. Metformin as anticancer agent and adjuvant in cancer combination therapy: Current progress and future prospect. Transl. Oncol. 2024, 44, 101945. [Google Scholar] [CrossRef]
- Muniyan, S.; Rachagani, S.; Parte, S.; Halder, S.; Seshacharyulu, P.; Kshirsagar, P.; Siddiqui, J.A.; Vengoji, R.; Rauth, S.; Islam, R.; et al. Sildenafil Potentiates the Therapeutic Efficacy of Docetaxel in Advanced Prostate Cancer by Stimulating NO-cGMP Signaling. Clin. Cancer Res. 2020, 26, 5720–5734. [Google Scholar] [CrossRef]
- von Lilienfeld-Toal, M.; Hahn-Ast, C.; Furkert, K.; Hoffmann, F.; Naumann, R.; Bargou, R.; Cook, G.; Glasmacher, A. A systematic review of phase II trials of thalidomide/dexamethasone combination therapy in patients with relapsed or refractory multiple myeloma. Eur. J. Haematol. 2008, 81, 247–252. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, Q.; Zhang, S.; Liu, H.; Zhao, B.; Du, B.; Wang, W.; Lin, P.; Zhang, Z.; Zhong, Y.; et al. Digoxin Enhances the Anticancer Effect on Non-Small Cell Lung Cancer While Reducing the Cardiotoxicity of Adriamycin. Front. Pharmacol. 2020, 11, 186. [Google Scholar] [CrossRef]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R.; et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Moura, C.; Correia, A.S.; Pereira, M.; Ribeiro, E.; Santos, J.; Vale, N. Atorvastatin and Nitrofurantoin Repurposed in the Context of Breast Cancer and Neuroblastoma Cells. Biomedicines 2023, 11, 903. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, M.J.; Ribeiro, E.; Vale, N. A Surprising Repurposing of Central Nervous System Drugs against Squamous Cell Carcinoma of the Bladder, UM-UC-5. Pharmaceutics 2024, 16, 212. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Lu, C.; Shahzad, M.M.; Pena, G.N.; Allen, J.K.; Stone, R.L.; Mangala, L.S.; Han, H.D.; Kim, H.S.; Farley, D.; et al. Dopamine blocks stress-mediated ovarian carcinoma growth. Clin. Cancer Res. 2011, 17, 3649–3659. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, D.; Sarkar, C.; Mitra, R.B.; Banerjee, S.; Dasgupta, P.S.; Basu, S. Depleted dopamine in gastric cancer tissues: Dopamine treatment retards growth of gastric cancer by inhibiting angiogenesis. Clin. Cancer Res. 2004, 10, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Dong, Q.; Zheng, M.; Xu, X.; Zou, G.; Ma, G.; Li, K. Antitumor activity of dobutamine on human osteosarcoma cells. Oncol. Lett. 2016, 11, 3676–3680. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Xu, Z.; Yang, G.; Chen, G.; Li, B.; Hu, L.; Xiao, W.; Sun, X.; Gao, M.; Gao, L.; et al. Antitumor effect of dobutamine on multiple myeloma via mitogen-activated protein kinase pathway in vitro. Acta Biochim. Et Biophys. Sin. 2016, 48, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Menger, L.; Vacchelli, E.; Kepp, O.; Eggermont, A.; Tartour, E.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Cardiac glycosides and cancer therapy. Oncoimmunology 2013, 2, e23082. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Wu, C.L.; Yeh, I.C.; Wu, S.N.; Sze, C.I.; Gean, P.W. Cilostazol eliminates radiation-resistant glioblastoma by re-evoking big conductance calcium-activated potassium channel activity. Am. J. Cancer Res. 2021, 11, 1148–1169. [Google Scholar]
- Di Cesare, M.; Perel, P.; Taylor, S.; Kabudula, C.; Bixby, H.; Gaziano, T.A.; McGhie, D.V.; Mwangi, J.; Pervan, B.; Narula, J.; et al. The Heart of the World. Glob. Heart 2024, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Wawer, I. Chapter 4—Solid-State Measurements of Drugs and Drug Formulations. In NMR Spectroscopy in Pharmaceutical Analysis; Holzgrabe, U., Wawer, I., Diehl, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 201–231. [Google Scholar]
