Abstract
Mutations in hormone receptors significantly influence infertility and the outcomes of assisted reproductive technologies (ART). This review explores the functional interplay among mutations in FSHR, LHCGR, AR, ESR1, and ESR2 hormone receptors and their combined effects on hormonal regulation, ovarian response, and implantation. Rather than analyzing receptor mutations in isolation, we explore how mutations in these genes interact within a complex hormonal signaling network, shaping reproductive outcomes. We detail the molecular mechanisms of receptor dysfunction, their associated clinical phenotypes, and the role of genetic screening in guiding personalized ART protocols. A comprehensive understanding of these interactions is crucial for optimizing treatment strategies, improving reproductive success, and advancing targeted therapeutic approaches in reproductive medicine.
1. Introduction
Infertility is a complex and multifactorial condition that affects a significant proportion of the population, with a growing number of couples seeking assisted reproductive technology [ART] as a treatment. While these technologies have revolutionized the management of infertility, success rates can be variable, often influenced by underlying genetic factors. One such factor involves mutations in the hormone receptors that regulate key aspects of reproductive physiology. These include the follicle-stimulating hormone receptor [FSHR], luteinizing hormone receptor [LHR], androgen receptor [AR], estrogen receptor [ER], and progesterone receptor [PGR] [1]. These receptors play crucial roles in regulating reproductive functions, including follicular development, ovulation, spermatogenesis, and uterine receptivity. Alterations in hormone receptors’ structure and function, induced by mutations, can significantly impair hormonal signaling pathways, leading to suboptimal responses to infertility therapies [2].
The FSHR gene is critical in regulating ovarian response to follicle-stimulating hormone [FSH], and mutations in this gene can disrupt ovarian reserve and response, influencing ART outcomes [3]. Similarly, mutations in the LHR can affect ovulation induction, testosterone production, and fertility in both males and females [4]. Mutations in the AR are commonly associated with androgen insensitivity syndrome, impacting spermatogenesis and male fertility [5]. The ER, through its regulation of estrogen signaling, is pivotal in follicular development, embryo implantation, and uterine receptivity, while mutations in the PGR can impair progesterone signaling, essential for the maintenance of pregnancy. The FSHR, AR, LHCGR, ESR1, and ESR2 genes encode receptors that mediate hormonal signals essential for sexual differentiation, fertility, and endocrine regulation [6,7]. Mutations in these genes can result in disrupted receptor function, leading to disorders of sexual development, infertility, and metabolic dysfunctions. We aimed to analyze selected receptors’ mutations type, functional domain involvement, and molecular implications, impacting fundamentals of infertility conditions and their clinical management.
Despite the accumulated evidence on these mutations, their specific impact on ART success remains underexplored [8]. Understanding the molecular mechanisms behind receptor mutations and their influence on reproductive processes is essential for optimizing infertility treatments. Genetic testing for these receptor mutations can offer valuable insights, enabling clinicians to tailor ART protocols to individual patients, ultimately improving treatment outcomes.
This review aims to synthesize the current knowledge on the genetic variations in FSHR, LHR, AR, ER, and PGR receptors, emphasizing their implications for infertility therapies and exploring future directions for personalized medicine in reproductive health.
2. Methodology
We have searched he ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, accessed 10 January 2024) for FSHR, AR, LHCGR, ESR1, ESR2, and PGR pathogenic or likely pathogenic mutations exclusively, affecting various aspects of infertility. There are in total 26 pathogenic/likely pathogenic mutations in the FSHR gene 248 in AR, 35 in LHCGR, five in ESR1, one in ESR2 and none in PGR. In Figure 1, we emphasized the differences in the number of mutations between the hormones, and in Table 1, we add detailed information for each hormone of interest. The PGR gene was excluded from the scope of the analysis. The reason for this is the disease-associated mutational co-occurrence of the PGR gene with several other genes.
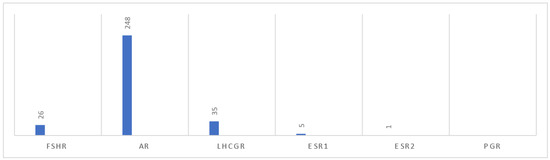
Figure 1.
Chart of reproductive hormones and number of likely pathogenic/pathogenic mutations.

Table 1.
Number of likely pathogenic/pathogenic mutations per referent gene.
For visualization of the mutation locations within protein domains, we used the Mutation3D (http://mutation3d.org, accessed 10 January 2024) [9] online tool and ALPHAFOLD (https://alphafold.com/, accessed 10 January 2024) [10,11] protein structure predictions. AlphaFold works with pLDDT confidence scores (0–100) reflecting the reliability of the predicted 3D protein structure, with 90–100 (blue) indicating very high confidence where atomic positions are very likely correct, 70–90 (cyan–blue) high confidence with backbone accuracy but minor side-chain uncertainty, 50–70 (yellow) low confidence in flexible or partly displayed regions, and <50 (orange–red) very low confidence in likely chaotic or unreliable regions, serving as a trust score for each part of the structure. These were used to illustrate how mutations are distributed across the ligand-binding domains, DNA-binding domains, transmembrane domains, and intracellular regions, depending on the specific receptor.
A systematic literature search was later performed using the three referent data bases, PUBMED (https://pmc.ncbi.nlm.nih.gov/ accessed 10 January 2024), Scopus (https://www.scopus.com/, accessed 10 January 2024), and Google Scholar (https://scholar.google.com/schhp?hl=en, accessed 10 January 2024), given the following combinations of keywords: gene ∈ {FSHR, LHR, AR, ER}, hormone receptor, mutation_id|from ClinVar database, effectiveness, infertility, therapy, assisted reproductive technology. A total of 103 studies met our inclusion criteria, with no language or period limitation.
To provide a comprehensive and clinically meaningful overview of hormone receptor mutations, we implemented a uniform structure for analyzing each receptor. Each hormone receptor discussed in this review is presented using two dedicated subchapters: one focused on the mutations and their molecular impact, and another on the clinical conditions associated with these mutations and their management.
In each case, we begin by identifying pathogenic and likely pathogenic mutations reported in ClinVar, prioritizing those with established associations to infertility-related phenotypes. Variants of uncertain significance (VUS) were excluded, unless they were consistently described in the literature in connection with reproductive dysfunction. Particular attention was given to the protein domains affected, as disruptions in different structural regions (e.g., extracellular, transmembrane, or intracellular) can lead to distinct alterations in receptor function.
By classifying mutations based on their molecular effect, such as loss of function or gain of function, we relate genetic changes to specific disruptions in receptor behavior, including impaired hormone binding, altered signal transduction, or constitutive activation. These mechanistic insights are essential for predicting disease severity and guiding personalized treatment strategies in assisted reproductive technology (ART).
In this review, we consider the “key mutations” for each receptor. The term “key mutation” refers to a pathogenic or likely pathogenic variant according to the current ClinVar database classification status, with available supporting evidence in the literature (at least one reference) that associates the concrete mutation with well-defined infertility phenotypes, based on the availability of functional or clinical characterization. They are intended to serve as representative examples of mutation–phenotype relationships with therapeutic significance.
3. Analysis of FSHR Gene Mutations: Detailed Overview and Clinical Implications
The FSHR (follicle-stimulating hormone receptor) gene encodes a G protein-coupled receptor (GPCR) that plays an essential role in mediating the actions of follicle-stimulating hormone (FSH), which is critical for regulating follicular development, gametogenesis, and overall reproductive hormone signaling [12]. This receptor is predominantly expressed in the ovaries, testes, and uterus, regulating processes such as folliculogenesis in females and spermatogenesis in males [13]. The FSHR gene is located on chromosome 2p21-p16, spans approximately 54 kb, and consists of 10 exons and 9 introns. The encoded receptor is a 75 kDa glycoprotein, consisting of 678 amino acids, with a 7-transmembrane structure, extracellular leucine-rich repeats for high-affinity FSH binding, and intracellular loops facilitating signal transduction through G proteins [14]. Upon FSH binding, the receptor undergoes a conformational change that activates the cAMP signaling pathway, ultimately leading to the activation of protein kinase A [PKA] and the regulation of gene expression via cAMP response elements (CREs) [15]. Additionally, FSHR signaling is modulated by prostaglandins and intracellular calcium, and receptor activation leads to downstream activation of extracellular signal-regulated kinases (ERK), contributing to further phosphorylation of the receptor [16].
In females, FSHR expression on granulosa cells is crucial for the development of ovarian follicles, while in males, it is expressed in Sertoli cells, where it regulates spermatogenesis. FSHR is also expressed in other tissues such as the placenta, prostate, bone, and ovarian epithelium, and its expression in tumor vasculature has been associated with metastasis. Receptor expression is regulated by estrogen, which promotes receptor upregulation, and prolonged exposure to FSH contributes to desensitization through receptor phosphorylation or uncoupling from the G protein, resulting in internalization and degradation or recycling of the receptor [17]. Additionally, FSHR expression remains a topic of debate, as Ponikwicka-Tyszko et al., 2016 [18], note, suggesting variations in its tissue distribution and regulatory mechanisms.
We use ALPHAFOLD to visualize structural features of the wild-type FSHR protein to better interpret the impact of pathogenic mutations (Figure 2).
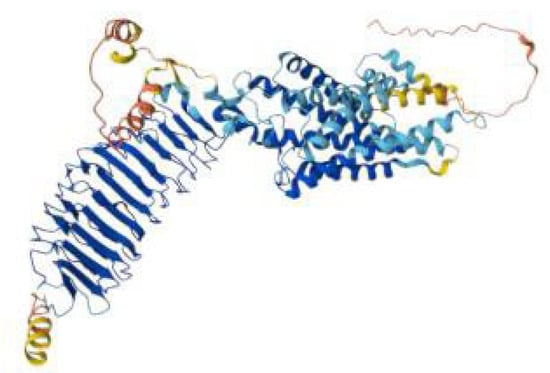
Figure 2.
AlphaFold model of the FSHR protein colored, showing the extracellular leucine-rich repeat hormone-binding domain in blue, the seven-transmembrane helical domain in cyan–blue, and the flexible intracellular and terminal regions in yellow to red.
FSHR mutations impair follicular development and response to gonadotropins by altering receptor-ligand binding, affecting intracellular signaling cascades, and modifying ovarian sensitivity to gonadotropins. The extracellular ligand-binding domain is critical for FSH binding, while the transmembrane and intracellular domains mediate signal transduction.
Understanding FSHR mutagenicity paves the way towards better diagnosis and personalized treatment options.
3.1. Mutations and Molecular Impact
We have identified 26 mutations in the ClinVar database that met our inclusion criteria: 24 missense, 1 nonsense, and 1 frameshift mutation (Table 2). Loss-of-function mutations (e.g., nonsense or frameshift) typically result in receptor inactivation, compromising FSH signaling and requiring higher exogenous hormone doses during ART. Gain-of-function mutations (e.g., certain missense variants) may increase receptor sensitivity or cause constitutive activation, leading to hormonal overstimulation and heightened OHSS risk.
Table 2.
