
Identification of Pharmacophore Groups with Antimalarial Potential in Flavonoids by QSAR-Based Virtual Screening

 , , , ,
, , , ,  , , and
, , and
Abstract
1. Introduction
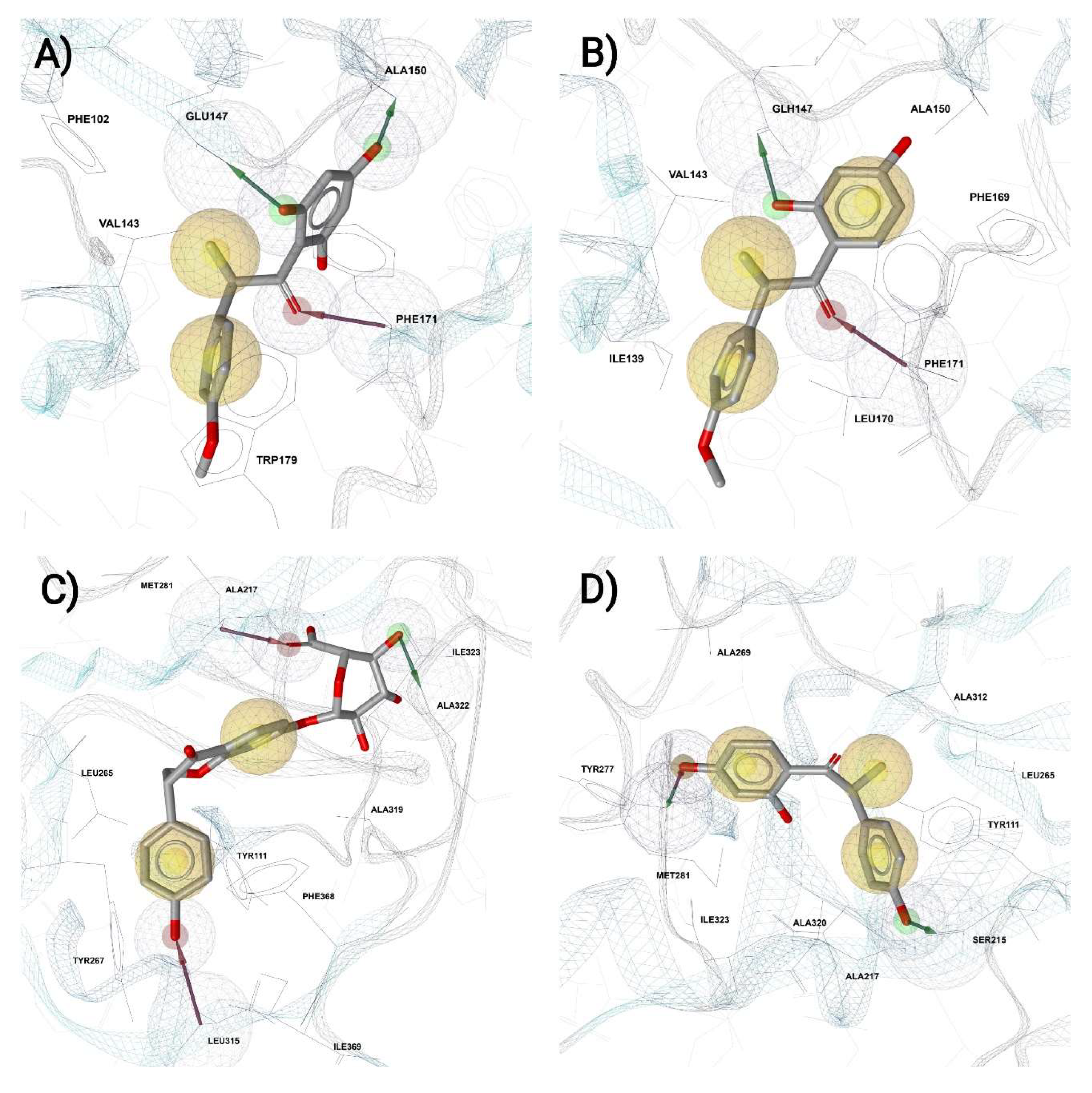
2. Results and Discussion
3. Materials and Methods
3.1. Database Building
3.2. Descriptors Calculation and Selection
3.3. Activity Prediction Model
3.4. Virtual Screening Development
3.5. Molecular Dynamics (MD) and Pharmacophore Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Basu, S.; Sahi, P.K. Malaria: An update. Indian J. Pediatr. 2017, 84, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.I.; Bora, K.; Kim, Y.J.; Kim, T.Y.; Cho, S.H.; Lee, S.E. Diagnosis and molecular analysis in imported Plasmodium ovale curtisi and P. ovale wallikeri malaria cases from West and South Africa during 2013–2016. Korean J. Parasitol. 2020, 58, 61–65. [Google Scholar] [CrossRef]
- Gozalo, A.S.; Robinson, C.K.; Holdridge, J.; Mahecha, O.F.L.; Elkins, W.R. Overview of Plasmodium spp. and Animal models in malaria research. Comp. Med. 2024, 74, 205–230. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2024; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Recht, J.; Siqueira, A.M.; Monteiro, W.M.; Herrera, S.M.; Herrera, S.; Lacerda, M.V.G. Malaria in Brazil, Colombia, Peru and Venezuela: Current challenges in malaria control and elimination. Malar. J. 2017, 16, 273. [Google Scholar] [CrossRef]
- Bittaye, S.O.; Jagne, A.; Jaiteh, L.E.; Nadjm, B.; Amambua-Ngwa, A.; Sesay, A.K.; Singhateh, Y.; Effa, E.; Nyan, O.; Njie, R. Clinical manifestations and outcomes of severe malaria in adult patients admitted to a tertiary hospital in the Gambia. Malar. J. 2022, 21, 270. [Google Scholar] [CrossRef]
- Dayananda, K.K.; Achur, R.N.; Gowda, D.C. Epidemiology, drug resistance, and pathophysiology of Plasmodium vivax malaria. J. Vector Borne Dis. 2018, 55, 1–8. [Google Scholar] [CrossRef]
- Daily, J.P.; Minuti, A.; Khan, N. Diagnosis, treatment, and prevention of malaria in the US: A review. JAMA 2022, 28, 460–471. [Google Scholar] [CrossRef]
- Tasdemir, D.; Lack, G.; Brun, R.; Rüedi, P.; Scapozza, L.; Perozzo, R. Inhibition of Plasmodium falciparum fatty acid biosynthesis: Evaluation of FabG, FabZ and FabI as drug targets for flavonoids. J. Med. Chem. 2006, 49, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.P.; Pandya, H.A.; Desai, V.H.; Jasrai, Y.T. Compound prioritization from inverse docking experiment using receptor-centric and ligand-centric methods: A case study on Plasmodium falciparum Fab enzymes. J. Mol. Recognit. 2014, 27, 215–229. [Google Scholar] [CrossRef]
- Hoelz, L.V.; Calil, F.A.; Nonato, M.C.; Pinheiro, L.C.S.; Boechat, N. Plasmodium falciparum dihydroorotate dehydrogenase: A drug target against malaria. Future Med. Chem. 2018, 10, 1853–1874. [Google Scholar] [CrossRef]