- Gillespie, N.D.; Witham, M.D.; Struthers, A.D. CHAPTER 40—Chronic Cardiac Failure. In Brocklehurst’s Textbook of Geriatric Medicine and Gerontology, 7th ed.; Fillit, H.M., Rockwood, K., Woodhouse, K., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2010; pp. 272–285. [Google Scholar]
- Sethi, N.J.; Nielsen, E.E.; Safi, S.; Feinberg, J.; Gluud, C.; Jakobsen, J.C. Digoxin for atrial fibrillation and atrial flutter: A systematic review with meta-analysis and trial sequential analysis of randomised clinical trials. PLoS ONE 2018, 13, e0193924. [Google Scholar] [CrossRef]
- Gona, S.R.; Rosenberg, J.; Fyffe-Freil, R.C.; Kozakiewicz, J.M.; Money, M.E. Review: Failure of current digoxin monitoring for toxicity: New monitoring recommendations to maintain therapeutic levels for efficacy. Front. Cardiovasc. Med. 2023, 10, 1179892. [Google Scholar] [CrossRef]
- Vivo, R.P.; Krim, S.R.; Perez, J.; Inklab, M.; Tenner, T., Jr.; Hodgson, J. Digoxin: Current use and approach to toxicity. Am. J. Med. Sci. 2008, 336, 423–428. [Google Scholar] [CrossRef] [PubMed]
- McCoshen, J.A.; Fernandes, P.A.; Boroditsky, M.L.; Allardice, J.G. Determinants of Reproductive Mortality and Preterm Childbirth. In Advances in Organ Biology; Bittar, E.E., Zakar, T., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 1, pp. 195–223. [Google Scholar]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, H.; Gao, H.; Kubo, H.; Berretta, R.M.; Chen, X.; Houser, S.R. {beta}1-Adrenergic receptor activation induces mouse cardiac myocyte death through both L-type calcium channel-dependent and -independent pathways. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H322–H331. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Beta-adrenoceptors on smooth muscle, nerves and inflammatory cells. Life Sci. 1993, 52, 2101–2109. [Google Scholar] [CrossRef]
- Dimitriadis, F.; Skouros, S.; Takenaka, A.; Sofikitis, N. Chapter 15—Beneficial or Detrimental Effects of Phosphodiesterase-5 (PDE5) Inhibitors on Semen Quality and Testicular Function? In Bioenvironmental Issues Affecting Men’s Reproductive and Sexual Health; Sikka, S.C., Hellstrom, W.J.G., Eds.; Academic Press: Boston, MA, USA, 2018; pp. 243–260. [Google Scholar]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Reiken, S.; Lacampagne, A.; Zhou, H.; Kherani, A.; Lehnart, S.E.; Ward, C.; Huang, F.; Gaburjakova, M.; Gaburjakova, J.; Rosemblit, N.; et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: Defective regulation in heart failure. J. Cell Biol. 2003, 160, 919–928. [Google Scholar] [CrossRef]
- Feil, R.; Lohmann, S.M.; de Jonge, H.; Walter, U.; Hofmann, F. Cyclic GMP-dependent protein kinases and the cardiovascular system: Insights from genetically modified mice. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef]
- Levy, J.H.; Ghadimi, K.; Bailey, J.M.; Ramsay, J.G. Chapter 30—Postoperative Cardiovascular Management. In Kaplan’s Essentials of Cardiac Anesthesia, 2nd ed.; Kaplan, J.A., Ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. 758–785. [Google Scholar]
- Orstavik, O.; Ata, S.H.; Riise, J.; Dahl, C.P.; Andersen, G.; Levy, F.O.; Skomedal, T.; Osnes, J.B.; Qvigstad, E. Inhibition of phosphodiesterase-3 by levosimendan is sufficient to account for its inotropic effect in failing human heart. Br. J. Pharmacol. 2014, 171, 5169–5181. [Google Scholar] [CrossRef] [PubMed]
- Grossini, E.; Molinari, C.; Caimmi, P.P.; Uberti, F.; Vacca, G. Levosimendan induces NO production through p38 MAPK, ERK and Akt in porcine coronary endothelial cells: Role for mitochondrial K(ATP) channel. Br. J. Pharmacol. 2009, 156, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Reyes-Resina, I.; Navarro, G. Dopamine in Health and Disease: Much More Than a Neurotransmitter. Biomedicines 2021, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Espinoza, S.; Gainetdinov, R.R. Dopamine receptors—IUPHAR Review 13. Br. J. Pharmacol. 2015, 172, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Nagy, J.A.; Pal, S.; Vasile, E.; Eckelhoefer, I.A.; Bliss, V.S.; Manseau, E.J.; Dasgupta, P.S.; Dvorak, H.F.; Mukhopadhyay, D. The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 2001, 7, 569–574. [Google Scholar] [CrossRef]