FSHR mutations’ types and molecular effects.
These were classified as the following:
- Pathogenic: 12 mutations (46%)
- Likely Pathogenic: 12 mutations (46%)
- Pathogenic/Likely Pathogenic: 2 mutations (8%) *
The majority of these mutations affect receptor structure and function through disruption of ligand binding, protein folding, or intracellular signaling. FSHR mutations are distributed across three principal structural domains, each with distinct functional roles (Figure 3):
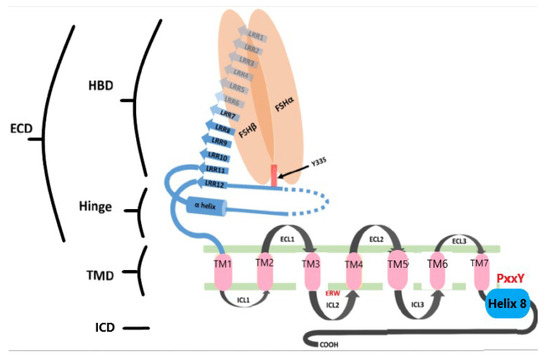
Figure 3.
Schematic representation of FSHR structure. The receptor consists of an N-terminal extracellular domain (ECD), transmembrane domain (TMD), and intracellular domain (ICD). The ECD includes the hormone-binding domain (HBD) with 10 leucine-rich repeats (LRRs) and a hinge region containing two additional LRRs, a hairpin loop, and an α-helix. A sulfated tyrosine at position 335 is essential for hormonal activation. The ICD, rich in serine and threonine residues, acts as a phosphorylation site in signal transduction. LRRs are shown as blue arrows, the α-helix in green, and the TMD helices (1–7) as pink boxes. FSH-α and FSH-β subunits are represented as orange ovoids. ECL: extracellular loop; ICL: intracellular loop; HBD: hormone-binding domain; ICD: intracellular domain.
- Extracellular domain (ECD): This domain includes leucine-rich repeat (LRR) motifs and is responsible for high-affinity binding to FSH. It plays a critical role in ligand recognition and initiation of receptor activation. Mutations such as p.Leu125Arg impair this binding, leading to defective receptor activation and impaired follicular development [19].
- Transmembrane domain (TMD): Composed of seven α-helical segments, the TMD is essential for conformational changes in the receptor that trigger intracellular signaling via G protein interaction. Mutations such as p.Asp567Asn and p.Ala462Pro destabilize this domain, altering signal transduction and in some cases causing constitutive receptor activation [20].
- Intracellular domain (ICD): This domain contains key phosphorylation sites involved in downstream signaling, especially through the cAMP pathway. Variants like p.Thr449Ile impair intracellular signaling, leading to disrupted granulosa cell activity and compromised folliculogenesis [21].
Understanding the type and domain location of mutations is essential for predicting disease severity and for guiding personalized treatment approaches in assisted reproductive technology (ART). Loss-of-function mutations (e.g., nonsense or frameshift) typically cause receptor inactivation, requiring higher exogenous hormone doses to achieve stimulation. Conversely, gain-of-function mutations (e.g., specific missense variants) enhance receptor sensitivity or lead to constitutive activity, increasing the risk for ovarian hyperstimulation syndrome (OHSS).
In Table 3, we summarize “FSHR key mutations” with information on pathogenicity, molecular effect, and clinical associations of each variant and schematically represented the locations of some mutations in the FSHR protein on the Figure 4 using Mutation3D.
Table 3.
Key FSHR mutations and their clinical impact.
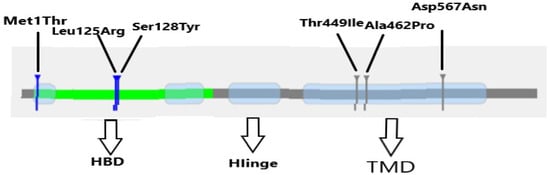
Figure 4.
Schematic representation of the locations of some mutations in FSHR according to Mutation3D.
3.2. Clinical Conditions and Management
FSHR mutations are associated with four major infertility phenotypes. Table 4 provides information on the distribution of FSHR mutations per clinical condition and the most common mutations, including their molecular implications.
Table 4.
Distribution of FSHR mutations per clinical condition.
Below is a summary of the underlying mechanisms, diagnostic criteria, and treatment options for each condition.
- o
- Ovarian Dysgenesis 1 is caused by mutations in the FSH receptor (FSHR) gene that result in receptor misfolding or structural instability, such as p.Leu125Arg and p.Pro348Arg [28,29,30,31]. Affected individuals typically present with delayed puberty, primary amenorrhea, and underdeveloped ovaries. Hormonal evaluation reveals elevated FSH and LH with low estrogen, and genetic confirmation through FSHR sequencing is crucial. Management includes hormone replacement therapy (HRT) to induce and sustain secondary sexual characteristics while preserving bone and cardiovascular health. Fertility preservation is not feasible due to the absence of functional ovarian follicles. Psychological support and genetic counseling are integral to patient care.
- o
- Genetic Non-Acquired Premature Ovarian Failure (POF) results from truncating mutations that inactivate the FSHR, such as p.Gln117Ter and p.Asn560fs [19,32]. Clinically, it manifests as menstrual irregularities or amenorrhea before the age of 40, accompanied by hypergonadotropic hypogonadism and diminished ovarian reserve. Genetic testing for FSHR mutations can confirm the diagnosis. Management may involve assisted reproductive technologies (ART) using high-dose gonadotropins if residual ovarian function exists, though oocyte donation is often the preferred option. Long-term HRT is necessary to manage hypoestrogenism and support overall systemic health.
- o
- Ovarian Hyperstimulation Syndrome (OHSS) is linked to gain-of-function FSHR mutations, such as p.Asp567Asn and p.Ala462Pro [20,21,22,23,24,25,32], which increase ovarian sensitivity to FSH. It commonly arises during ART cycles and is characterized by an exaggerated ovarian response, ascites, and elevated estradiol levels. It leads to an exaggerated response to follicle-stimulating hormone (FSH), which is the primary cause of OHSS [26]. Diagnosis is typically clinical, with genetic testing used to confirm predisposition. Management includes using low-dose gonadotropin regimens and GnRH antagonists and replacing hCG with GnRH agonists to trigger ovulation. Preventive strategies may involve cycle segmentation, coasting, or elective embryo freezing. Preconception genetic screening is recommended in patients with a history of OHSS.
- o
- Amenorrhea due to FSHR mutations, such as the p.Met1Thr variant [ClinVar; VCV000523337.3], is caused by a complete loss of receptor translation. This condition presents as primary amenorrhea with elevated FSH, low estrogen, and absent follicular activity. Diagnosis involves hormonal profiling and imaging, with genetic testing confirming FSHR dysfunction. Lifelong HRT is required for the development of secondary sexual characteristics and maintenance of endometrial health. Fertility treatment typically relies on oocyte donation and assisted reproductive techniques, and patients benefit from comprehensive reproductive and psychological support.
Identification of pathogenic FSHR variants enables genotype-based risk stratification and supports the optimization of gonadotropin stimulation protocols in controlled ovarian hyperstimulation (COH), thereby facilitating individualized ART strategies, preventing complications such as OHSS, and improving overall fertility outcomes [27].
4. Analysis of AR Gene Mutations: Detailed Overview and Clinical Implications
The androgen receptor (AR), shown in Figure 5, according to ALPHAFOLD, also known as NR3C4, is a nuclear receptor activated by binding to androgens like testosterone and dihydrotestosterone in the cytoplasm, after which it translocates to the nucleus [33]. The AR, closely related to the progesterone receptor, can be inhibited by high doses of progestins [34]. It functions primarily as a DNA-binding transcription factor that regulates gene expression and plays an important role in developing and maintaining male sexual characteristics. In development, testosterone can directly bind to AR or be converted to dihydrotestosterone (DHT) via 5-alpha-reductase, a more potent activator of the receptor. Testosterone regulates primary male sexual characteristics, while DHT is responsible for secondary characteristics. Androgens also influence bone maturation, partly through estrogen produced via aromatization, and can impact growth, as seen in androgen or estrogen insensitivity syndromes. Studies indicate that AR is essential for female fertility, aiding ovarian follicle development and ovulation through intra-ovarian and neuroendocrine mechanisms [35,36]. The receptor also plays a role in skeletal integrity in males, affecting both osteoblasts and osteocytes, and regulates various functions in females, including cardiac and bone growth [37]. Disruptions in AR signaling can lead to conditions like androgen insensitivity syndrome (AIS).
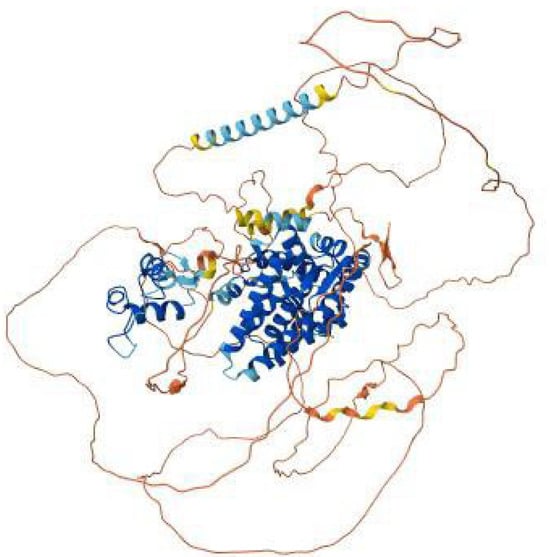
Figure 5.
AlphaFold model of the AR (androgen receptor) protein colored, showing the well-structured ligand-binding and DNA-binding domains in blue, connecting α-helices in cyan–blue, and the flexible N- and C-terminal regions in yellow to red.
The primary mechanism of AR involves direct gene transcription regulation. When androgens bind to AR in the cytoplasm, the receptor translocates to the nucleus, where it dimerizes and binds androgen-responsive elements on DNA, regulating genes such as the insulin-like growth factor 1 receptor (IGF-1R), which is involved in altering cell behavior [38]. Following androgen binding, AR undergoes conformational changes that release heat shock proteins and associates with hormone response elements, recruiting co-regulators for rapid gene regulation. AR can also influence cellular processes independent of DNA binding by interacting with other DNA-binding proteins, promoting muscle growth [39]. Post-translational modifications, such as acetylation, enhance AR transactivation and cancer cell proliferation, with regulation by histone deacetylases and long non-coding RNAs. Recently, AR has demonstrated non-genomic actions, where cytoplasmic ARs influence signal transduction proteins, leading to rapid cellular responses that can indirectly affect gene transcription by phosphorylating transcription factors. The AR gene is located on the X chromosome at Xq11–12, with numerous disease-causing mutations identified [40]. Androgen insensitivity syndrome results from AR gene mutations and affects neuron physiology, linking to conditions such as Kennedy’s disease [41]. Point mutations and polymorphisms in the AR gene have been also associated with trinucleotide repeat disorders. For instance, variations in CAG repeats may potentially influence receptor sensitivity and prostate cancer incidence and mortality, particularly noted in studies comparing African Americans and non-Hispanic whites [42,43].
