
Biophysical Dissection of Isolated GPCRs: The Adenosine A2A Receptor under the Bistouries


 , , , , , , , , ,
, , , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Foreword: Biophysical Approaches to Answer Which Questions?
2. Background and Introduction
3. Mass Production of A2A Receptors
3.1. Expression Systems
3.2. Molecular Constructs
4. Extraction and Purification
5. Isolation in Lipid Nanoparticles
6. Quality Controls on Isolated Receptors
7. Functionality of the Isolated A2A
7.1. Ligand Binding
7.1.1. Radioactive Ligand Binding
7.1.2. Surface Plasmon Resonance
7.1.3. Calorimetry
7.1.4. Receptor Fluorescent Labeling
7.1.5. Ligand-Based NMR
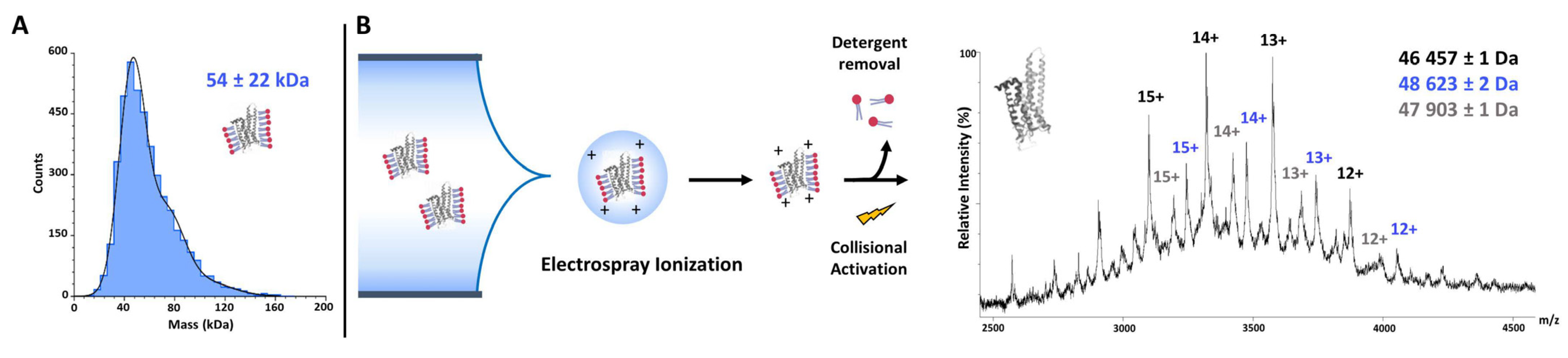
7.1.6. Native Mass Spectrometry
7.2. Activity
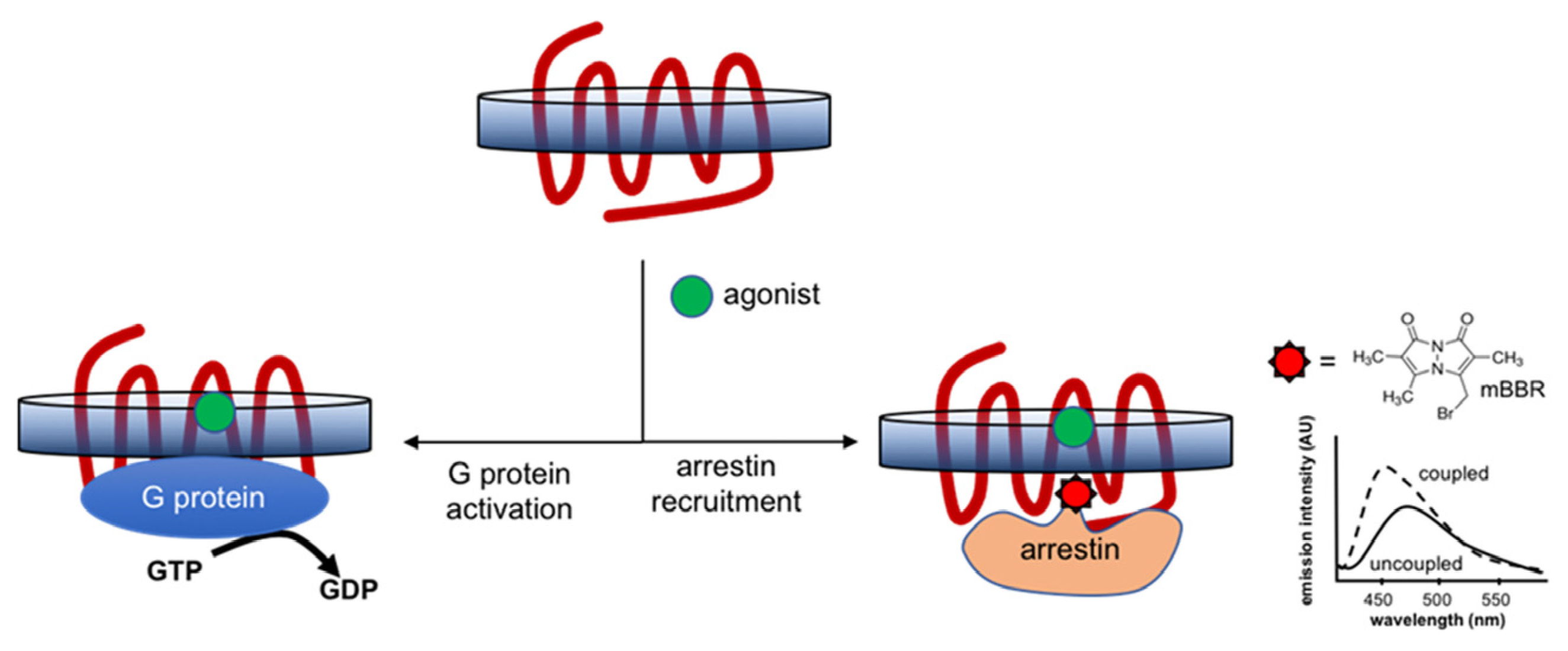
7.2.1. G-Protein Coupling and Arrestin Recruitment
7.2.2. Functionality of Isolated Receptors: Activity of Receptor Dimers and Oligomers
8. Structure of A2A
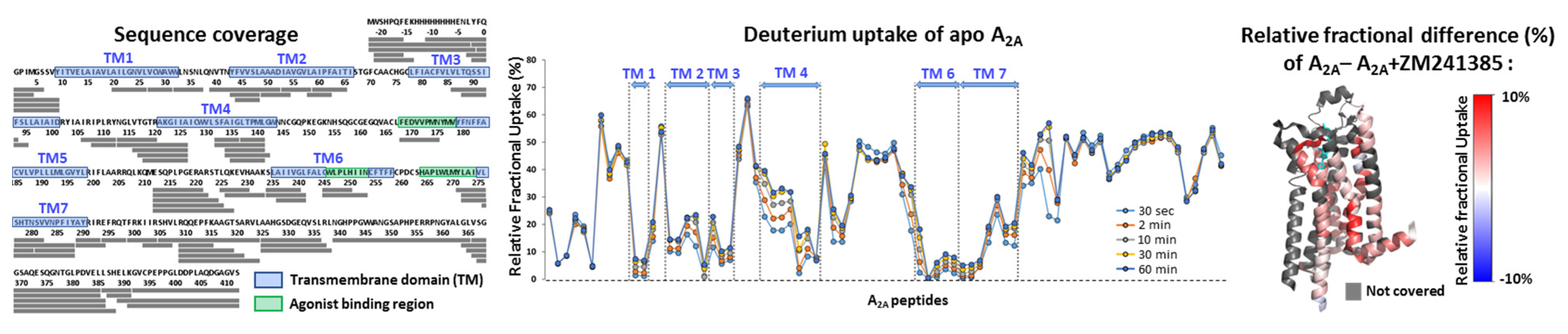
8.1. Structural Mass Spectrometry Approaches
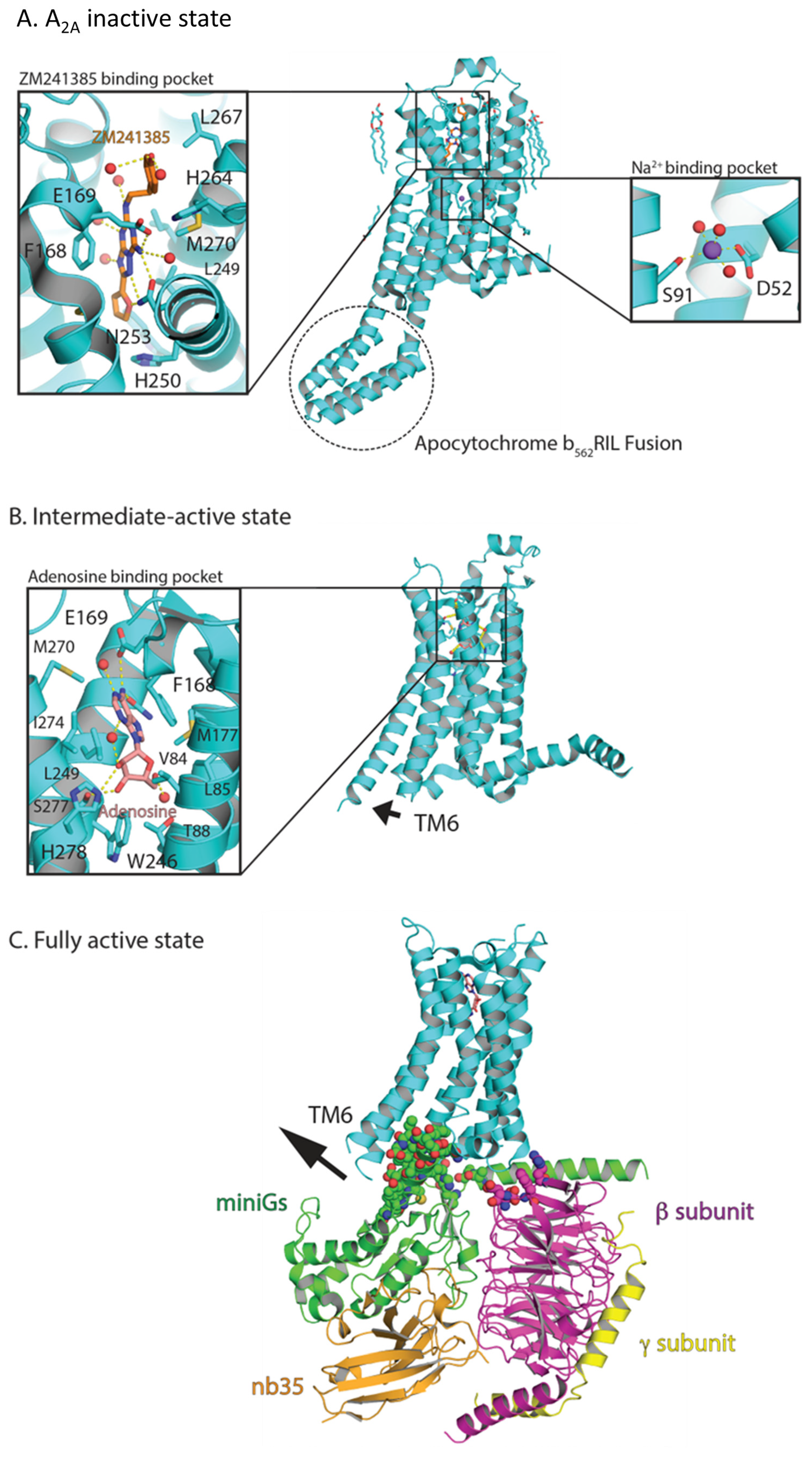
8.2. X-ray Crystallography of GPCRs: Uncovering Conserved Activation Mechanisms Using A2A as a Model
8.3. Cryo-Electron Microscopy
8.4. NMR Approaches
8.5. Future Trends for GPCR Structure Studies
8.5.1. Cryo-Electron Tomography
8.5.2. Structural Flexibility of GPCR Investigated Using Single Molecule Fluorescence and Atomic Force Microscopy
9. Pharmacology of the Isolated Protein A2A
9.1. Background
9.2. Background on Ligand Chemistry
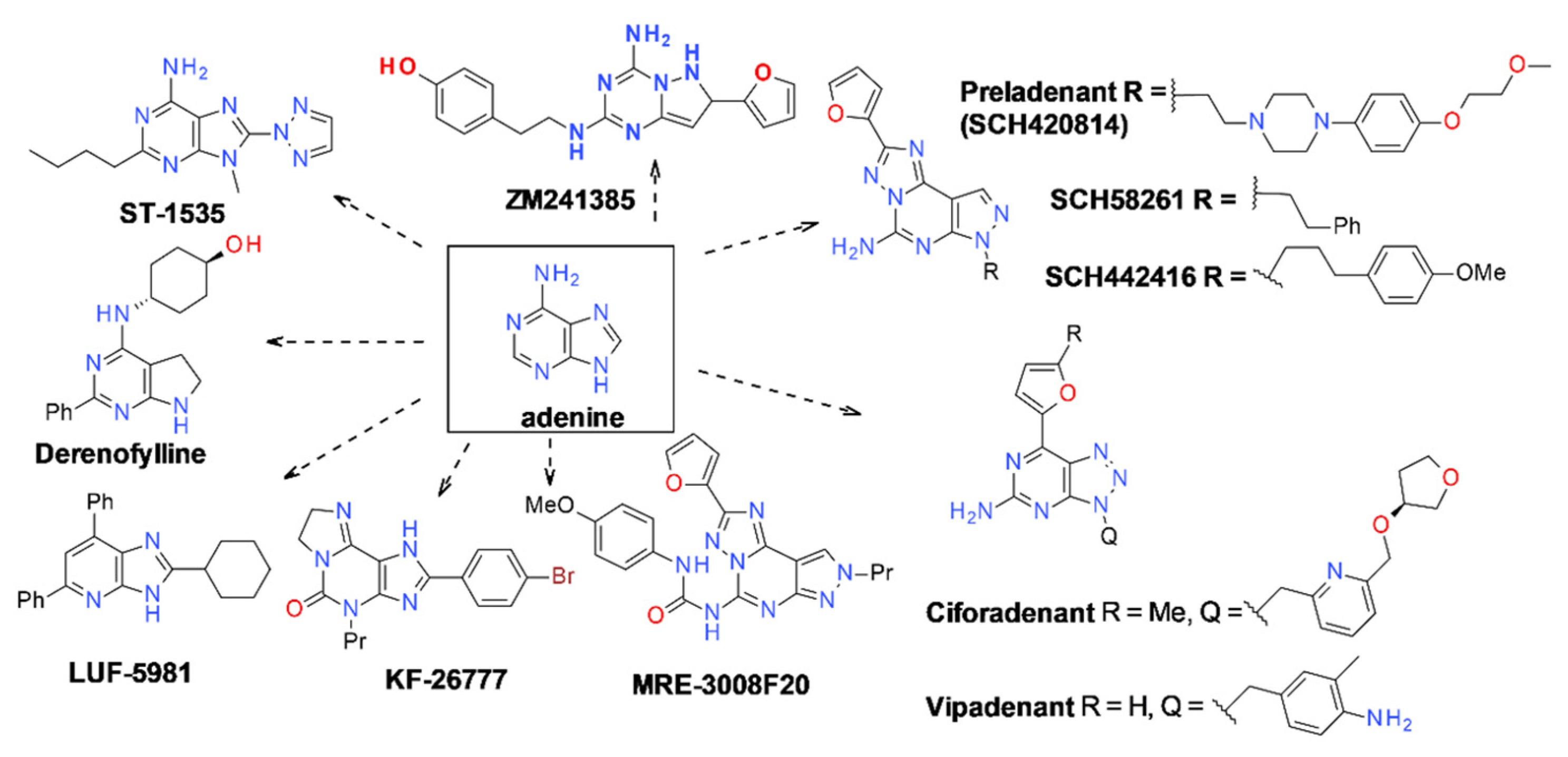
9.2.1. A2A Receptor Agonists
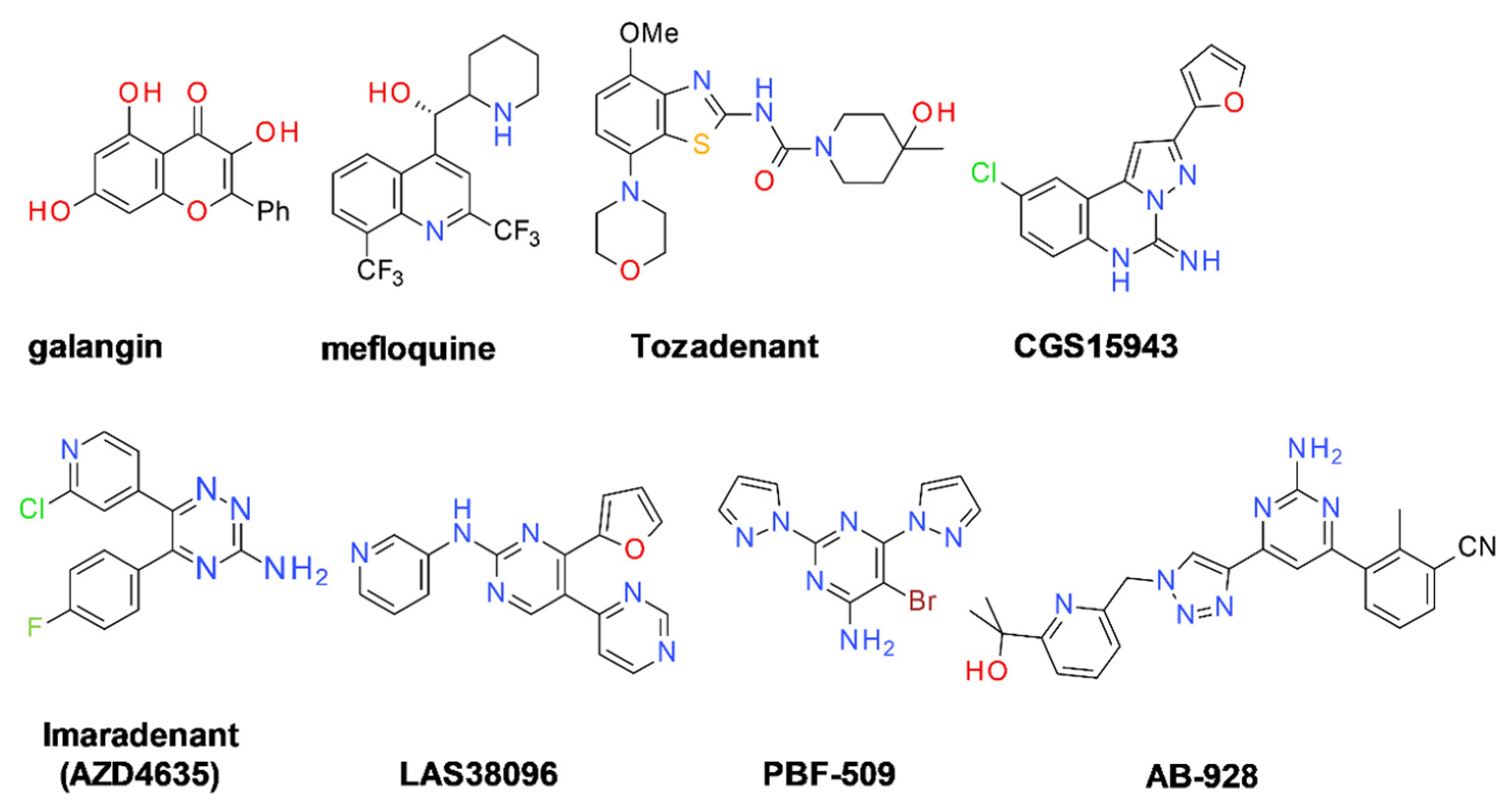
9.2.2. A2A Receptor Antagonists
9.3. Allosteric Modulators
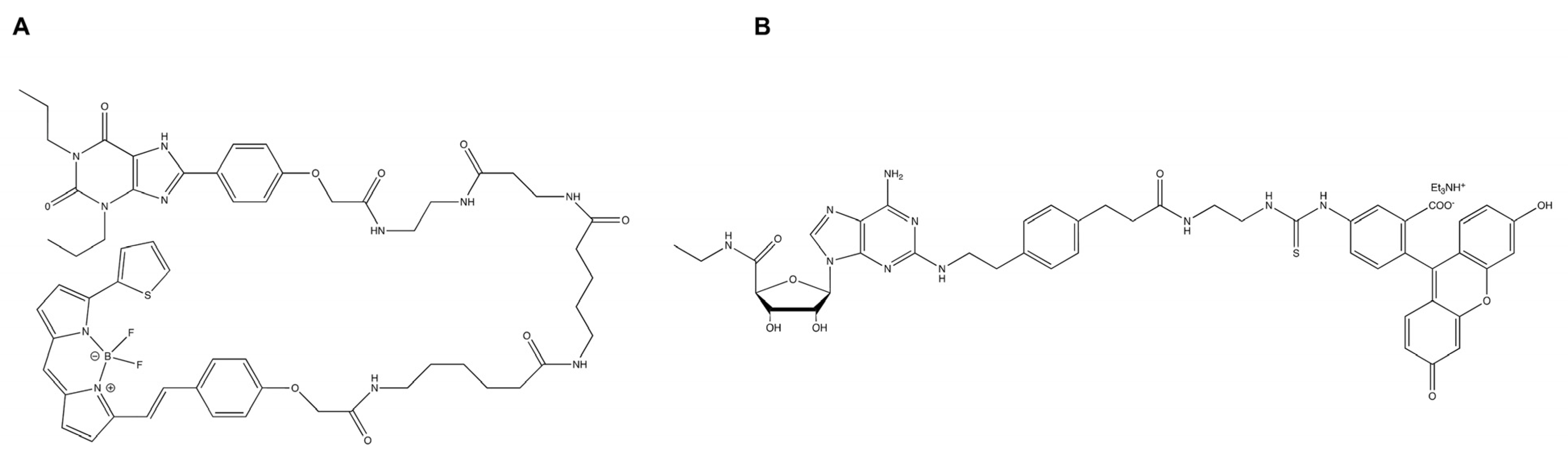
9.4. Fluorescent Ligands
9.5. Structure-Based Ligand Discovery for A2A
9.6. Fragment-Based Ligand Discovery for A2A
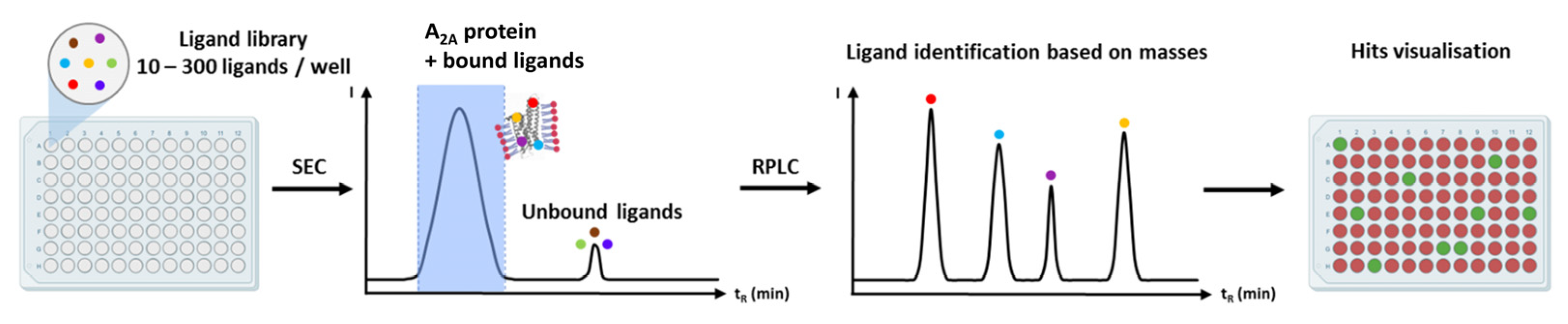
9.7. Mass Spectrometry-Based Ligand Screening