- Sarkar, C.; Chakroborty, D.; Chowdhury, U.R.; Dasgupta, P.S.; Basu, S. Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 2008, 14, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- De, D.; Upadhyay, P.; Das, A.; Ghosh, A.; Adhikary, A.; Goswami, M.M. Studies on cancer cell death through delivery of dopamine as anti-cancer drug by a newly functionalized cobalt ferrite nano-carrier. Colloids Surf. A Physicochem. Eng. Asp. 2021, 627, 127202. [Google Scholar] [CrossRef]
- Nguyen, L.; Roth, D.M.; Shanewise, J.S.; Kaplan, J.A. Chapter 28—Discontinuing Cardiopulmonary Bypass. In Kaplan’s Essentials of Cardiac Anesthesia, 2nd ed.; Kaplan, J.A., Ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. 715–740. [Google Scholar]
- Ruffolo, R.R. Review: The Pharmacology of Dobutamine. Am. J. Med. Sci. 1987, 294, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Nakagawa, K.; Yang, Z.; Ikeda, M.; Withanage, K.; Ishigami-Yuasa, M.; Okuno, Y.; Hata, S.; Nishina, H.; Hata, Y. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J. Biochem. 2011, 150, 199–208. [Google Scholar] [CrossRef]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.; Wu, T.C. How will HPV vaccines affect cervical cancer? Nat. Rev. Cancer 2006, 6, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Castle, P.E.; Dockter, J.; Giachetti, C.; Garcia, F.A.; McCormick, M.K.; Mitchell, A.L.; Holladay, E.B.; Kolk, D.P. A cross-sectional study of a prototype carcinogenic human papillomavirus E6/E7 messenger RNA assay for detection of cervical precancer and cancer. Clin. Cancer Res. 2007, 13, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.X.; Wu, L.N.; Xiao, H.; Du, Q.; Liang, J.F. Inhibitory effects of dobutamine on human gastric adenocarcinoma. World J. Gastroenterol. 2014, 20, 17092–17099. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Eskiocak, U.; Gill, J.G.; Yuan, S.; Ramesh, V.; Froehlich, T.W.; Ahn, C.; Morrison, S.J. Digoxin Plus Trametinib Therapy Achieves Disease Control in BRAF Wild-Type Metastatic Melanoma Patients. Neoplasia 2017, 19, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Garrett Injac, S.; Cui, K.; Braun, F.; Lin, Q.; Du, Y.; Zhang, H.; Kogiso, M.; Lindsay, H.; Zhao, S.; et al. Systems biology-based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci. Transl. Med. 2018, 10, eaat0150. [Google Scholar] [CrossRef]
- Platz, E.A.; Yegnasubramanian, S.; Liu, J.O.; Chong, C.R.; Shim, J.S.; Kenfield, S.A.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E.; Nelson, W.G. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011, 1, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.H.; Kim, S.; Kuh, H.-J.; Lee, J.-H. Downregulation of BIS sensitizes A549 cells for digoxin-mediated inhibition of invasion and migration by the STAT3-dependent pathway. Biochem. Biophys. Res. Commun. 2020, 524, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Shen, J.; Wang, W.; Li, M.; Li, H.; Chen, C.; Zhao, H.; Zhang, M.; Xue, T.; Liu, Q.; et al. Sodium chloride (NaCl) potentiates digoxin-induced anti-tumor activity in small cell lung cancer. Cancer Biol. Ther. 2019, 20, 52–64. [Google Scholar] [CrossRef]
- Shen, S.S.; Hamamoto, S.T.; Bern, H.A.; Steinhardt, R.A. Alteration of sodium transport in mouse mammary epithelium associated with neoplastic transformation. Cancer Res. 1978, 38, 1356–1361. [Google Scholar]
- Cove, D.H.; Barker, G.A. Digoxin and hormone receptors. Lancet 1979, 2, 204. [Google Scholar] [CrossRef]