- Kingston, D.G.I.; Cassera, M.B. Antimalarial natural products. Prog. Chem. Org. Nat. Prod. 2022, 117, 1–106. [Google Scholar] [CrossRef]
- White, N.J.; Chotivanich, K. Artemisinin-resistant malaria. Clin. Microbiol. Rev. 2024, 37, e0010924. [Google Scholar] [CrossRef] [PubMed]
- Tajuddeen, N.; van Heerden, F.R. Antiplasmodial natural products: An update. Malar. J. 2019, 18, 404. [Google Scholar] [CrossRef]
- Rakha, A.; Umar, N.; Rabail, R.; Butt, M.S.; Kieliszek, M.; Hassoun, A.; Aadil, R.M. Anti-inflammatory and anti-allergic potential of dietary flavonoids: A review. Biomed. Pharmacother. 2022, 153, 113945. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Ahmad, J.; Ahamad, J.; Kundu, S.; Goel, A.; Mishra, A. Flavonoids as promising anticancer therapeutics: Contemporary research, nanoantioxidant potential, and future scope. Phytother. Res. 2023, 37, 5159–5192. [Google Scholar] [CrossRef]
- Adewole, K.E. Nigerian antimalarial plants and their anticancer potential: A review. J. Integr. Med. 2020, 18, 92–113. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Castillo, M.; Pacheco-Yepez, J.; Flores-Huerta, N.; Guzmán-Téllez, P.; Jarillo-Luna, R.A.; Cárdenas-Jaramillo, L.M.; Campos-Rodríguez, R.; Shibayama, M. Flavonoids as natural treatment against Entamoeba histolytica. Front. Cell. Infect. Microbiol. 2018, 8, 209. [Google Scholar] [CrossRef]
- Araújo, M.V.; Queiroz, A.C.; Silva, J.F.M.; Silva, A.E.; Silva, J.K.S.; Silva, G.R.; Silva, E.C.O.; Souza, S.T.; Fonseca, E.J.S.; Camara, C.A.; et al. Flavonoids induce cell death in Leishmania amazonensis: In vitro characterization by flow cytometry and Raman spectroscopy. Analyst 2019, 144, 5232–5244. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Sureda, A.; Daglia, M.; Izadi, M.; Rastrelli, L.; Nabavi, S.M. Flavonoids and Chagas’ disease: The story so far! Curr. Top. Med. Chem. 2017, 17, 460–466. [Google Scholar] [CrossRef]
- Boniface, P.K.; Ferreira, E.I. Flavonoids as efficient scaffolds: Recent trends for malaria, leishmaniasis, Chagas disease and dengue. Phytother. Res. 2019, 33, 2473–2517. [Google Scholar] [CrossRef]
- Baldim, J.L.; Alcântara, B.G.V.; Domingos, O.S.; Soares, M.G.; Caldas, I.S.; Noaves, R.D.; Oliveira, T.B.; Lago, J.H.G.; Chagas-Paula, D.A. The correlation between chemical structures and antioxidant, prooxidant and antitrypanosomatid properties of flavonoids. Oxid. Med. Cell. Longev. 2017, 2017, 3789856. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Delgado, J.; Ty, M.; Orengo, J.M.; Van Der Hoef, D.; Rodriguez, A. A surprising role for uric acid: The inflammatory malaria response. Curr. Rheumatol. Rep. 2014, 16, 401. [Google Scholar] [CrossRef]