The androgen receptor (AR) gene encodes a nuclear receptor that mediates the actions of androgens [e.g., testosterone and dihydrotestosterone], playing a key role in male sexual differentiation, development of secondary sexual characteristics, and fertility. AR gene mutations can lead to a spectrum of disorders, based on the type of mutation and its location in the gene. These include Complete Androgen Insensitivity Syndrome [CAIS], Partial Androgen Insensitivity Syndrome [PAIS], Spinal and Bulbar Muscular Atrophy [SBMA], Androgen-Related Infertility, Prostate Cancer Susceptibility, and other androgen-related conditions. AR mutations disrupt androgen receptor signaling, leading to a variety of clinical manifestations ranging from androgen insensitivity syndromes [AIS] to disorders affecting muscles, cognition, and fertility. ALPHAFOLD modeling (Figure 5) was used to assess structural impacts.
4.1. Mutations and Molecular Impact
The AR gene encodes the androgen receptor, a ligand-dependent nuclear transcription factor that mediates the biological effects of androgens such as testosterone and dihydrotestosterone. It plays a key role in male sexual differentiation, spermatogenesis, prostate development, and neuromuscular function. The AR protein contains several critical domains: the N-terminal trans activation domain (NTD), DNA-binding domain (DBD), hinge region, and ligand-binding domain (LBD). It also includes a polyglutamine (polyQ) tract encoded by a polymorphic CAG repeat.
Reported AR mutations are primarily of single nucleotide variant (181), deletion (39), and duplication (19) and three indels, insertions, and microsatellites, as seen in Figure 6. Most of the AR gene mutations are missense variants, with a smaller portion of frameshift and nonsense mutations. These variations contribute to different molecular implications, impacting the receptor’s structure and function, as seen in Table 5.
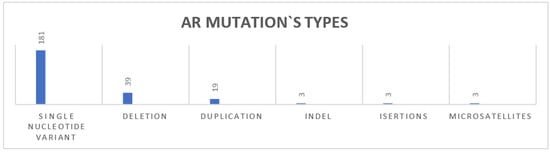
Figure 6.
AR gene mutation types.
Table 5.
AR gene mutations’ molecular effects.
The AR protein consists of several key domains, each playing a critical role in its function, as seen in Figure 7.
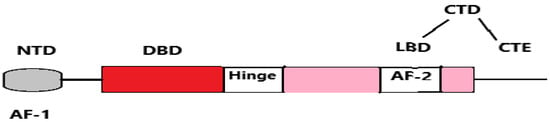
Figure 7.
Schematic representation of the human androgen receptor (AR) structure, showing its key domains: NTD (N-terminal domain), DBD (DNA-binding domain), LBD (ligand-binding domain), CTE (C-terminal extension), and CTD (C-terminal domain). It also includes the activation function regions: AF-1 (activation function-1) and AF-2 (activation function-2).
Mutations in these regions may drive different functional impairments:
- Ligand-Binding Domain (LBD): Mutations here can impair androgen binding or receptor activation, leading to conditions such as Complete Androgen Insensitivity Syndrome (CAIS) Such is the p.Val904Leu mutation [44].
- DNA-Binding Domain (DBD): Essential for AR-DNA interaction; mutations such as p.Cys580Phe reduce transcriptional activity [45].
- N-Terminal Domain (NTD): Critical for transcriptional activation; premature stop mutations such as p.Ter921Arg truncate the receptor, disabling activation [46].
- PolyQ Region (CAG repeats): Expansion mutations (e.g., c.172_174 CAG (38_68)) result in toxic gain-of-function effects and can cause Spinal and Bulbar Muscular Atrophy (SBMA) [47].
Pathogenic classification (ClinVar):
- Pathogenic: 162 mutations (65%)
- Likely pathogenic: 62 mutations (25%)
- Pathogenic/likely pathogenic: 24 mutations (10%)
* Mutations that in different protein changes have different pathogenic status.
Approximately 65% of the total number of AR gene mutations are already classified as pathogenic, which underlines their clinical relevance, contributing directly to the phenotypic expression of androgen insensitivity and other associated disorders.
Functional implications of AR pathogenic gene mutations range from receptor loss-of-function, resulting in androgen insensitivity, to gain-of-function effects, which can enhance AR activity and contribute to disease phenotypes such as prostate cancer or SBMA. In clinical contexts, understanding the molecular behavior of these mutations is crucial for interpreting disease mechanisms and designing personalized treatment protocols.
In Table 6 we summarize the “AR key mutations,” the associated clinical conditions, the current ClinVar classification status, mutations’ type, and molecular implications:
Table 6.
Key AR gene mutations and their clinical impact.
4.2. Clinical Conditions and Management
Table 7 provides information on the distribution of AR mutations per clinical condition and the most common key mutations, including their molecular implications.
Table 7.
Distribution of AR gene mutations per clinical condition.
AR mutations are associated with several androgen-related disorders:
- o
- Androgen Insensitivity Syndrome (AIS) is caused by loss-of-function mutations in the androgen receptor (AR) gene that impair receptor binding or downstream signaling. The condition presents along a spectrum: in complete AIS (CAIS) [48], individuals with an XY karyotype exhibiting a typical female phenotype [54], while in partial AIS (PAIS), the phenotype ranges from ambiguous genitalia to varying degrees of undervirilization [55]. Clinical signs include primary amenorrhea, absence of Müllerian structures, or underdeveloped genitalia. Hormonal analysis typically shows elevated testosterone levels with minimal androgenic effects. Confirmation requires sequencing of the AR gene. Management of CAIS involves bilateral gonadectomy to mitigate malignancy risk, followed by lifelong estrogen replacement for feminization. PAIS management includes surgical correction of genital ambiguity, androgen therapy, and psychological support.
- o
- Spinal and Bulbar Muscular Atrophy (SBMA), also known as Kennedy’s disease, results from a CAG trinucleotide repeat expansion in the AR gene, leading to a toxic gain of function [50,51]. It typically manifests in adulthood with progressive muscle weakness, bulbar symptoms, gynecomastia, and infertility [56]. Diagnosis is confirmed by genetic testing showing CAG repeat expansion in the AR gene. Management is primarily supportive and includes physical therapy, speech and swallowing interventions, and participation in clinical trials exploring treatments aimed at reducing AR expression or preventing protein aggregation.
- o
- Androgen-Related Infertility is attributed to AR mutations that disrupt androgen-mediated gene regulation in Sertoli cells, impairing spermatogenesis [52,53]. Clinically, it presents as azoospermia or oligospermia despite normal hormone levels, indicating defective AR function [57]. Diagnosis involves gene sequencing of the AR gene. Management strategies include assisted reproductive technologies such as in vitro fertilization (IVF) or intracytoplasmic sperm injection (ICSI), often in combination with testicular sperm extraction (TESE). Hormonal modulation may be explored in select cases.
- o
- Prostate Cancer Susceptibility is linked to gain-of-function mutations or amplification of the AR gene, which result in increased AR activity and tumor progression [49,58]. Diagnosis relies on elevated PSA levels, prostate biopsy, and molecular profiling, with AR mutation analysis contributing to risk stratification. Treatment options include androgen deprivation therapy (ADT), use of second-generation AR antagonists like enzalutamide, and precision medicine approaches targeting AR signaling pathways.
- o
- Other Androgen-Related Disorders may involve disruptions in the neurodevelopmental, metabolic, or endocrine functions of AR due to various mutations [59,60]. The clinical presentation is diverse and depends on the affected system. Diagnosis requires a multidisciplinary approach and may include neurological, endocrine, and genetic evaluations. Management is individualized and can involve hormone replacement therapy, metabolic or neurologic monitoring, and targeted endocrine interventions based on the specific manifestations.
Recognition of AR mutations helps diagnosis of AIS subtypes, infertility, or neuromuscular disorders, guiding androgen therapy, surgical management, and fertility preservation strategies.
5. Analysis of LHCGR Gene Mutations: Detailed Overview and Clinical Implications
The LHCGR gene encodes the luteinizing hormone/choriogonadotropin receptor, an important G protein-coupled receptor (GPCR) responsible for mediating the biological effects of luteinizing hormone (LH) and human chorionic gonadotropin (hCG), Figure 8. Located on chromosome 2p21, the LHCGR gene spans approximately 70 kilobases and produces a receptor comprising 674 amino acids [61,62]. Structurally, the receptor includes three critical regions: a highly glycosylated extracellular domain for hormone binding, a seven-transmembrane α-helical region characteristic of GPCRs, and a cytoplasmic tail enriched with serine and threonine residues, which facilitates phosphorylation and downstream signaling [63].
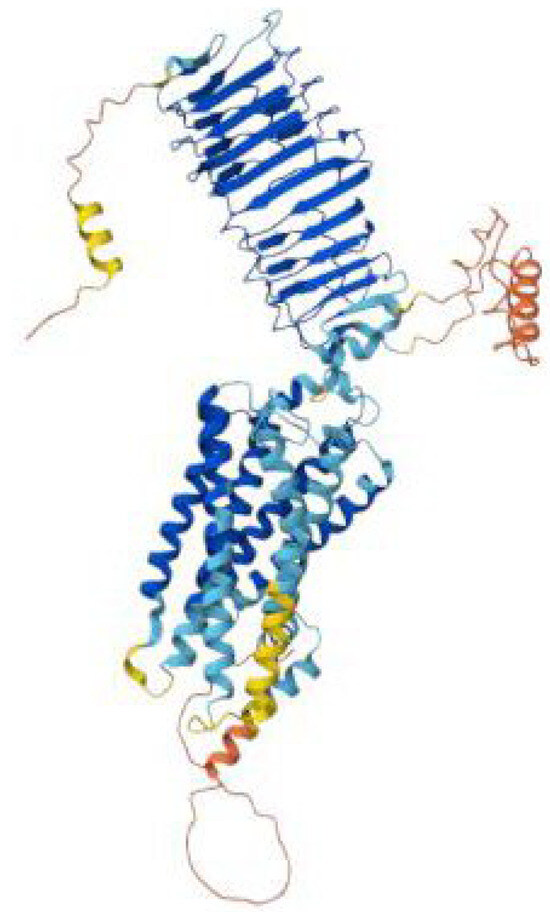
Figure 8.
AlphaFold model of the LHCGR (luteinizing hormone/choriogonadotropin receptor) protein colored, showing the extracellular leucine-rich repeat hormone-binding domain in blue, the seven-transmembrane helical domain in cyan–blue, and the flexible N- and C-terminal regions in yellow to red.