- Prassas, I.; Karagiannis, G.S.; Batruch, I.; Dimitromanolakis, A.; Datti, A.; Diamandis, E.P. Digitoxin-induced cytotoxicity in cancer cells is mediated through distinct kinase and interferon signaling networks. Mol. Cancer Ther. 2011, 10, 2083–2093. [Google Scholar] [CrossRef]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, Y.; Chen, L.; Qian, Z.; Zhang, Y. Associations between HIFs and tumor immune checkpoints: Mechanism and therapy. Discov. Oncol. 2024, 15, 2. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chang, H.H.; Lai, Y.H.; Lin, C.H.; Chen, M.H.; Chang, G.C.; Tsai, M.F.; Chen, J.J. Digoxin Suppresses Tumor Malignancy through Inhibiting Multiple Src-Related Signaling Pathways in Non-Small Cell Lung Cancer. PLoS ONE 2015, 10, e0123305. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Peng, J.J.; Gao, H.; Li, H.; Li, D.; Tan, Y.; Zhang, T. Digoxin downregulates NDRG1 and VEGF through the inhibition of HIF-1α under hypoxic conditions in human lung adenocarcinoma A549 cells. Int. J. Mol. Sci. 2013, 14, 7273–7285. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, Q.; Shen, J.J.; Li, D.D.; Jiang, X.J.; Si, S.Y.; Shao, R.G.; Wang, Z. Cardiac glycosides induce autophagy in human non-small cell lung cancer cells through regulation of dual signaling pathways. Int. J. Biochem. Cell Biol. 2012, 44, 1813–1824. [Google Scholar] [CrossRef]
- Kherallah, R.Y.; Khawaja, M.; Olson, M.; Angiolillo, D.; Birnbaum, Y. Cilostazol: A Review of Basic Mechanisms and Clinical Uses. Cardiovasc. Drugs Ther. 2022, 36, 777–792. [Google Scholar] [CrossRef]
- Ikeda, Y.; Matsumata, T.; Takenaka, K.; Yamagata, M.; Sugimachi, K. Effects of doxorubicin and/or cilostazol on cancer cells during liver regeneration after two-thirds hepatectomy in rats. Oncology 1998, 55, 354–356. [Google Scholar] [CrossRef]
- Murata, K.; Kameyama, M.; Fukui, F.; Ohigashi, H.; Hiratsuka, M.; Sasaki, Y.; Kabuto, T.; Mukai, M.; Mammoto, T.; Akedo, H.; et al. Phosphodiesterase type III inhibitor, cilostazol, inhibits colon cancer cell motility. Clin. Exp. Metastasis 1999, 17, 525–530. [Google Scholar] [CrossRef]
- Sim, K.H.; Shu, M.-S.; Kim, S.; Kim, J.-Y.; Choi, B.-H.; Lee, Y.J. Cilostazol Induces Apoptosis and Inhibits Proliferation of Hepatocellular Carcinoma Cells by Activating AMPK. Biotechnol. Bioprocess. Eng. 2021, 26, 776–785. [Google Scholar] [CrossRef]
- Feneck, R. Phosphodiesterase inhibitors and the cardiovascular system. Contin. Educ. Anaesth. Crit. Care Pain. 2007, 7, 203–207. [Google Scholar] [CrossRef]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Begum, N.; Shen, W.; Manganiello, V. Role of PDE3A in regulation of cell cycle progression in mouse vascular smooth muscle cells and oocytes: Implications in cardiovascular diseases and infertility. Curr. Opin. Pharmacol. 2011, 11, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Cyclic GMP Phosphodiesterases. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 567–573. [Google Scholar]
- Zimmerman, N.P.; Roy, I.; Hauser, A.D.; Wilson, J.M.; Williams, C.L.; Dwinell, M.B. Cyclic AMP regulates the migration and invasion potential of human pancreatic cancer cells. Mol. Carcinog. 2015, 54, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, J.J.; Huang, X.Y. cAMP inhibits cell migration by interfering with Rac-induced lamellipodium formation. J. Biol. Chem. 2008, 283, 13799–13805. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Yang, W.L.; Ravatn, R.; Kita, T.; Reitman, E.; Vettori, D.; Cvijic, M.E.; Shin, M.; Iacono, L. Reinventing the wheel of cyclic AMP: Novel mechanisms of cAMP signaling. Ann. N. Y Acad. Sci. 2002, 968, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Chiaradonna, F.; Balestrieri, C.; Gaglio, D.; Vanoni, M. RAS and PKA pathways in cancer: New insight from transcriptional analysis. Front. Biosci. 2008, 13, 5257–5278. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Rangarajan, S.; Enserink, J.M.; Kuiperij, H.B.; de Rooij, J.; Price, L.S.; Schwede, F.; Bos, J.L. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the beta 2-adrenergic receptor. J. Cell Biol. 2003, 160, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Enserink, J.M.; Price, L.S.; Methi, T.; Mahic, M.; Sonnenberg, A.; Bos, J.L.; Taskén, K. The cAMP-Epac-Rap1 pathway regulates cell spreading and cell adhesion to laminin-5 through the alpha3beta1 integrin but not the alpha6beta4 integrin. J. Biol. Chem. 2004, 279, 44889–44896. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, M.R.; Corada, M.; Dejana, E.; Bos, J.L. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005, 579, 4966–4972. [Google Scholar] [CrossRef] [PubMed]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Kiermayer, S.; Biondi, R.M.; Imig, J.; Plotz, G.; Haupenthal, J.; Zeuzem, S.; Piiper, A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol. Biol. Cell 2005, 16, 5639–5648. [Google Scholar] [CrossRef] [PubMed]
- Grandoch, M.; Rose, A.; ter Braak, M.; Jendrossek, V.; Rübben, H.; Fischer, J.W.; Schmidt, M.; Weber, A.A. Epac inhibits migration and proliferation of human prostate carcinoma cells. Br. J. Cancer 2009, 101, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Prasad, P.; Jash, E.; Saini, M.; Husain, A.; Goldman, A.; Sehrawat, S. Insights into exchange factor directly activated by cAMP (EPAC) as potential target for cancer treatment. Mol. Cell Biochem. 2018, 447, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Bortner, C.D.; Cidlowski, J.A. Potential roles of electrogenic ion transport and plasma membrane depolarization in apoptosis. J. Membr. Biol. 2006, 209, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Sifre, M.I.; Cidlowski, J.A. Cationic gradient reversal and cytoskeleton-independent volume regulatory pathways define an early stage of apoptosis. J. Biol. Chem. 2008, 283, 7219–7229. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death. J. Biol. Chem. 2003, 278, 39176–39184. [Google Scholar] [CrossRef]
- Yurinskaya, V.; Goryachaya, T.; Guzhova, I.; Moshkov, A.; Rozanov, Y.; Sakuta, G.; Shirokova, A.; Shumilina, E.; Vassilieva, I.; Lang, F.; et al. Potassium and sodium balance in U937 cells during apoptosis with and without cell shrinkage. Cell Physiol. Biochem. 2005, 16, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hirst, D.G.; Robson, T. Nitric oxide physiology and pathology. Methods Mol. Biol. 2011, 704, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef] [PubMed]
- Falke, D.; Fisher, M.; Ye, D.; Juliano, R.L. Design of artificial transcription factors to selectively regulate the pro-apoptotic bax gene. Nucleic Acids Res. 2003, 31, e10. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; De Nardo, D.; Gao, W.; Vince, A.J.; Hall, C.; McArthur, K.; Simpson, D.; Vijayaraj, S.; Lindqvist, L.M.; Bouillet, P.; et al. The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1β Activation. Cell Rep. 2018, 25, 2339–2353.e2334. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Choy, G.; Deng, X.; Bhatia, B.; Daniel, P.; Tang, D.G. Association of active caspase 8 with the mitochondrial membrane during apoptosis: Potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. Mol. Cell Biol. 2004, 24, 6592–6607. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxidative Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Lee, J.D.; Cai, Q.; Shu, X.O.; Nechuta, S.J. The Role of Biomarkers of Oxidative Stress in Breast Cancer Risk and Prognosis: A Systematic Review of the Epidemiologic Literature. J. Womens Health 2017, 26, 467–482. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Gao, G.; Wei, G.; Zheng, Y.; Wang, C.; Gao, N.; Zhao, Y.; Deng, J.; Chen, H.; et al. Elevation of GPRC5A expression in colorectal cancer promotes tumor progression through VNN-1 induced oxidative stress. Int. J. Cancer 2017, 140, 2734–2747. [Google Scholar] [CrossRef] [PubMed]