10. Concluding Remarks and Perspectives
Funding
Conflicts of Interest
Abbreviations
References
- Fredholm, B.B.; Frenguelli, B.G.; Hills, R.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.-N.; Linden, J.; Müller, C.E.; Schwabe, U.; Stiles, G.L. Adenosine receptors in GtoPdb v.2021.2. IUPHAR/BPS Guide Pharmacol. CITE 2021, 2021. [Google Scholar] [CrossRef]
- Blackmore, R.V.; Linn, S. Partial purification and characterization of four endodeoxyribonuclease activities from Escherichia coli K-12. Nucleic Acids Res. 1974, 1, 1–17. [Google Scholar] [CrossRef]
- Pierce, K.D.; Furlong, T.J.; Selbie, L.A.; Shine, J. Molecular cloning and expression of an adenosine A2b receptor from human brain. Biochem. Biophys. Res. Commun. 1992, 187, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Michalke, K.; Huyghe, C.; Lichière, J.; Gravière, M.-E.; Siponen, M.; Sciara, G.; Lepaul, I.; Wagner, R.; Magg, C.; Rudolph, R.; et al. Mammalian G protein-coupled receptor expression in Escherichia coli: II. Refolding and biophysical characterization of mouse cannabinoid receptor 1 and human parathyroid hormone receptor 1. Anal. Biochem. 2010, 401, 74–80. [Google Scholar] [CrossRef] [PubMed]
- IJzerman, A.P.; Jacobson, K.A.; Müller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef]
- Yu, L.; Frith, M.C.; Suzuki, Y.; Peterfreund, R.A.; Gearan, T.; Sugano, S.; Schwarzschild, M.A.; Weng, Z.; Fink, J.S.; Chen, J.-F. Characterization of genomic organization of the adenosine A2A receptor gene by molecular and bioinformatics analyses. Brain Res. 2004, 1000, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, M.; Rosin, D.L.; Hayashi, N.; Linden, J.; Sitkovsky, M.V. Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol. Pharmacol. 1999, 55, 614–624. [Google Scholar]
- Cooper, J.A.; Hill, S.J.; Alexander, S.P.; Rubin, P.C.; Horn, E.H. Adenosine receptor-induced cyclic AMP generation and inhibition of 5-hydroxytryptamine release in human platelets. Br. J. Clin. Pharmacol. 1995, 40, 43–50. [Google Scholar] [CrossRef]