- Chen, H.; Kogej, T.; Engkvist, O. Cheminformatics in drug discovery, an industrial perspective. Mol. Inform. 2018, 37, e1800041. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From machine learning to deep learning: Progress in machine intelligence for rational drug design. Drug Discov. Today. 2017, 22, 1680–1685. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo Filho, C.C.; Moreira Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Deterministic pharmacophore detection via multiple flexible alignment of drug-like molecules. J. Comput. Biol. 2008, 15, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Nour, A.M.M.; Khalid, S.A.; Kaiser, M.; Brun, R. Quantitative-antiprotozoal relationships of sesquiterpene lactones. Molecules 2009, 14, 2062–2076. [Google Scholar] [CrossRef]
- Sadgrove, N.J.; Oliveira, T.B.; Khumalo, G.P.; van Vuuren, S.F.; van Wyk, B.-E. Antimicrobial isoflavones and derivatives from Erythrina (Fabaceae): Structure activity perspective (SAR & QSAR) on experimental and mined values against Staphylococcus aureus. Antibiotics 2020, 9, 223. [Google Scholar] [CrossRef]
- Lee, Y.W.; Choi, J.W.; Shin, E.H. Machine learning model for predicting malaria using clinical information. Comput. Biol. Med. 2021, 129, 1041151. [Google Scholar] [CrossRef] [PubMed]
- Mbaye, O.; Ba, M.L.; Sy, A. On the efficiency of machine learning models in malaria prediction. Stud. Health Technol. Inform. 2021, 27, 437–441. [Google Scholar] [CrossRef]
- Eze, P.U.; Asogwa, C.O. Deep machine learning model trade-offs for malaria elimination in resource-constrained locations. Bioengineering 2021, 8, 150. [Google Scholar] [CrossRef]
- Ford, C.T.; Janie, D. Ensemble machine learning modelling for the prediction of artemisinin resistance in malaria. F100Res 2020, 9, 62. [Google Scholar] [CrossRef]
- Borba, J.V.B.; Salazar-Alvarez, L.C.; Ferreira, L.T.; Silva-Mendonca, S.; Silva, M.F.B.D.; Sanches, I.H.; Clementino, L.D.C.; Magalhaes, M.L.; Rimoldi, A.; Calit, J.; et al. Innovative multistage ML-QSAR models for malaria: From data to discovery. ACS Med. Chem. Lett. 2024, 15, 1386–1395. [Google Scholar] [CrossRef]
- Hesping, E.; Chua, M.J.; Pflieger, M.; Qian, Y.; Dong, L.; Bachu, P.; Liu, L.; Kurz, T.; Fisher, G.M.; Skinner-Adams, T.S.; et al. QSAR classification models for prediction of hydroxamate histone deacetylase inhibitor activity against malaria parasites. ACS Infect. Dis. 2020, 8, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Ncube, N.B.; Tukulula, M.; Govender, K.G. Leveraging computational tools to combat malaria: Assessment and development of new therapeutics. J. Cheminform. 2024, 16, 50. [Google Scholar] [CrossRef]