The activation of LHCGR by LH or hCG induces a conformational change that triggers the adenylate cyclase-cAMP signaling pathway, subsequently activating protein kinase A (PKA). This signaling cascade regulates steroidogenic enzyme activity, enabling the conversion of cholesterol into androgen and estrogen precursors essential for gonadal steroidogenesis [64]. In females, LHCGR controls ovarian follicle maturation, ovulation, and maintenance of the luteal phase. In males, it is expressed in Leydig cells, which support testosterone production and spermatogenesis [65]. During pregnancy, hCG binds to LHCGR to prolong luteal function, supporting early gestation. Extragonadal expression of LHCGR has been detected in tissues such as the uterus and breast, although its roles in these sites remain less well-defined [66]. LHCGR expression is regulated by estrogen and follicle-stimulating hormone (FSH). Upregulation enhances receptor sensitivity, whereas prolonged LH exposure induces desensitization via phosphorylation and clathrin-mediated endocytosis. Following internalization, the receptor is either recycled to the membrane or targeted for lysosomal degradation [67]. Pharmacological advances have enabled the development of selective LHCGR agonists and antagonists, offering therapeutic potential in fertility treatments and hormonal regulation [64].
Mutations in LHCGR result in a spectrum of reproductive disorders. Gain-of-function mutations lead to Gonadotropin-Independent Familial Sexual Precocity (GDFSP) by constitutively activating the receptor. Conversely, loss-of-function mutations are associated with Leydig Cell Agenesis and LH Resistance Syndromes, resulting in gonadal dysfunction and impaired sexual development [68]. These mutations highlight the receptor’s critical role in reproductive physiology and hormone-dependent processes.
In addition, we provide detailed analysis of LHCGR mutations, the associated clinical conditions, the affected receptor domains, and their molecular implications, further elucidating its indispensable role in reproductive health.
5.1. Mutations and Molecular Impact
There are 35 pathogenic/likely pathogenic mutations of the LHCGR gene available on ClinVar (Table 1). These mutations are associated with diverse phenotypes, primarily related to sexual development and gonadal function. They are classified based on the variant type, molecular consequences, and pathogenicity. Several types of LHCGR mutations have been reported to ClinVar, Table 8.
Table 8.
LHCGR gene mutations and their molecular effect.
Following data distribution in Table 8, missense variants are the most frequent, accounting for 65% of the total number of mutations. In addition, we provide statistics on their pathogenic status:
- Pathogenic: 22 mutations (65%)
- Likely Pathogenic: 8 mutations (24%)
- Pathogenic/Likely pathogenic: 4 mutations (11%) *
* Mutations that in different protein changes have different pathogenic status.
The predominance of pathogenic mutations highlights the necessity of fundamental understanding of the molecular implications of LHCGR gene mutations and their impact on reproductive function.
LHCGR mutations are distributed across key protein domains:
- Extracellular domain (ligand-binding): Mutations such as p.Leu457Arg and p.Ala568Val impair ligand recognition and binding, altering signal initiation [69,70].
- Transmembrane domain: Variants such as p.Val144Phe and p.Ser616Tyr compromise signal transduction or cause constitutive receptor activity [71,72].
- Intracellular domain: Mutations like p.Cys131Arg impair G protein interaction, blocking downstream signaling [73].
- Hinge region: Structural changes from mutations like p.Cys343Ser disrupt receptor stability and folding [74].
The location of the mutation on specific protein domains is shown in Figure 9. according to Mutation3D.
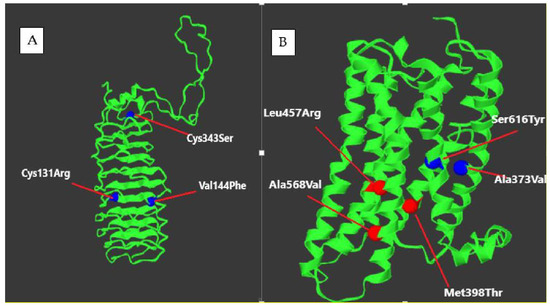
Figure 9.
Location of some LHCGR mutation in (A) extracellular domain (B) transmembrane domain.
In Table 9, we summarize the “key LHCGR mutations,” the associated clinical manifestations, the current ClinVar pathogenic status, mutations’ type, and molecular implications.
Table 9.
Key LHCGR gene mutations and their clinical impact.
5.2. Clinical Conditions and Management
LHCGR mutations are associated with several well-defined clinical conditions, with distinct molecular mechanisms and therapeutic approaches. Table 10 provides information on the distribution of LHCGR mutations per clinical condition and the most common key mutations, including their molecular implications.
Table 10.
Distribution of LHCGR mutations per clinical condition.
- o
- Gonadotropin-Independent Familial Sexual Precocity (GDFSP) Gain-of-function mutations in the LHCGR gene result in constitutive receptor activation, leading to autonomous testosterone production without necessity for LH stimulation. Clinically, this condition manifests as gonadotropin-independent familial sexual precocity (GDFSP), where affected boys present with early-onset puberty, accelerated growth, advanced bone age, and macroorchidism, despite prepubertal gonadotropin levels. Diagnosis is based on clinical features, biochemical findings (elevated testosterone with suppressed LH and FSH), and molecular confirmation of activating LHCGR mutations [75,77,79]. Management strategies include the use of GnRH analogs to suppress the hypothalamic-pituitary axis, anti-androgens to block androgen receptors, or aromatase inhibitors to reduce estrogen conversion, all aimed at mitigating the effects of premature androgen exposure and minimizing psychosocial consequences [75,79].
- o
- Leydig Cell Agenesis Conversely, loss-of-function mutations in the LHCGR gene lead to Leydig cell agenesis or hypoplasia, depending on the extent of receptor dysfunction. These mutations impair LH signaling and disrupt the development and function of Leydig cells, resulting in androgen deficiency. Clinically, the phenotype may range from ambiguous genitalia and pseudohermaphroditism in 46, XY individuals to complete gonadal dysgenesis and delayed or absent puberty. Laboratory findings often reveal low testosterone levels with elevated LH concentrations. Molecular diagnosis is confirmed by identifying biallelic inactivating mutations in LHCGR [76,78,80]. Management protocol includes testosterone replacement therapy for pubertal induction and maintenance, surgical correction of genital anomalies when necessary, and fertility counseling in adolescence or adulthood.
- o
- Luteinizing Hormone (LH) Resistance represents a partial loss-of-function spectrum, where receptor mutations reduce sensitivity to LH or impair surface receptor expression. Affected individuals may present with primary or secondary infertility, menstrual irregularities in females, or undermasculinized genitalia in males. Biochemical findings include high LH with normal or low sex steroid levels, and the condition can be confirmed via genetic testing [81]. Treatment involves hormone replacement therapy (HRT) in females to induce or maintain secondary sexual characteristics, while in males, individualized ART strategies may be employed depending on residual Leydig cell function.
- o
- Leydig Cell Hypoplasia is associated with severe receptor dysfunction, preventing normal Leydig cell development [76]. Individuals with 46,XY karyotype may show signs of undervirilization. Genetic confirmation and endocrine evaluation are essential. Management includes lifelong HRT, surgical correction, and fertility counseling.
- o
- Leydig Cell Adenoma with Male-Limited Precocious Puberty is associated with Somatic gain-of-function mutations in LHCGR. These mutations cause autonomous activation of the receptor within localized testicular tissue, leading to excessive testosterone production and precocious puberty in affected males. Patients may present with testicular enlargement, early secondary sexual characteristics, and suppressed gonadotropins [82]. Management is based on clinical presentation and may include observation, hormonal suppression, or surgical intervention in cases with significant mass effect or uncontrolled androgen excess.
- o
- Mosaic Trisomy 2 can affect the gonads, disrupting gamete production and leading to infertility or recurrent pregnancy loss [83]. Diagnosis is made through cytogenetic testing. Management includes fertility preservation and genetic counseling.
- o
- Pseudohermaphroditism is a disorder in which chromosomal and phenotypic sex do not align, often due to LHCGR-related hormonal disruptions [84]. Diagnosis requires a multidisciplinary workup including karyotyping, hormone profiling, and genetic sequencing. Treatment involves individualized endocrine, surgical, and psychological care.
The identification of pathogenic LHCGR mutations aids in distinguishing between gain- and loss-of-function conditions, enabling precision in managing early puberty, gonadal dysfunction, and infertility through hormonal modulation or ART protocols.
6. Analysis of ERs Gene Mutation: Detailed Overview and Clinical Implications
Estrogen receptors [ERs] are essential mediators of the biological effects of estrogen, particularly 17β-estradiol, throughout the body. They are classified into nuclear receptors [ERα and ERβ] and membrane-bound receptors, such as the G protein-coupled estrogen receptor [GPER] and ER-X. Upon estrogen binding, the nuclear receptors translocate to the nucleus, where they bind to estrogen response elements [EREs] on DNA and act as transcription factors, regulating gene expression. ERα and ERβ exhibit distinct tissue-specific distributions: ERα is primarily expressed in the breast tissue, the hypothalamus, and the endometrium, while ERβ is abundant in the ovaries, prostate, kidneys, lungs, and bone. These receptors can form homodimers or heterodimers, enabling complex cellular responses to estrogen [85].
ERs mediate both genomic and non-genomic effects. The genomic effects involve direct regulation of transcription through DNA binding, whereas non-genomic effects occur via rapid signaling cascades initiated at the cell membrane, activating pathways such as MAPK/ERK and PI3K/AKT. These mechanisms influence various physiological processes, including those in reproductive tissues, the brain, cardiovascular system, bones, and immune cells [86].
Estrogen receptors (ERs) play a pivotal role in cancer biology, notably in ER-positive breast cancers, where estrogen signaling promotes tumor growth. Therapeutic interventions, such as selective estrogen receptor modulators (SERMs) like tamoxifen, function by blocking estrogen receptors on breast cancer cells, thereby inhibiting estrogen from stimulating cancer cell proliferation [87]. ERα, in particular, is crucial for fat distribution and bone maintenance. Studies have shown that variations in the estrogen receptor alpha gene are related to differences in bone mineral density and fat distribution, underscoring its therapeutic potential in addressing metabolic and skeletal disorders [88]. Beyond cancer, ERs are implicated in other malignancies, including ovarian and prostate cancers, as well as in metabolic and skeletal disorders such as obesity and osteoporosis. Estrogen deficiency, especially post-menopause, is associated with increased adiposity and metabolic dysfunction, predisposing individuals to conditions like type 2 diabetes and cardiovascular diseases [89].
A comprehensive understanding of the diverse roles and mechanisms of ERs mutations is essential for developing targeted treatments across a range of estrogen-related conditions. This knowledge facilitates the design of therapies that can modulate ER activity, offering potential benefits in cancer treatment, metabolic regulation, and bone health.
6.1. ESR1 [Estrogen Receptor 1]
The ESR1 [estrogen receptor 1] gene encodes the estrogen receptor alpha, a nuclear receptor that mediates the effects estrogen, Figure 10. This receptor regulates key physiological processes, including reproductive development, bone density maintenance, cardiovascular health, and cellular proliferation. Mutations in the ESR1 gene disrupt these key functions, leading to a spectrum of clinical disorders such as Estrogen Resistance Syndrome and temperature-sensitive estrogen receptor variants. These conditions arise due to impaired estrogen binding, disrupted signal transduction, or receptor instability, significantly impacting patient outcomes.