- Schulte, G.; Fredholm, B.B. Human adenosine A1, A2A, A2B, and A3 receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase ½. Mol. Pharmacol. 2000, 58, 477–482. [Google Scholar] [CrossRef]
- Yang, S.N.; Dasgupta, S.; Lledo, P.M.; Vincent, J.D.; Fuxe, K. Reduction of dopamine D2 receptor transduction by activation of adenosine A2a receptors in stably A2a/D2 (long-form) receptor co-transfected mouse fibroblast cell lines: Studies on intracellular calcium levels. Neuroscience 1995, 68, 729–736. [Google Scholar] [CrossRef]
- Ikeuchi, Y.; Nishizaki, T.; Mori, M.; Okada, Y. Adenosine activates the K+ channel and enhances cytosolic Ca2+ release via a P2Y purinoceptor in hippocampal neurons. Eur. J. Pharmacol. 1996, 304, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, F.M. cAMP-producing chemogenetic and adenosine A2a receptor activation inhibits the inwardly rectifying potassium current in striatal projection neurons. Neuropharmacology 2019, 148, 229–243. [Google Scholar] [CrossRef]
- Varani, K.; Gessi, S.; Dalpiaz, A.; Borea, P.A. Pharmacological and biochemical characterization of purified A2a adenosine receptors in human platelet membranes by [3H]-CGS 21680 binding. Br. J. Pharmacol. 1996, 117, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.; Burnstock, G. Autoradiographic demonstration of peripheral adenosine binding sites using [3H]NECA. Brain Res. 1983, 269, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Schütz, W.; Brugger, G. Characterization of [3H]-adenosine binding to media membranes of hog carotid arteries. Pharmacology 1982, 24, 26–34. [Google Scholar] [CrossRef]
- Ji, X.D.; Stiles, G.L.; van Galen, P.J.; Jacobson, K.A. Characterization of human striatal A2-adenosine receptors using radioligand binding and photoaffinity labeling. J. Recept. Res. 1992, 12, 149–169. [Google Scholar] [CrossRef]