- Yousefinejad, S.; Mahboubifar, M.; Eskandari, R. Quantitative structure-activity relationship to predict the antimalarial activity in a set of new imidazolpiperazines based on artificial neural networks. Malar. J. 2019, 18, 310. [Google Scholar] [CrossRef] [PubMed]
- Kalafi, E.Y.; Nor, N.A.M.; Taib, N.A.; Ganggayah, M.D.; Town, C.; Dhillon, S.K. Machine learning and deep learning approaches in breast cancer survival prediction using clinical data. Folia Biol. 2019, 65, 212–220. [Google Scholar] [CrossRef]
- Tuenter, E.; Segers, K.; Kang, K.B.; Viaene, J.; Sung, S.H.; Cos, P.; Maes, L.; Heyde, Y.V.; Pieters, L. Antiplasmodial activity, cytotoxicity and structure-activity relationship study of cyclopetide alkaloids. Molecules 2017, 22, 224. [Google Scholar] [CrossRef]
- Alkadri, S.; Ledwos, N.; Mirchi, N.; Reich, A.; Yilmaz, R.; Driscoll, M.; Del Maestro, R.F. Utilizing a multilayer perceptron artificial neural network to assess a virtual reality surgical procedure. Comput. Biol. Med. 2021, 136, 104770. [Google Scholar] [CrossRef] [PubMed]
- Castro, W.; Oblitas, J.; Santa-Cruz, R.; Avila-George, H. Multilayer perceptron architecture optimisation using parallel computing techniques. PLoS ONE 2017, 12, e0189369. [Google Scholar] [CrossRef]
- Stathakis, D. How many hidden layers and nodes? Int. J. Remote Sens. 2009, 30, 2133–2147. [Google Scholar] [CrossRef]
- Lee, J.G.; Jun, S.; Cho, Y.W.; Lee, H.; Kim, G.B.; Seo, J.B.; Kim, N. Deep learning in medical imaging: General overview. Korean J. Radiol. 2017, 18, 570–584. [Google Scholar] [CrossRef]
- Majumdar, A.; Gupta, M. Recurrent transforming learning. Neural Netw. 2019, 118, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Danishuddin, G.M.; Malik, M.Z.; Subbarao, N. Development and rigorous validation of antimalarial predictive models using machine learning approaches. SAR QSAR Environ. Res. 2019, 30, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Deng, J.; Liu, M. Classification models for predicting the antimalarial activity against Plasmodium falciparum. SAR QSAR Environ. Res. 2020, 31, 312–324. [Google Scholar] [CrossRef]
- Grisoni, F.; Consonni, V.; Todeschini, R. Impact of molecular descriptors on computational models. Methods Mol. Biol. 2018, 1825, 171–209. [Google Scholar] [CrossRef]
- Leach, A.R.; Gillet, V.J. Cheminformatics. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2006; Volume 3, pp. 235–264. [Google Scholar] [CrossRef]
- Araya-Coutier, C.; Vincken, J.P.; Van de Schans, M.G.M.; Hageman, J.; Schaftenaar, G.; Den Besten, H.M.W.; Gruppen, H. QSAR-based molecular signatures of prenylated (iso)flavonoids underlying antimicrobial potency against and membrane-disruption in Gram positive and Gram negative bacteria. Sci. Rep. 2018, 8, 9267. [Google Scholar] [CrossRef]