- Saijo, H.; Hirohashi, Y.; Torigoe, T.; Horibe, R.; Takaya, A.; Murai, A.; Kubo, T.; Kajiwara, T.; Tanaka, T.; Shionoya, Y.; et al. Plasticity of lung cancer stem-like cells is regulated by the transcription factor HOXA5 that is induced by oxidative stress. Oncotarget 2016, 7, 50043–50056. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxid. Med. Cell Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef]
- Oh, B.; Figtree, G.; Costa, D.; Eade, T.; Hruby, G.; Lim, S.; Elfiky, A.; Martine, N.; Rosenthal, D.; Clarke, S.; et al. Oxidative stress in prostate cancer patients: A systematic review of case control studies. Prostate Int. 2016, 4, 71–87. [Google Scholar] [CrossRef]
- Saed, G.M.; Diamond, M.P.; Fletcher, N.M. Updates of the role of oxidative stress in the pathogenesis of ovarian cancer. Gynecol. Oncol. 2017, 145, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Namkaew, J.; Noisa, P. Curcumin attenuates paraquat-induced cell death in human neuroblastoma cells through modulating oxidative stress and autophagy. Neurosci. Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef]
- Bailo, P.S.; Martín, E.L.; Calmarza, P.; Breva, S.M.; Gómez, A.B.; Giráldez, A.P.; Callau, J.J.S.-P.; Santamaría, J.M.V.; Khialani, A.D.; Micó, C.C.; et al. The role of oxidative stress in neurodegenerative diseases and potential antioxidant therapies. Adv. Lab. Med. / Av. En Med. De Lab. 2022, 3, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-mediated oncogenic signaling pathways in human malignancies. Semin. Cancer Biol. 2019, 54, 1–13. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2018, 142, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2021, 21, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, D.L.; Powis, G. Clinically Evaluated Cancer Drugs Inhibiting Redox Signaling. Antioxid. Redox Signal 2017, 26, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Kirtonia, A.; Sethi, G.; Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef]
- Ralph, S.J.; Nozuhur, S.; RA, A.L.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. Repurposing drugs as pro-oxidant redox modifiers to eliminate cancer stem cells and improve the treatment of advanced stage cancers. Med. Res. Rev. 2019, 39, 2397–2426. [Google Scholar] [CrossRef] [PubMed]
- Mannino, F.; Urzì Brancati, V.; Lauro, R.; Pirrotta, I.; Rottura, M.; Irrera, N.; Cavallini, G.M.; Pallio, G.; Gitto, E.; Manti, S. Levosimendan and Dobutamin Attenuate LPS-Induced Inflammation in Microglia by Inhibiting the NF-κB Pathway and NLRP3 Inflammasome Activation via Nrf2/HO-1 Signalling. Biomedicines 2024, 12, 1009. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Han, S.N.; Arumugam, S.; Yousaf, M.N.; Qin, Y.; Jiang, J.X.; Torok, N.J.; Chen, Y.; Mankash, M.S.; Liu, J.; et al. Digoxin improves steatohepatitis with differential involvement of liver cell subsets in mice through inhibition of PKM2 transactivation. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G387–G397. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; An, B.; Lin, Y.; Ni, Y.; Zhao, X.; Liang, X. Molecular mechanisms of ROS-modulated cancer chemoresistance and therapeutic strategies. Biomed. Pharmacother. 2023, 165, 115036. [Google Scholar] [CrossRef]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef]
- Begley, C.G.; Ashton, M.; Baell, J.; Bettess, M.; Brown, M.P.; Carter, B.; Charman, W.N.; Davis, C.; Fisher, S.; Frazer, I.; et al. Drug repurposing: Misconceptions, challenges, and opportunities for academic researchers. Sci. Transl. Med. 2021, 13, eabd5524. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Drug Name | Original Indication | New Indication | References |
|---|---|---|---|
| Verapamil | Heart rhythm problems | Fungal biofilms | [15] |
| Sildenafil | Angina | Erectile dysfunction | [16] |
| Carboplatin | Antitumor | Fungal biofilms | [15] |