- Alexander, S.P.; Millns, P.J. [3H]ZM241385—An antagonist radioligand for adenosine A2A receptors in rat brain. Eur. J. Pharmacol. 2001, 411, 205–210. [Google Scholar] [CrossRef]
- Yu, J.C.; Lin, G.; Field, J.J.; Linden, J. Induction of antiinflammatory purinergic signaling in activated human iNKT cells. JCI Insight 2018, 3, e91954. [Google Scholar] [CrossRef]
- Klammt, C.; Schwarz, D.; Eifler, N.; Engel, A.; Piehler, J.; Haase, W.; Hahn, S.; Dötsch, V.; Bernhard, F. Cell-free production of G protein-coupled receptors for functional and structural studies. J. Struct. Biol. 2007, 158, 482–493. [Google Scholar] [CrossRef]
- Kögler, L.M.; Stichel, J.; Beck-Sickinger, A.G. Structural investigations of cell-free expressed G protein-coupled receptors. Biol. Chem. 2019, 401, 97–116. [Google Scholar] [CrossRef]
- Banères, J.-L.; Popot, J.-L.; Mouillac, B. New advances in production and functional folding of G-protein-coupled receptors. Trends Biotechnol. 2011, 29, 314–322. [Google Scholar] [CrossRef]
- André, N.; Cherouati, N.; Prual, C.; Steffan, T.; Zeder-Lutz, G.; Magnin, T.; Pattus, F.; Michel, H.; Wagner, R.; Reinhart, C. Enhancing functional production of G protein-coupled receptors in Pichia pastoris to levels required for structural studies via a single expression screen. Protein Sci. 2006, 15, 1115–1126. [Google Scholar] [CrossRef]
- Krettler, C.; Reinhart, C.; Bevans, C.G. Expression of GPCRs in Pichia pastoris for structural studies. Methods Enzymol. 2013, 520, 1–29. [Google Scholar] [CrossRef]
- Blocker, K.M.; Britton, Z.T.; Naranjo, A.N.; McNeely, P.M.; Young, C.L.; Robinson, A.S. Recombinant G protein-coupled receptor expression in Saccharomyces cerevisiae for protein characterization. Methods Enzymol. 2015, 556, 165–183. [Google Scholar] [CrossRef]
- Errey, J.C.; Fiez-Vandal, C. Production of membrane proteins in industry: The example of GPCRs. Protein Expr. Purif. 2020, 169, 105569. [Google Scholar] [CrossRef] [PubMed]