- Damale, M.; Harke, S.N.; Khan, F.A.K.; Shinde, D.B.; Sangshetti, J.N. Recent advances in multidimensional QSAR (4D-6D): A critical review. Mini Rev. Med. Chem. 2014, 14, 35–55. [Google Scholar] [CrossRef]
- Hall, L.H.; Mohney, B.; Kier, L.B. The electrotopological state: An atom index for QSAR. Quant. Struct. Act. Relatsh. 1991, 10, 43–51. [Google Scholar] [CrossRef]
- Korjus, K.; Hebart, M.N.; Vicente, R. An efficient data partitioning to improve classification performance while keeping parameters interpretable. PLoS ONE 2016, 11, e0161788. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.B.; Gobbo-Neto, L.; Schmidt, T.J.; Da Costa, F.B. Study of chromatographic retention of natural terpenoids by cheminformatics tools. J. Chem. Inf. Model. 2015, 55, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Puzyn, T.; Mostrag-Szlichtyng, A.; Gajewicz, A.; Skrynski, M.; Worth, A.P. Investigating the influence of data splitting on the predictive ability of QSAR/QSPR models. Struct. Chem. 2011, 22, 795–804. [Google Scholar] [CrossRef]
- Landrum, G.A.; Riniker, S. Combining IC50 or Ki values from different sources is a source of significant noise. J. Chem. Inf. Model. 2024, 64, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, R. Estrogenic flavonoids and their molecular mechanisms of action. J. Nutr. Biochem. 2023, 114, 109250. [Google Scholar] [CrossRef]
- Pfitscher, A.; Reiter, E.; Jungbauer, A. Receptor binding and transactivation activities of red clover isoflavones and their metabolites. J. Steroid Biochem. Mol. Biol. 2008, 112, 87–94. [Google Scholar] [CrossRef]
- Happi, G.M.; Ngadjui, B.T.; Green, I.R.; Kouam, S.F. Phytochemistry and pharmacology of the genus Entandrophragma over the 50 years from 1967 to 2018: A ‘golden’ overview. J. Pharm. Pharmacol. 2018, 70, 1431–1460. [Google Scholar] [CrossRef]
- Padmavathi, G.; Roy, N.K.; Bordoloi, D.; Arfuso, F.; Mishra, S.; Sethi, G.; Bishayee, A.; Kunnumakkara, A.B. Butein in health and disease: A comprehensive review. Phytomedicine 2017, 25, 118–127. [Google Scholar] [CrossRef]
- Okoye, I.; Yu, S.; Caruso, F.; Rossi, M. X-ray structure determination, antioxidant voltammetry studies of Butein and 2′,4′-Dihydroxy-3,4-dimethoxychalcone. Computational studies of 4 structurally related 2′,4′-diOH chalcones to examine their antimalarial activity by binding to Falcipain-2. Molecules 2021, 26, 6511. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Chun, W. Virtual screening of flavonoids against Plasmodium vivax Duffy binding protein utilizing molecular docking and molecular dynamics simulation. Curr. Comput. Aided Drug Des. 2024, 20, 616–627. [Google Scholar] [CrossRef]