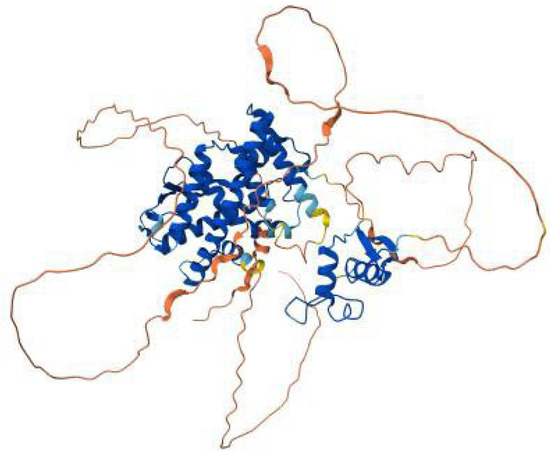
Figure 10.
AlphaFold model of the ESR1 (estrogen receptor α) protein, showing the well-structured ligand-binding and DNA-binding domains in blue, the connecting α-helices in cyan–blue, and the intrinsically disordered N- and C-terminal regions in yellow to red.
6.1.1. Mutations and Molecular Impact
The ClinVar database tracks 10 distinct mutations of the ESR1 gene. These mutations impact the receptor’s ability to bind estrogen or mediate downstream signaling, leading to a variety of estrogen-related dysfunctions.
Three types of ESR1 mutations, with diverse molecular implications, have been reported so far, as seen in Table 11.
Table 11.
ESR1 mutations’ types and molecular effects.
Missense variants are the most prevalent, comprising 80% of the total number of mutations, which highlights the sensitivity of the ESR1 protein to even subtle amino acid changes.
The following numbers are based on pathogenic classification:
- Pathogenic: seven mutations (70%)
- Likely pathogenic: one mutation (10%)
- Pathogenic/likely pathogenic: one mutation (10%)
The ESR1 protein consists of three structural domains, crucial for its function and activity. The identified mutations impact the following domains, and Mutation3D visualization (Figure 11) illustrates their domain-specific distribution:
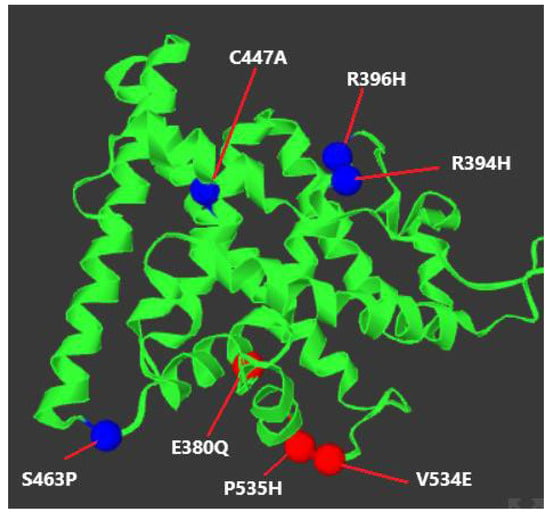
Figure 11.
Locations of the key ESR1 mutations (visualized in Mutation3D).
- DNA-Binding Domain [DBD]: Mutations, such as R394H and R396H (Table 12), impair the receptor’s ability to interact with DNA at estrogen response elements [EREs], leading to a failure in gene transcription regulation.
Table 12. List of key ESR1 mutations and molecular implications.
- Ligand-Binding Domain [LBD]: This domain is essential for binding estrogen and initiating receptor activation. Mutations, such as Q375H and Q377H (Table 12), reduce the receptor’s estrogen-binding affinity, rendering it inactive.
- Hinge Region: The hinge region facilitates receptor flexibility and its interaction with co-regulatory proteins. Mutations like C447A (Table 12) may destabilize the receptor, particularly under temperature fluctuations, contributing to temperature-sensitive estrogen resistance.
The ESR1 mutations lead to impaired estrogen signaling via two major mechanisms:
- Loss-of-Function Mutations [e.g., R157*, R394H]: These mutations impair receptor binding to estrogen or disrupt downstream signal transduction pathways. The functional absence of a receptor working properly leads to conditions such as estrogen resistance syndrome, with symptoms including delayed puberty, infertility, and poor bone health.
- Structural Instability Mutations [e.g., C447A]: Mutations which destabilize receptor conformation, rendering it temperature-sensitive or prone to functional loss under specific physiological conditions. These mechanisms contribute to variable clinical manifestations depending on environmental factors.
6.1.2. Clinical Conditions and Management
- o
- Estrogen Resistance Syndrome (ERS) is caused by loss-of-function ESR1 mutations that impair receptor function. Mutations, such as R394H, Q375H, and R157*, reduce ligand binding or disrupt downstream signaling. Affected individuals often exhibit delayed puberty, infertility, and metabolic disturbances despite high serum estrogen levels [90,91,92]. Diagnosis involves hormone testing and confirmation via ESR1 gene sequencing. Management includes high-dose estrogen therapy to overcome receptor insensitivity and hormone replacement therapy (HRT) to mitigate long-term consequences such as osteoporosis and infertility. Genetic testing can aid in the identification of atypical cases and guide personalized treatment.
- o
- Breast Neoplasm is associated with somatic ESR1 mutations, particularly within the ligand-binding domain. Mutations like L536R, V534E, and E380Q confer ligand-independent activation of ERα, contributing to estrogen receptor-positive breast cancer and resistance to endocrine therapies such as aromatase inhibitors [93,94,95]. Diagnosis is achieved through molecular profiling of tumor samples. Treatment strategies involve the use of selective estrogen receptor degraders (SERDs), estrogen receptor modulators, or investigational drugs targeting mutant ERα. These mutations may also impact reproductive function by impairing folliculogenesis, endometrial development, and ovulation—factors essential for fertility.
- o
- Temperature-Sensitive Estrogen Receptor Mutants involve structural instability under physiological conditions. Mutations, such as C447A in the LBD, render the receptor thermosensitive, impairing estrogen binding and transcriptional activity at normal body temperatures [96]. Clinical features may include estrogen resistance and infertility. Diagnosis requires molecular confirmation, and the treatment focuses on customized hormone therapy and potential use of ligand stabilizers to restore receptor function.
Characterizing ESR1 mutations supports targeted endocrine therapy in cancer and estrogen resistance syndromes, with implications for bone health, puberty induction, and fertility preservation.
6.2. ESR2 [Estrogen Receptor 2]
The ESR2 [estrogen receptor beta] (Figure 12) gene encodes a receptor that mediates the effects of estrogen in a variety of tissues, including reproductive organs, the cardiovascular system, and the central nervous system. Mutations in this gene are linked to conditions such as ovarian dysgenesis, infertility, and other estrogen-related dysfunctions. There is one available pathogenic, missense ESR2 mutation available on ClinVar, shown in Table 13.
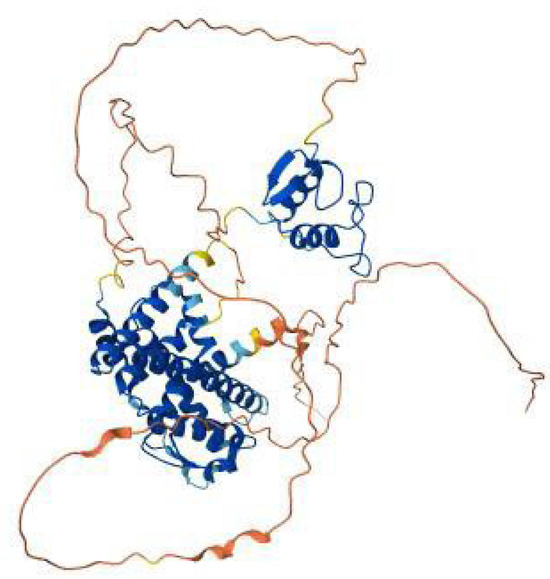
Figure 12.
AlphaFold model of the ESR2 (estrogen receptor β) protein colored, showing the ligand-binding and DNA-binding domains in blue, the connecting α-helices in cyan–blue, and the flexible N- and C-terminal regions in yellow to red.
Table 13.
ESR2 SNV associated clinical condition.
This mutation involves the substitution of lysine [K] with arginine [R] at position 314, altering protein’s structure and disrupting estrogen signaling, Figure 13. K314R is associated with Ovarian Dysgenesis 8, a condition marked by the failure of ovarian development, primary amenorrhea, infertility, and low estrogen levels. The mutation is likely to impair the receptor’s ability to bind estrogen or interact with co-regulatory proteins, leading to insufficient estrogenic signaling [97].
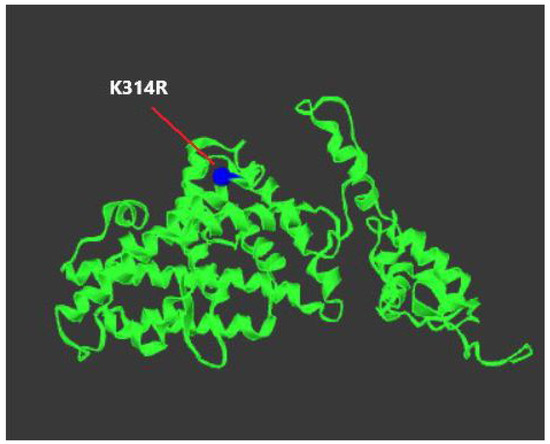
Figure 13.
Location of K314R mutation in ESR2 (visualized in Mutation3D).
Genetic testing for ESR2 mutations should be considered in individuals presenting with suspected ovarian dysgenesis or other estrogen-related disorders, as they can significantly impact estrogen signaling and reproductive health. In particular, K314R mutation is associated with impaired receptor function. Functional assays can provide further insight into its effect on estrogen binding and interaction with co-regulatory proteins, thereby confirming mutation’s pathogenicity.
Management of affected individuals often requires a multi-faceted approach. Hormone Replacement Therapy (HRT) is commonly prescribed to address estrogen deficiency, which helps to facilitate the development of secondary sexual characteristics, alleviate symptoms of estrogen insufficiency, and mitigate long-term risks such as osteoporosis and cardiovascular issues. Additionally, fertility preservation strategies may be necessary, particularly for individuals with impaired ovarian function or infertility. Assisted reproductive technologies (ART), including in vitro fertilization (IVF) or egg freezing, could be considered to support reproductive goals in patients facing fertility challenges due to estrogen signaling defects.
Although rare, ESR2 mutations, such as K314R, underscore the importance of receptor testing in unexplained ovarian failure, guiding HRT and fertility intervention decisions.
7. Discussion
Infertility remains a significant challenge in reproductive medicine [98], and the genetic basis underlying infertility has become a key area of focus in improving treatment outcomes [99]. Mutations in the hormone receptors that mediate critical reproductive processes, including follicle-stimulating hormone receptor [FSHR], luteinizing hormone receptor [LHR], androgen receptor [AR], estrogen receptor [ER], and progesterone receptor [PGR], can greatly influence the success of infertility therapies.