- Panneels, V.; Kock, I.; Krijnse-Locker, J.; Rezgaoui, M.; Sinning, I. Drosophila photoreceptor cells exploited for the production of eukaryotic membrane proteins: Receptors, transporters and channels. PLoS ONE 2011, 6, e18478. [Google Scholar] [CrossRef]
- Zhang, L.; Salom, D.; He, J.; Okun, A.; Ballesteros, J.; Palczewski, K.; Li, N. Expression of functional G protein-coupled receptors in photoreceptors of transgenic Xenopus laevis. Biochemistry 2005, 44, 14509–14518. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Sasaki, K.; Wakimoto, Y.; Toyooka, M.; Motohashi, T.; Shimojima, T.; Takeda, S.; Park, E.Y.; Maenaka, K. Efficient silkworm expression of human GPCR (nociceptin receptor) by a Bombyx mori bacmid DNA system. Biochem. Biophys. Res. Commun. 2009, 385, 375–379. [Google Scholar] [CrossRef]
- Michalke, K.; Gravière, M.-E.; Huyghe, C.; Vincentelli, R.; Wagner, R.; Pattus, F.; Schroeder, K.; Oschmann, J.; Rudolph, R.; Cambillau, C.; et al. Mammalian G-protein-coupled receptor expression in Escherichia coli: I. High-throughput large-scale production as inclusion bodies. Anal. Biochem. 2009, 386, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.M.; Grisshammer, R. Purification and characterization of the human adenosine A2a receptor functionally expressed in Escherichia coli. Eur. J. Biochem. 2002, 269, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Shibata, Y.; Serrano-Vega, M.J.; Tate, C.G. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 10744–10749. [Google Scholar] [CrossRef]
- Lebon, G.; Bennett, K.; Jazayeri, A.; Tate, C.G. Thermostabilisation of an Agonist-Bound Conformation of the Human Adenosine A2A Receptor. J. Mol. Biol. 2011, 409, 298–310. [Google Scholar] [CrossRef]
- Niebauer, R.T.; Robinson, A.S. Exceptional total and functional yields of the human adenosine (A2a) receptor expressed in the yeast Saccharomyces cerevisiae. Protein Expr. Purif. 2006, 46, 204–211. [Google Scholar] [CrossRef]