- Chabir, N.; Ibrahim, H.; Romdhane, M.; Valentin, A.; Moukarzel, B.; Mars, M.; Bouajila, J. Seeds of Peganum harmala L. chemical analysis, antimalarial and antioxidant activities, and cytotoxicity against human breast cancer cells. Med. Chem. 2015, 11, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.B.; Mäser, P.; Kaiser, M.; Hamburger, M.; Khalid, S. Mining sudanese medicinal plants for antiprotozoal agents. Front. Pharmacol. 2020, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Chepkirui, C.; Ochieng, P.J.; Sarkar, B.; Hussain, A.; Pal, C.; Yang, L.J.; Coghi, P.; Akala, H.M.; Derese, S.; Ndakala, A.; et al. Antiplasmodial and antileishmanial flavonoids from Mundulea sericea. Fitoterapia 2021, 149, 104796. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, S.; Vásquez-Ocmín, P.G.; Cojean, S.; Bouzidi, C.; Michel, S.; Figadère, B.; Grougnet, R.; Boutefnouchet, S.; Maciuk, A. Correlation study on methoxylation pattern of flavonoids and their heme-targeted antiplasmodial activity. Bioorg. Chem. 2020, 104, 104243. [Google Scholar] [CrossRef]
- Zininga, T.; Ramatsui, L.; Makhado, P.B.; Makumire, S.; Achlinou, I.; Hoppe, H.; Dirr, H.; Shonhai, A. (−)-Epigallocatechin-3-gallate inhibits the chaperone activity of Plasmodium falciparum Hsp70 chaperones and abrogates their association with functional partners. Molecules 2017, 22, 2139. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 19, 616–628. [Google Scholar] [CrossRef]
- Dahlin, J.L.; Walters, M.A. How to triage PAINS-Full Research. Assay Drug Dev. Technol. 2016, 14, 168–174. [Google Scholar] [CrossRef]
- MarvinSketch, v. 19.22; ChemAxon Ltd.: Budapest, Hungary, 2022. Available online: https://chemaxon.com/products/marvin (accessed on 30 June 2025).
- Luo, J.; Cisler, R.A. Discovering outliers or potential drug toxicities using a large-scale data-driven approach. Cancer Inform. 2016, 15, 211–217. [Google Scholar] [CrossRef]
- KNIME, v. 4.0.2; KNIME AG: Zürich, Switzerland, 2022. Available online: https://www.knime.com/downloads (accessed on 30 June 2025).
- Molina-Cruz, A.; DeJong, R.J.; Ortega, C.; Haile, A.; Abban, E.; Rodrigues, J.; Jaramillo-Gutierrez, G.; Barillas-Mury, C. Some strains of Plasmodium falciparum, a human malaria parasite, evade complement-like system of Anopheles gambiae mosquitoes. Proc. Natl. Acad. Sci. USA 2012, 109, E1957–E1962. [Google Scholar] [CrossRef]
- Awandare, G.A.; Nyarko, P.B.; Aniweh, Y.; Ayivor-Djanie, R.; Stoute, J.A. Plasmodium falciparum strains spontaneously switch evasion phenotype in suspension culture. Sci. Rep. 2018, 8, 5782. [Google Scholar] [CrossRef] [PubMed]
- MOPAC2016. Paddington Circle, USA. 2022. Available online: http://openmopac.net/downloads.html (accessed on 30 June 2025).