The complex interaction between FSHR, LHR, AR, ER, and PGR mutations is crucial in determining ART success, as these receptors do not function in isolation but rather in an integrated hormonal network. While previous sections have described individual receptor mutations, their combined impact shapes reproductive outcomes. FSHR and LHR mutations disrupt ovarian follicular development, with FSHR primarily regulating early follicular growth and LHR being crucial for ovulation induction [100,101]. Mutations in FSHR that lower follicular sensitivity to FSH can lead to suboptimal LH response, as the FSH prime effect on LHR expression is reduced. Conversely, LHR mutations that impair hCG-mediated signaling can affect corpus luteum function, altering progesterone levels and impacting implantation success. AR mutations influence ovarian steroidogenesis by modulating FSHR and LHR sensitivity to gonadotropins [57]. Reduced AR function leads to altered testosterone conversion to estrogen, affecting FSHR-driven follicular recruitment [57]. In males, AR mutations disrupt LH-stimulated Leydig cell function, leading to testosterone deficiency, which can impair spermatogenesis and ART outcomes [102]. Estrogen receptor (ER) and progesterone receptor (PGR) mutations impact endometrial receptivity, crucial for implantation success in ART cycles. FSHR and LHR mutations that alter estrogen and progesterone secretion indirectly affect ER and PGR activity, leading to failed implantation despite successful fertilization. Recent studies highlight the role of ER and PGR mutations in conditions such as adenomyosis and endometriosis, which further contribute to implantation failure and ART resistance [103]. Cross-talk between ER, AR, and FSHR is crucial in coordinating follicular growth, ovulation, and luteal phase support, illustrating the need for a comprehensive approach in ART treatment strategies. Understanding the interplay between hormone receptor mutations enables personalized ART protocols. For patients with FSHR mutations, adjusting FSH dosage while considering LHR function can optimize ovarian response. Patients with AR mutations affecting androgen-mediated FSHR sensitization may benefit from testosterone priming before ovarian stimulation. PGR mutations that impair progesterone responsiveness might require enhanced luteal phase support to improve implantation rates in ART cycles.
Future studies should focus on integrating multi-receptor genetic profiling into ART workflows and validating mutation-specific treatment responses to develop standardized, mutation-guided fertility protocols.
8. Conclusions
The complex interplay between hormone receptors’ variations is a crucial determinant of infertility and ART success. Understanding how mutations in FSHR, LHCGR, AR, ESR1, and PGR disrupt hormonal signaling pathways enables clinicians to assess treatment resistance more accurately and develop optimized therapeutic strategies. These mutations exert diverse structural and functional effects, resulting in distinct clinical phenotypes that require personalized ART protocols. Genetic screening plays a crucial role in identifying patients who may benefit from tailored interventions, such as modified gonadotropin stimulation, testosterone priming, or enhanced luteal support.
Further research into receptor-specific therapies and their integration into ART workflows will be essential to improve reproductive outcomes. Continued exploration of the molecular mechanisms underlying hormone receptor dysfunctions will advance the application of precision medicine in reproductive health and refine clinical approaches to infertility management.
Author Contributions
Conceptualization, D.H. and D.S.; methodology, D.H. and D.S.; software, D.H.; validation, D.S.; formal analysis, D.H.; investigation, D.H.; resources, D.H.; data curation, D.H.; writing—original draft preparation, D.H. and D.S.; writing—review and editing, D.H. and D.S.; visualization, D.H.; supervision, D.S.; project administration, D.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, [D.H.], upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Huhtaniemi, I.T.; Themmen, A.P.N. Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine 2005, 26, 207–217. [Google Scholar] [CrossRef]
- Simoni, E.M.; Nieschlag, J. Gromoll, Isoforms and single nucleotide polymorphisms of the FSH receptor gene: Implications for human reproduction. Hum. Reprod. Update 2002, 8, 413–421. [Google Scholar] [CrossRef]
- Polyzos, N.P.; Neves, A.R.; Drakopoulos, P.; Spits, C.; Mercadal, B.A.; Garcia, S.; Ma, P.Q.M.; Le, L.H.; Ho, M.T.; Mertens, J.; et al. The effect of polymorphisms in FSHR and FSHB genes on ovarian response: A prospective multicenter multinational study in Europe and Asia. Hum. Reprod. 2021, 36, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Themmen, A.P.N.; Huhtaniemi, I.T. Mutations of Gonadotropins and Gonadotropin Receptors: Elucidating the Physiology and Pathophysiology of Pituitary-Gonadal Function. Endocr. Rev. 2000, 21, 551–583. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutatio ns database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef]
- Lindgren, I.; Bååth, M.; Uvebrant, K.; Dejmek, A.; Kjaer, L.; Henic, E.; Bungum, M.; Bungum, L.; Cilio, C.; Leijonhufvud, I.; et al. Combined assessment of polymorphisms in the LHCGR and FSHR genes predict chance of pregnancy after in vitro fertilization. Hum. Reprod. 2016, 31, 672–683. [Google Scholar] [CrossRef]
- Agwuegbo, U.T.; Colley, E.; Albert, A.P.; Butnev, V.Y.; Bousfield, G.R.; Jonas, K.C. Differential FSH Glycosylation Modulates FSHR Oligomerization and Subsequent cAMP Signaling. Front. Endocrinol. 2021, 12, 765727. [Google Scholar] [CrossRef]
- Alviggi, C.; Longobardi, S.; Papaleo, E.; Santi, D.; Alfano, S.; Vanni, V.S.; Campitiello, M.R.; De Rosa, P.; Strina, I.; Huhtaniemi, I.; et al. Genetic Variants of Gonadotropins and Their Receptors Could Influence Controlled Ovarian Stimulation: IVF Data from a Prospective Multicenter Study. Genes 2023, 14, 1269. [Google Scholar] [CrossRef]
- Meyer, M.J.; Lapcevic, R.; Romero, A.E.; Yoon, M.; Das, J.; Beltrán, J.F.; Mort, M.; Stenson, P.D.; Cooper, D.N.; Paccanaro, A.; et al. mutation3D: Cancer Gene Prediction Through Atomic Clustering of Coding Variants in the Structural Proteome. Hum. Mutat. 2016, 37, 447–456. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Velankar, S. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.R.; Wang, Y.; Lu, N.; Matzuk, M.M. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 1997, 15, 201–204. [Google Scholar] [CrossRef]
- Heckert, L.L.; Griswold, M.D. The Expression of the Follicle-stimulating Hormone Receptor in Spermatogenesis. Recent Prog. Horm. Res. 2002, 57, 129–148. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ulloa-Aguirre, A.; Zariñán, T.; Jardón-Valadez, E.; Gutiérrez-Sagal, R.; Dias, J.A. Structure-Function Relationships of the Follicle-Stimulating Hormone Receptor. Front. Endocrinol. 2018, 9, 707. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Menon, K.; Menon, B. Structure, function and regulation of gonadotropin receptors—A perspective. Mol. Cell. Endocrinol. 2012, 356, 88–97. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haldar, S.; Agrawal, H.; Saha, S.; Straughn, A.R.; Roy, P.; Kakar, S.S. Overview of follicle stimulating hormone and its receptors in reproduction and in stem cells and cancer stem cells. Int. J. Biol. Sci. 2022, 18, 675–692. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Das, N.; Kumar, T.R. Molecular regulation of follicle-stimulating hormone synthesis, secretion and action. J. Mol. Endocrinol. 2018, 60, R131–R155. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ponikwicka-Tyszko, D.; Chrusciel, M.; Stelmaszewska, J.; Bernaczyk, P.; Sztachelska, M.; Sidorkiewicz, I.; Doroszko, M.; Tomaszewski, J.; Tapanainen, J.S.; Huhtaniemi, I.; et al. Functional Expression of FSH Receptor in Endometriotic Lesions. J. Clin. Endocrinol. Metab. 2016, 101, 2905–2914. [Google Scholar] [CrossRef]
- Yoo, S.; Yoon, J.Y.; Keum, C.; Cheon, C.K. The first case of novel variants of the FSHR mutation causing primary amenorrhea in 2 siblings in Korea. Ann. Pediatr. Endocrinol. Metab. 2023, 28, 54–60. [Google Scholar] [CrossRef]
- Chen, X.; Chen, L.; Wang, Y.; Shu, C.; Zhou, Y.; Wu, R.; Jin, B.; Yang, L.; Sun, J.; Qi, M.; et al. Identification and characterization of novel compound heterozygous variants in FSHR causing primary ovarian insufficiency with resistant ovary syndrome. Front. Endocrinol. 2023, 13, 1013894. [Google Scholar] [CrossRef]
- Vasseur, C.; Rodien, P.; Beau, I.; Desroches, A.; Gérard, C.; de Poncheville, L.; Chaplot, S.; Savagner, F.; Croué, A.; Mathieu, E.; et al. A Chorionic Gonadotropin–Sensitive Mutation in the Follicle-Stimulating Hormone Receptor as a Cause of Familial Gestational Spontaneous Ovarian Hyperstimulation Syndrome. N. Engl. J. Med. 2003, 349, 753–759. [Google Scholar] [CrossRef] [PubMed]
- De Leener, A.; Caltabiano, G.; Erkan, S.; Idil, M.; Vassart, G.; Pardo, L.; Costagliola, S. Identification of the first germline mutation in the extracellular domain of the follitropin receptor responsible for spontaneous ovarian hyperstimulation syndrome. Hum. Mutat. 2008, 29, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Olatunbosun, O.A.; Gilliland, B.; Brydon, L.A.; Chizen, D.R.; Pierson, R.A. Spontaneous ovarian hyper- stimulation syndrome in four consecutive pregnancies. Clin. Exp. Obstet. Gynecol. 1996, 23, 127–132. [Google Scholar] [PubMed]