- O’Malley, M.A.; Lazarova, T.; Britton, Z.T.; Robinson, A.S. High-level expression in Saccharomyces cerevisiae enables isolation and spectroscopic characterization of functional human adenosine A2a receptor. J. Struct. Biol. 2007, 159, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Schonenbach, N.S.; Rieth, M.D.; Han, S.; O’Malley, M.A. Adenosine A2a receptors form distinct oligomers in protein detergent complexes. FEBS Lett. 2016, 590, 3295–3306. [Google Scholar] [CrossRef] [PubMed]
- Fraser, N.J. Expression and functional purification of a glycosylation deficient version of the human adenosine 2a receptor for structural studies. Protein Expr. Purif. 2006, 49, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Gras, A.; Fiez-Vandal, C.; Ruprecht, J.; Rana, R.; Martinez, M.; Strange, P.G.; Wagner, R.; Byrne, B. Large-scale functional expression of WT and truncated human adenosine A2A receptor in Pichia pastoris bioreactor cultures. Microb. Cell Fact. 2008, 7, 28. [Google Scholar] [CrossRef]
- Hino, T.; Arakawa, T.; Iwanari, H.; Yurugi-Kobayashi, T.; Ikeda-Suno, C.; Nakada-Nakura, Y.; Kusano-Arai, O.; Weyand, S.; Shimamura, T.; Nomura, N.; et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 2012, 482, 237–240. [Google Scholar] [CrossRef]
- Jamshad, M.; Charlton, J.; Lin, Y.-P.; Routledge, S.J.; Bawa, Z.; Knowles, T.J.; Overduin, M.; Dekker, N.; Dafforn, T.R.; Bill, R.M.; et al. G-protein coupled receptor solubilization and purification for biophysical analysis and functional studies, in the total absence of detergent. Biosci. Rep. 2015, 35, e00188. [Google Scholar] [CrossRef]
- Lengger, B.; Jensen, M.K. Engineering G protein-coupled receptor signalling in yeast for biotechnological and medical purposes. FEMS Yeast Res. 2020, 20, foz087. [Google Scholar] [CrossRef]