- RDKit, v. 2021.03.1; Greg Landrum: Dallas, TX, USA, 2022. Available online: https://www.rdkit.org (accessed on 30 June 2025).
- Yap, C.W. PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Akoglu, H. User’s guide to correlation coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef]
- Saunders, L.J.; Russel, R.A.; Crabb, D.P. The coefficient of determination: What determines a useful R2 statistic? Investig. Ophtalmol. Vis. Sci. 2010, 53, 6830–6832. [Google Scholar] [CrossRef]
- Russo, D.P.; Zorn, K.M.; Clark, A.M.; Zhu, H.; Ekins, S. Comparing multiple machine learning algorithms and metrics for estrogen receptor binding prediction. Mol. Pharm. 2018, 15, 4361–4370. [Google Scholar] [CrossRef] [PubMed]
- Taraji, M.; Haddad, P.R.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Error measures in quantitative structure-activity relationship studies. J. Chromatogr. A. 2017, 1524, 298–302. [Google Scholar] [CrossRef]
- Enalos, v. 2021; NovaMechanics Ltd.: Nicosia, Cyprus, 2022. Available online: http://www.novamechanics.com (accessed on 30 June 2025).
- Kar, S.; Roy, K.; Leszczynski, J. Applicability domain: A step toward confident predictions and decidability for QSAR modelling. Methods Mol. Biol. 2018, 1800, 141–169. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.J. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, 1214–1219. [Google Scholar] [CrossRef]
- Neveu, V.; Pérez-Jiménez, J.; Vos, F.; Crespy, V.; Du Chaffaut, L.; Mennen, L.; Knox, C.; Eisner, R.; Cruz, J.; Wishart, D.; et al. Phenol-Explorer: An online comprehensive database on polyphenol contents in foods. Database 2010, 2010, bap024. [Google Scholar] [CrossRef]
- GOLD, v. 5.4; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2022. Available online: https://www.ccdc.cam.ac.uk (accessed on 30 June 2025).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Kaur, A.; Kaur, I. An empirical evaluation of classification algorithms for fault prediction in open source projects. J. King Saud Univ. Comput. Info. Sci. 2018, 30, 2–17. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), v. 2020.0901; Chemical Computing Group ULC: Montreal, QC, Canada, 2025.
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2004, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FlavStr Classification | ||||
|---|---|---|---|---|
| ATS8m | minsCH3 | maxHdsCH | MDEO-22 | |
| ATS8m | 1.00 | −0.02 | −0.21 | 0.61 |
| minsCH3 | −0.02 | 1.00 | 0.22 | 0.34 |
| maxHdsCH | −0.20 | 0.22 | 1.00 | −0.13 |
| MDEO-22 | 0.61 | 0.34 | −0.13 | 1.00 |
| FlavStr regression | ||||
| smr_VSA10 | minsCH3 | MDEO-22 | GGI8 | |
| smr_VSA10 | 1.00 | 0.11 | 0.38 | 0.27 |
| minsCH3 | 0.11 | 1.00 | 0.34 | −0.08 |
| MDEO-22 | 0.38 | 0.34 | 1.00 | 0.51 |
| GGI8 | 0.27 | −0.08 | 0.51 | 1.00 |
| FabG | ||||
| AATS8m | VE3_Dzv | FNSA-1 | ||
| AATS8m | 1.00 | 0.48 | 0.48 | |
| VE3_Dzv | 0.48 | 1.00 | −0.06 | |
| FNSA-1 | 0.48 | −0.06 | 1.00 | |