- Smits, G.; Olatunbosun, O.; Delbaere, A.; Pierson, R.; Vassart, G.; Costagliola, S. Ovarian Hyperstimulation Syndrome Due to a Mutation in the Follicle-Stimulating Hormone Receptor. N. Engl. J. Med. 2003, 349, 760–766. [Google Scholar] [CrossRef]
- Gromoll, J.; Simoni, M. Follicle-stimulating-hormone receptor and twinning. Lancet 2001, 357, 230–232. [Google Scholar] [CrossRef]
- De Leener, A.; Montanelli, L.; Van Durme, J.; Chae, H.; Smits, G.; Vassart, G.; Costagliola, S. Presence and Absence of Follicle-Stimulating Hormone Receptor Mutations Provide Some Insights into Spontaneous Ovarian Hyperstimulation Syndrome Physiopathology. J. Clin. Endocrinol. Metab. 2006, 91, 555–562. [Google Scholar] [CrossRef]
- Aittomäki, K.; Lucena, J.D.; Pakarinen, P.; Sistonen, P.; Tapanainen, J.; Gromoll, J.; Kaskikari, R.; Sankila, E.-M.; Lehväslaiho, H.; Engel, A.R.; et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 1995, 82, 959–968. [Google Scholar] [CrossRef]
- Doherty, E.; Pakarinen, P.; Tiitinen, A.; Kiilavuori, A.; Huhtaniemi, I.; Forrest, S.; Aittomäki, K. A Novel Mutation in the FSH Receptor Inhibiting Signal Transduction and Causing Primary Ovarian Failure. J. Clin. Endocrinol. Metab. 2002, 87, 1151–1155. [Google Scholar] [CrossRef]
- Meduri, G.; Touraine, P.; Beau, I.; Lahuna, O.; Desroches, A.; Vacher-Lavenu, M.C.; Kuttenn, F.; Misrahi, M. Delayed Puberty and Primary Amenorrhea Associated with a Novel Mutation of the Human Follicle-Stimulating Hormone Receptor: Clinical, Histological, and Molecular Studies. J. Clin. Endocrinol. Metab. 2003, 88, 3491–3498. [Google Scholar] [CrossRef]
- Allen, L.A.; Achermann, J.C.; Pakarinen, P.; Kotlar, T.J.; Huhtaniemi, I.T.; Jameson, J.L.; Cheetham, T.D.; Ball, S.G. A novel loss of function mutation in exon 10 of the FSH receptor gene causing hypergonadotrophic hypogonadism: Clinical and molecular characteristics. Hum. Reprod. 2003, 18, 251–256. [Google Scholar] [CrossRef]
- Bhartiya, D.; Patel, H. An overview of FSH-FSHR biology and explaining the existing conundrums. J. Ovarian Res. 2021, 14, 114. [Google Scholar] [CrossRef]
- Beck-Peccoz, P.; Persani, L. Premature ovarian failure. Orphanet J. Rare Dis. 2006, 1, 9. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. The Roles of Androgen Receptors and Androgen-Binding Proteins in Nongenomic Androgen Actions. Mol. Endocrinol. 2002, 16, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- García-Sáenz, M.; Ibarra-Salce, R.; Pozos-Varela, F.J.; Mena-Ureta, T.S.; Flores-Villagómez, S.; Santana-Mata, M.; Santos-Aguilar, R.G.D.L.; Uribe-Cortés, D.; Ferreira-Hermosillo, A. Understanding Progestins: From Basics to Clinical Applicability. J. Clin. Med. 2023, 12, 3388. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, M.; Woodruff, T.K. Involvement of androgens in ovarian health and disease. Mol. Hum. Reprod. 2013, 19, 828–837. [Google Scholar] [CrossRef]
- Astapova, O.; Minor, B.M.N.; Hammes, S.R. Physiological and Pathological Androgen Actions in the Ovary. Endocrinology 2019, 160, 1166–1174. [Google Scholar] [CrossRef]
- Chen, J.-F.; Lin, P.-W.; Tsai, Y.-R.; Yang, Y.-C.; Kang, H.-Y. Androgens and Androgen Receptor Actions on Bone Health and Disease: From Androgen Deficiency to Androgen Therapy. Cells 2019, 8, 1318. [Google Scholar] [CrossRef]
- Yavuz, S.; Abraham, T.E.; Houtsmuller, A.B.; van Royen, M.E. Phase Separation Mediated Sub-Nuclear Compartmentalization of Androgen Receptors. Cells 2024, 13, 1693. [Google Scholar] [CrossRef]
- Yin, L.; Lu, L.; Lin, X.; Wang, X. Crucial role of androgen receptor in resistance and endurance trainings-induced muscle hypertrophy through IGF-1/IGF-1R- PI3K/Akt- mTOR pathway. Nutr. Metab. 2020, 17, 26. [Google Scholar] [CrossRef]
- Brown, C.; Goss, S.; Lubahn, D.; Joseph; Wilson, E.; French, F.; Willard, H. Androgen receptor locus on the human X chromosome: Regional localization to Xq11-12 and description of a DNA polymorphism. Am. J. Hum. Genet. 1989, 44, 264–269. [Google Scholar]
- Listyasari, N.A.; Juniarto, A.Z.; Robevska, G.; Ayers, K.L.; Sinclair, A.H.; Faradz, S.M.H. Analysis of the androgen receptor (AR) gene in a cohort of Indonesian undermasculinized 46, XY DSD patients. Egypt. J. Med. Hum. Genet. 2021, 22, 14. [Google Scholar] [CrossRef]
- Sartor, O.; Zheng, Q.; Eastham, J.A. Androgen receptor gene CAG repeat length varies in a race-specific fashion in men without prostate cancer. Urology 1999, 53, 378–380. [Google Scholar] [CrossRef]
- Bennett, C.L.; Price, D.K.; Kim, S.; Liu, D.; Jovanovic, B.D.; Nathan, D.; Johnson, M.E.; Montgomery, J.S.; Cude, K.; Brockbank, J.C.; et al. Racial Variation in CAG Repeat Lengths Within the Androgen Receptor Gene Among Prostate Cancer Patients of Lower Socioeconomic Status. J. Clin. Oncol. 2002, 20, 3599–3604. [Google Scholar] [CrossRef]
- De Bellis, A.; Quigley, C.A.; Cariello, N.F.; el-Awady, M.K.; Sar, M.; Lane, M.V.; Wilson, E.M.; French, F.S. Single base mutations in the human androgen receptor gene causing complete androgen insensitivity: Rapid detection by a modified denaturing gradient gel electrophoresis technique. Mol. Endocrinol. 1992, 6, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Imasaki, K.; Okabe, T.; Murakami, H.; Tanaka, Y.; Haji, M.; Takayanagi, R.; Nawata, H. Androgen insensitivity syndrome due to new mutations in the DNA-binding domain of the androgen receptor. Mol. Cell. Endocrinol. 1996, 120, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Teng, Y.; Chen, X.; Liang, N.; Li, Z.; Liang, D.; Wu, L. Six novel Mutation analysis of the androgen receptor gene in 17 Chinese patients with androgen insensitivity syndrome. Clin. Chim. Acta 2020, 506, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Philibert, P.; Audran, F.; Pienkowski, C.; Morange, I.; Kohler, B.; Flori, E.; Heinrich, C.; Dacou-Voutetakis, C.; Joseph, M.-G.; Guedj, A.-M.; et al. Complete androgen insensitivity syndrome is frequently due to premature stop codons in exon 1 of the androgen receptor gene: An international collaborative report of 13 new mutations. Fertil. Steril. 2010, 94, 472–476. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, C.-H. A novel arg616Cys mutation in the DNA-binding domain of complete androgen insensitivity syndrome in a Chinese family. Chin. Med. J. 2013, 126, 4192–4193. [Google Scholar] [CrossRef]
- Taplin, M.-E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef]
- Lund, A.; Udd, B.; Juvonen, V.; Andersen, P.M.; Cederquist, K.; Davis, M.; Gellera, C.; Kölmel, C.; Ronnevi, L.-O.; Sperfeld, A.-D.; et al. Multiple founder effects in spinal and bulbar muscular atrophy (SBMA, Kennedy disease) around the world. Eur. J. Hum. Genet. 2001, 9, 431–436. [Google Scholar] [CrossRef]
- La Spada, A. Spinal and Bulbar Muscular Atrophy. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1999. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1333/ (accessed on 22 December 2024).
- Lillepea, K.; Juchnewitsch, A.-G.; Kasak, L.; Valkna, A.; Dutta, A.; Pomm, K.; Poolamets, O.; Nagirnaja, L.; Tamp, E.; Mahyari, E.; et al. Toward clinical exomes in diagnostics and management of male infertility. Am. J. Hum. Genet. 2024, 111, 877–895. [Google Scholar] [CrossRef]
- Ledig, S.; Jakubiczka, S.; Neulen, J.; Aulepp, U.; Burck-Lehmann, U.; Mohnike, K.; Thiele, H.; Zierler, H.; Brewer, C.; Wieacker, P. Novel and recurrent mutations in patients with androgen insensitivity syndromes. Horm. Res. Paediatr. 2005, 63, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Oakes, M.B.; Eyvazzadeh, A.D.; Quint, E.; Smith, Y.R. Complete Androgen Insensitivity Syndrome—A Review. J. Pediatr. Adolesc. Gynecol. 2008, 21, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Fan, L.; Wang, Y.; Li, L.; Ren, X.; Sui, S.; Song, Y.; Cheng, M.; Cao, B.; Gong, C. Analysis of genetic and clinical characteristics of androgen insensitivity syndrome: A cohort study including 12 families. Eur. J. Endocrinol. 2024, 191, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P.; Nirmalananthan, N.; Masset, L.; Skorupinska, I.; Collins, T.; Cortese, A.; Pemble, S.; Malaspina, A.; Fisher, E.M.; Greensmith, L.; et al. Correlation of clinical and molecular features in spinal bulbar muscular atrophy. Neurology 2014, 82, 2077–2084. [Google Scholar] [CrossRef]
- Yong, E.; Loy, C.; Sim, K. Androgen receptor gene and male infertility. Hum. Reprod. Updat. 2003, 9, 1–7. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Klein, D.A.; Emerick, J.E.; Sylvester, J.E.; Vogt, K.S. Disorders of Puberty: An Approach to Diagnosis and Management. Am. Fam. Physician 2017, 96, 590–599. [Google Scholar]
- Batista, R.L.; Costa, E.M.F.; Rodrigues, A.d.S.; Gomes, N.L.; Faria, J.A.; Nishi, M.Y.; Arnhold, I.J.P.; Domenice, S.; de Mendonca, B.B. Androgen insensitivity syndrome: A review. Arch. Endocrinol. Metab. 2018, 62, 227–235. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ascoli, M.; Fanelli, F.; Segaloff, D.L. The Lutropin/Choriogonadotropin Receptor, A 2002 Perspective. Endocr. Rev. 2002, 23, 141–174. [Google Scholar] [CrossRef]
- Richards, J.S. New signaling pathways for hormones and cyclic adenosine 3′,5′-monophosphate action in endocrine cells. Mol. Endocrinol. 2001, 15, 209–218. [Google Scholar] [CrossRef]
- Dufau, M.L. The luteinizing hormone receptor. Annu. Rev. Physiol. 1998, 60, 461–496. [Google Scholar] [CrossRef]
- Shpakov, A.O. Hormonal and Allosteric Regulation of the Luteinizing Hormone/Chorionic Gonadotropin Receptor. Front. Biosci. 2024, 29, 313. [Google Scholar] [CrossRef]