- Shiroishi, M.; Tsujimoto, H.; Makyio, H.; Asada, H.; Yurugi-Kobayashi, T.; Shimamura, T.; Murata, T.; Nomura, N.; Haga, T.; Iwata, S.; et al. Platform for the rapid construction and evaluation of GPCRs for crystallography in Saccharomyces cerevisiae. Microb. Cell Fact. 2012, 11, 78. [Google Scholar] [CrossRef]
- Bertheleme, N.; Strege, A.; Bunting, S.E.; Dowell, S.J.; Byrne, B. Arginine 199 and leucine 208 have key roles in the control of adenosine A2A receptor signalling function. PLoS ONE 2014, 9, e89613. [Google Scholar] [CrossRef]
- Mitsumoto, M.; Sugaya, K.; Kazama, K.; Nakano, R.; Kosugi, T.; Murata, T.; Koga, N. State-Targeting Stabilization of Adenosine A2A Receptor by Fusing a Custom-Made de Novo Designed α-Helical Protein. Int. J. Mol. Sci. 2021, 22, 12906. [Google Scholar] [CrossRef]
- Wang, X.; van Westen, G.J.P.; Heitman, L.H.; IJzerman, A.P. G protein-coupled receptors expressed and studied in yeast. The adenosine receptor as a prime example. Biochem. Pharmacol. 2021, 187, 114370. [Google Scholar] [CrossRef]
- Meltzer, M.; Zvagelsky, T.; Hadad, U.; Papo, N.; Engel, S. Yeast-based directed-evolution for high-throughput structural stabilization of G protein-coupled receptors (GPCRs). Sci. Rep. 2022, 12, 8657. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yasuda, S.; Kasai, R.S.; Nakano, R.; Hikiri, S.; Sugaya, K.; Hayashi, T.; Ogasawara, S.; Shiroishi, M.; Fujiwara, T.K.; et al. A methodology for creating mutants of G-protein coupled receptors stabilized in active state by combining statistical thermodynamics and evolutionary molecular engineering. Protein Sci. 2022, 31, e4425. [Google Scholar] [CrossRef] [PubMed]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 2018, 172, 68–80.e12. [Google Scholar] [CrossRef]