| FabZ | ||||
| ATS3m | nHBint7 | SIC3 | ||
| ATS3m | 1.00 | 0.48 | −0.58 | |
| nHBint7 | 0.48 | 1.00 | −0.14 | |
| SIC3 | −0.58 | −0.14 | 1.00 | |
| FabI | ||||
| VE3_Dzv | maxHBint9 | TDB6u | ||
| VE3_Dzv | 1.00 | 0.46 | −0.45 | |
| maxHBint9 | 0.46 | 1.00 | −0.53 | |
| TDB6u | −0.45 | −0.53 | 1.00 | |
| Algorithm | Database | Dataset | A | K | E |
|---|---|---|---|---|---|
| MLP | FlavStr | Tr | 86.53% | 0.72 | 13.46% |
| Ts | 92.85% | 0.85 | 7.14% | ||
| CV | 73.07% | 0.45 | 26.92% | ||
| RF | FabG | Tr | 100.00% | 1.00 | 0.00% |
| Ts | 100.00% | 1.00 | 0.00% | ||
| CV | 87.50% | 0.71 | 12.50% | ||
| SVM | FabZ | Tr | 100.00% | 1.00 | 0.00% |
| Ts | 100.00% | 1.00 | 0.00% | ||
| CV | 100.00% | 1.00 | 0.00% | ||
| KNN | FabI | Tr | 100.00% | 1.00 | 0.00% |
| Ts | 100.00% | 1.00 | 0.00% | ||
| CV | 87.50% | 0.75 | 12.50% |
| Algorithm | Database | Dataset | R2 | RMSE | Acceptability Criteria |
|---|---|---|---|---|---|
| SVM | FlavStr | Tr | 0.65 | 0.72 | |
| Ts | 0.64 | 0.86 | Passed | ||
| CV | 0.54 | 0.83 | |||
| MLP | FabG | Tr | 0.97 | 0.13 | |
| Ts | 0.95 | 0.09 | Passed | ||
| CV | 0.94 | 0.19 | |||
| MLP | FabZ | Tr | 0.99 | 0.03 | |
| Ts | 0.97 | 0.10 | Passed | ||
| CV | 0.99 | 0.03 | |||
| PLS | FabI | Tr | 0.99 | 0.07 | |
| Ts | 0.94 | 0.14 | Passed | ||
| CV | 0.99 | 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, A.d.O.; Paixão, V.V.M.; Santos, Y.J.A.; Alves, E.B.; Rodrigues, R.P.; Chagas-Paula, D.A.; Faraoni, A.S.; Casoti, R.; Batista, M.V.d.A.; Bermudez, M.; et al. Identification of Pharmacophore Groups with Antimalarial Potential in Flavonoids by QSAR-Based Virtual Screening. Drugs Drug Candidates 2025, 4, 33. https://doi.org/10.3390/ddc4030033
Fernandes AdO, Paixão VVM, Santos YJA, Alves EB, Rodrigues RP, Chagas-Paula DA, Faraoni AS, Casoti R, Batista MVdA, Bermudez M, et al. Identification of Pharmacophore Groups with Antimalarial Potential in Flavonoids by QSAR-Based Virtual Screening. Drugs and Drug Candidates. 2025; 4(3):33. https://doi.org/10.3390/ddc4030033
Chicago/Turabian StyleFernandes, Adriana de Oliveira, Valéria Vieira Moura Paixão, Yria Jaine Andrade Santos, Eduardo Borba Alves, Ricardo Pereira Rodrigues, Daniela Aparecida Chagas-Paula, Aurélia Santos Faraoni, Rosana Casoti, Marcus Vinicius de Aragão Batista, Marcel Bermudez, and et al. 2025. "Identification of Pharmacophore Groups with Antimalarial Potential in Flavonoids by QSAR-Based Virtual Screening" Drugs and Drug Candidates 4, no. 3: 33. https://doi.org/10.3390/ddc4030033
APA StyleFernandes, A. d. O., Paixão, V. V. M., Santos, Y. J. A., Alves, E. B., Rodrigues, R. P., Chagas-Paula, D. A., Faraoni, A. S., Casoti, R., Batista, M. V. d. A., Bermudez, M., Dolabella, S. S., & Oliveira, T. B. (2025). Identification of Pharmacophore Groups with Antimalarial Potential in Flavonoids by QSAR-Based Virtual Screening. Drugs and Drug Candidates, 4(3), 33. https://doi.org/10.3390/ddc4030033