- Oduwole, O.O.; Huhtaniemi, I.T.; Misrahi, M. The roles of luteinizing hormone, follicle-stimulating hormone and testosterone in spermatogenesis and folliculogenesis revisited. Int. J. Mol. Sci. 2021, 22, 12735. [Google Scholar] [CrossRef]
- Gridelet, V.; Perrier d’Hauterive, S.; Polese, B.; Foidart, J.M.; Nisolle, M.; Geenen, V. Human chorionic gonadotrophin: New pleiotropic functions for an “old” hormone during pregnancy. Front. Immunol. 2020, 11, 343. [Google Scholar] [CrossRef]
- Menon, B.; Gulappa, T.; Menon, K. Molecular regulation of LHCGR expression by miR-122 during follicle growth in the rat ovary. Mol. Cell. Endocrinol. 2017, 442, 81–89. [Google Scholar] [CrossRef]
- Huhtaniemi, I.; Rivero-Müller, A. Mutations and polymorphisms, and their functional consequences, in gonadotropin and gonadotropin receptor genes. In The Ovary; Academic Press: Cambridge, MA, USA, 2019; pp. 127–148. [Google Scholar]
- Latronico, A.C.; Abell, A.N.; Arnhold, I.J.P.; Liu, X.; Lins, T.S.S.; Brito, V.N.; Billerbeck, A.E.; Segaloff, D.L.; Mendonca, B.B. A unique constitutively activating mutation in third transmembrane helix of luteinizing hormone receptor causes sporadic male gonadotropin-independent precocious puberty. J. Clin. Endocrinol. Metab. 1998, 83, 2435–2440. [Google Scholar] [CrossRef]
- Latronico, A.C.; Shinozaki, H.; Guerra, G., Jr.; Pereira, M.A.A.; Marini, S.H.V.L.; Baptista, M.T.M.; Arnhold, I.J.P.; Fanelli, F.; Mendonca, B.B.; Segaloff, D.L. Gonadotropin-Independent Precocious Puberty Due to Luteinizing Hormone Receptor Mutations in Brazilian Boys: A Novel Constitutively Activating Mutation in the First Transmembrane Helix 1. J. Clin. Endocrinol. Metab. 2000, 85, 4799–4805. [Google Scholar] [CrossRef][Green Version]
- Richter-Unruh, A.; Verhoef-Post, M.; Malak, S.; Homoki, J.; Hauffa, B.P.; Themmen, A.P.N. Leydig Cell Hypoplasia: Absent Luteinizing Hormone Receptor Cell Surface Expression Caused by a Novel Homozygous Mutation in the Extracellular Domain. J. Clin. Endocrinol. Metab. 2004, 89, 5161–5167. [Google Scholar] [CrossRef][Green Version]
- Vezzoli, V.; Duminuco, P.; Vottero, A.; Kleinau, G.; Schülein, R.; Minari, R.; Bassi, I.; Bernasconi, S.; Persani, L.; Bonomi, M. A new variant in signal peptide of the human luteinizing hormone receptor (LHCGR) affects receptor biogenesis causing leydig cell hypoplasia. Hum. Mol. Genet. 2015, 24, 6003–6012. [Google Scholar] [CrossRef]
- Newton, C.L.; Anderson, R.C.; Katz, A.A.; Millar, R.P. Loss-of-Function Mutations in the Human Luteinizing Hormone Receptor Predominantly Cause Intracellular Retention. Endocrinology 2016, 157, 4364–4377. [Google Scholar] [CrossRef]
- Martens, J.W.M.; Lumbroso, S.; Verhoef-Post, M.; Georget, V.; Richter-Unruh, A.; Szarras-Czapnik, M.; Romer, T.E.; Brunner, H.G.; Themmen, A.P.N.; Sultan, C. Mutant Luteinizing Hormone Receptors in a Compound Heterozygous Patient with Complete Leydig Cell Hypoplasia: Abnormal Processing Causes Signaling Deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 2506–2513. [Google Scholar] [CrossRef]
- Gromoll, J.; Partsch, C.-J.; Simoni, M.; Nordhoff, V.; Sippell, W.G.; Nieschlag, E.; Saxena, B.B. A mutation in the first transmembrane domain of the lutropin receptor causes male precocious puberty. J. Clin. Endocrinol. Metab. 1998, 83, 476–480. [Google Scholar] [CrossRef][Green Version]
- Laue, L.L.; Wu, S.M.; Kudo, M.; Bourdony, C.J.; Cutler, G.B., Jr.; Hsueh, A.J.; Chan, W.Y. Compound heterozygous mutations of the luteinizing hormone receptor gene in Leydig cell hypoplasia. Mol. Endocrinol. 1996, 10, 987–997. [Google Scholar]
- Evans, B.A.; Bowen, D.J.; Smith, P.J.; Clayton, P.E.; Gregory, J.W. A new point mutation in the luteinising hormone receptor gene in familial and sporadic male limited precocious puberty: Genotype does not always correlate with phenotype. J. Med. Genet. 1996, 33, 143–147. [Google Scholar] [CrossRef][Green Version]
- Hassan, H.A.; Essawi, M.L.; Mekkawy, M.K.; Mazen, I. Novel mutations of the LHCGR gene in two families with 46,XY DSD causing Leydig cell hypoplasia I. Hormones 2020, 19, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, S.M.; Grumbach, M.M.; Kaplan, S.L. Gonadotropin-Independent Familial Sexual Precocity with Premature Leydig and Germinal Cell Maturation (Familial Testotoxicosis): Effects of a Potent Luteinizing Hormone-Releasing Factor Agonist and Medroxyprogesterone Acetate Therapy in Four Cases *. J. Clin. Endocrinol. Metab. 1983, 57, 571–579. [Google Scholar] [CrossRef]
- Berthezene, F.; Forest, M.G.; Grimaud, J.A.; Claustrat, B.; Mornex, R. LEYDIG-CELL AGENESIS. A CAUSE OF MALE PSEUDOHERMAPHRODITISM. Obstet. Gynecol. Surv. 1977, 32, 167–169. [Google Scholar] [CrossRef]
- Latronico, A.C.; Anasti, J.; Arnhold, I.J.; Rapaport, R.; Mendonca, B.B.; Bloise, W.; Castro, M.; Tsigos, C.; Chrousos, G.P. Testicular and Ovarian Resistance to Luteinizing Hormone Caused by Inactivating Mutations of the Luteinizing Hormone–Receptor Gene. N. Engl. J. Med. 1996, 334, 507–512. [Google Scholar] [CrossRef]
- Yano, K.; Kohn, L.D.; Saji, M.; Kataoka, N.; Okuno, A.; Cutler, J.G.B. A Case of Male-Limited Precocious Puberty Caused by a Point Mutation in the Second Transmembrane Domain of the Luteinizing Hormone Choriogonadotropin Receptor Gene. Biochem. Biophys. Res. Commun. 1996, 220, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.S.; Zhu, Y.-S.; Cai, L.-Q.; Katz, M.D.; Herrera, C.; DeFillo-Ricart, M.; Imperato-McGinley, J. A Novel Mutation of the Human Luteinizing Hormone Receptor in 46XY and 46XX Sisters1. J. Clin. Endocrinol. Metab. 1998, 83, 2091–2098. [Google Scholar] [CrossRef]
- Suzuki, E.; Miyado, M.; Kuroki, Y.; Fukami, M. Genetic variants of G-protein coupled receptors associated with pubertal disorders. Reprod. Med. Biol. 2023, 22, e12515. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilkenfeld, S.R.; Lin, C.; Frigo, D.E. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids 2018, 133, 2–7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Mechanisms of anticarcino- genesis and drug resistance. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 591, 247–263. [Google Scholar] [CrossRef]
- Jian, W.-X.; Yang, Y.-J.; Long, J.-R.; Li, Y.-N.; Deng, F.-Y.; Jiang, D.-K.; Deng, H.-W. Estrogen receptor α gene relationship with peak bone mass and body mass index in Chinese nuclear families. J. Hum. Genet. 2005, 50, 477–482. [Google Scholar] [CrossRef]
- Lizcano, F.; Guzmán, G. Estrogen Deficiency and the Origin of Obesity during Menopause. Bio. Med. Res. Int. 2014, 2014, 757461. [Google Scholar] [CrossRef]
- Bernard, V.; Kherra, S.; Francou, B.; Fagart, J.; Viengchareun, S.; Guéchot, J.; Ladjouze, A.; Guiochon-Mantel, A.; Korach, K.S.; Binart, N.; et al. Familial Multiplicity of Estrogen Insensitivity Associated with a Loss-of-Function ESR1 Mutation. J. Clin. Endocrinol. Metab. 2017, 102, 93–99. [Google Scholar] [CrossRef]
- Quaynor, S.D.; Stradtman, E.W.J.; Kim, H.-G.; Shen, Y.; Chorich, L.P.; Schreihofer, D.A.; Layman, L.C. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N. Engl. J. Med. 2013, 369, 164–171. [Google Scholar] [CrossRef]
- Smith, E.P.; Boyd, J.; Frank, G.R.; Takahashi, H.; Cohen, R.M.; Specker, B.; Williams, T.C.; Lubahn, D.B.; Korach, K.S. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N. Engl. J. Med. 1994, 331, 1056–1061. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor–positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Reese, J.C.; Katzenellenbogen, B. Characterization of a temperature-sensitive mutation in the hormone binding domain of the human estrogen receptor. Studies in cell extracts and intact cells and their implications for hormone-dependent transcriptional activation. J. Biol. Chem. 1992, 267, 9868–9873. [Google Scholar] [CrossRef]
- Lang-Muritano, M.; Sproll, P.; Wyss, S.; Kolly, A.; Hürlimann, R.; Konrad, D.; Biason-Lauber, A. Early-Onset Complete Ovarian Failure and Lack of Puberty in a Woman with Mutated Estrogen Receptor β (ESR2). J. Clin. Endocrinol. Metab. 2018, 103, 3748–3756. [Google Scholar] [CrossRef]
- Hristov, D.; Stojanov, D. Exploring Regulatory Properties of Genes Associated with Nonsyndromic Male Infertility. Reprod. Med. 2024, 5, 136–153. [Google Scholar] [CrossRef]
- Stojanov, D.; Bogdanova, A.M.; Orzechowski, T.M. TMO: Time and memory optimized algorithm applicable for more accurate alignment of trinucleotide repeat disorders associated genes. Biotechnol. Biotechnol. Equip. 2015, 30, 388–403. [Google Scholar] [CrossRef]
- Gerasimova, T.; Thanasoula, M.N.; Zattas, D.; Seli, E.; Sakkas, D.; Lalioti, M.D. Identification and in Vitro Characterization of Follicle Stimulating Hormone (FSH) Receptor Variants Associated with Abnormal Ovarian Response to FSH. J. Clin. Endocrinol. Metab. 2010, 95, 529–536. [Google Scholar] [CrossRef]
- Latronico, A.C.; Segaloff, D.L. Naturally occurring mutations of the luteinizing-hormone receptor: Lessons learned about reproductive physiology and G protein–coupled receptors. Am. J. Hum. Genet. 1999, 65, 949–958. [Google Scholar] [CrossRef]
- Hiort, O.; Holterhus, P.-M.; Horter, T.; Schulze, W.; Kremke, B.; Bals-Pratsch, M.; Sinnecker, G.H.G.; Kruse, K. Significance of Mutations in the Androgen Receptor Gene in Males with Idiopathic Infertility1. J. Clin. Endocrinol. Metab. 2000, 85, 2810–2815. [Google Scholar] [CrossRef]
- Sztachelska, M.; Ponikwicka-Tyszko, D.; Martínez-Rodrigo, L.; Bernaczyk, P.; Palak, E.; Półchłopek, W.; Bielawski, T.; Wołczyński, S. Functional Implications of Estrogen and Progesterone Receptors Expression in Adenomyosis, Potential Targets for Endocrinological Therapy. J. Clin. Med. 2022, 11, 4407. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).