| Amiloride | Hypertension | Secondary progressive multiple sclerosis (SPMS) | [17] |
| Minoxidil | Hypertension | Hair loss | [18] |
| Aspirin | Analgesia | Colorectal cancer | [19] |
| Thalidomide | Morning sickness | Multiple myeloma | [20] |
| Zidovudine | Cancer | HIV | [21] |
| Rituximab | Cancer | Rheumatoid arthritis | [22] |
| Raloxifene | Osteoporosis | Breast cancer | [23] |
| Category | Drug | Indication | Mechanism of Action |
|---|---|---|---|
| Cardiac glycosides | Digoxin | Mild-to-moderate heart failure, increased myocardial contraction, and maintained control ventricular rate | Na-K ATPase enzyme inhibition |
| Phosphodiesterase III (PDE3) inhibitor | Enoximone | Congestive heart failure | PDE3 inhibition Stimulates NO production Increases cAMP levels |
| Milrinone | Acute decompensated heart failure | PDE3 inhibition Increases cAMP levels | |
| Amrinone | Congestive heart failure | PDE3 inhibition Increases cAMP levels | |
| β-agonists | Dobutamine | Cardiac decompensation | Increases cAMP levels |
| Dopamine | Hypotension | Increases cAMP levels | |
| Isoetharine | Asthma | Increases cAMP levels | |
| Ritodrine | Premature labor | Increases cAMP levels | |
| Terbutaline | Asthma and premature labor | Increases cAMP levels | |
| Calcium sensitizer | Levosimendan | Chronic heart failure | PDE3 inhibition Stimulates NO production Decreases ROS levels Increases cAMP levels |
| Drug | Type of Cancer | Anticancer Activity | Doses | Maximum Dose Used (Cardiovascular Diseases) | References |
|---|---|---|---|---|---|
| Dopamine | Breast and colon | Inhibits angiogenesis and tumor growth | 50 mg/kg/day | 50 µg/kg/min | [62] |
| Dobutamine | Gastric | Inhibits cell growth, migration, cell colony formation, and cell invasion, arrests the cell cycle at the G1 or S phase, and increases the rate of apoptosis | 30 μmol/L | 40 µg/kg/min | [71] |
| Bone | Inhibits cell growth, migration, and cell invasion, augments cell apoptosis, and arrests the cell cycle in the G2 or M phase | 10 µM | [38] | ||
| Digoxin | NSCLC | Reduces the cell viability, increases DNA damage by promoting ROS generation, and inhibitis both DNA double-strand break (DSB) and single-strand break (SSB) repair | 0.2 µM– 1.0 mg/kg/day | 0.75–1.5 mg/day | [31] |
| Cilostazol | Colon | Suppress migration | 50 µM | 200 mg/day | [87] |
| Liver | Inhibits proliferation, induces apoptosis, induces G0 and G1 cell cycle arrest, and decreases the expression of cyclin D1 and nuclear antigen in proliferating cells | 100 µM | [88] | ||
| Levosimendan | Bladder and prostate | Inhibits cell migration, cell colony formation, and proliferation | 100 µM | 6–12 µg/kg/10 min | Our Lab |
| Drug | Type of Cancer | Doses |
|---|---|---|
| Doxorubicin | Breast, bone, and liver | 60–75 mg/m2/d |
| 5-FU | Colon and gastric | 500–2600 mg/m2/d |
| Cisplatin and pemetrexed | NSCLC | 75 mg/m2/d + 500 mg/m2/d |
| Gemcitabine and cisplatin | Bladder | 1000 mg/m2/d + 70 mg/m2/d |
| Docetaxel and prednisone | Prostate | 75 mg/m2/d + 10 mg/d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, E.; Vale, N. Positive Inotropic Agents in Cancer Therapy: Exploring Potential Anti-Tumor Effects. Targets 2024, 2, 137-156. https://doi.org/10.3390/targets2020009
Ribeiro E, Vale N. Positive Inotropic Agents in Cancer Therapy: Exploring Potential Anti-Tumor Effects. Targets. 2024; 2(2):137-156. https://doi.org/10.3390/targets2020009
Chicago/Turabian StyleRibeiro, Eduarda, and Nuno Vale. 2024. "Positive Inotropic Agents in Cancer Therapy: Exploring Potential Anti-Tumor Effects" Targets 2, no. 2: 137-156. https://doi.org/10.3390/targets2020009
APA StyleRibeiro, E., & Vale, N. (2024). Positive Inotropic Agents in Cancer Therapy: Exploring Potential Anti-Tumor Effects. Targets, 2(2), 137-156. https://doi.org/10.3390/targets2020009