- Ye, L.; van Eps, N.; Zimmer, M.; Ernst, O.P.; Prosser, R.S. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, T.; Kondo, K.; Kurita, M.; Kofuku, Y.; Natsume, M.; Imai, S.; Shiraishi, Y.; Ueda, T.; Shimada, I. Activation of adenosine A2A receptor by lipids from docosahexaenoic acid revealed by NMR. Sci. Adv. 2020, 6, eaay8544. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.K.; Pandey, A.; Tran, D.P.; Villanueva, N.L.; Kitao, A.; Sunahara, R.K.; Sljoka, A.; Prosser, R.S. Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell 2021, 184, 1884–1894.e14. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.W.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Miyagi, H.; Asada, H.; Suzuki, M.; Takahashi, Y.; Yasunaga, M.; Suno, C.; Iwata, S.; Saito, J.-I. The discovery of a new antibody for BRIL-fused GPCR structure determination. Sci. Rep. 2020, 10, 11669. [Google Scholar] [CrossRef]
- Claff, T.; Klapschinski, T.A.; Tiruttani Subhramanyam, U.K.; Vaaßen, V.J.; Schlegel, J.G.; Vielmuth, C.; Voß, J.H.; Labahn, J.; Müller, C.E. Single Stabilizing Point Mutation Enables High-Resolution Co-Crystal Structures of the Adenosine A2A Receptor with Preladenant Conjugates. Angew. Chem. Int. Ed Engl. 2022, 61, e202115545. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Edwards, P.C.; Leslie, A.G.W.; Tate, C.G. Molecular Determinants of CGS21680 Binding to the Human Adenosine A2A Receptor. Mol. Pharmacol. 2015, 87, 907–915. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Eddy, M.T.; Gao, Z.-G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269.e5. [Google Scholar] [CrossRef]
- McGraw, C.; Koretz, K.S.; Oseid, D.; Lyman, E.; Robinson, A.S. Cholesterol Dependent Activity of the Adenosine A2A Receptor Is Modulated via the Cholesterol Consensus Motif. Molecules 2022, 27, 3529. [Google Scholar] [CrossRef]
- Yeliseev, A.; van den Berg, A.; Zoubak, L.; Hines, K.; Stepnowski, S.; Williston, K.; Yan, W.; Gawrisch, K.; Zmuda, J. Thermostability of a recombinant G protein-coupled receptor expressed at high level in mammalian cell culture. Sci. Rep. 2020, 10, 16805. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, H.; Hoppe, N.; Manglik, A.; Cheng, Y. Fusion protein strategies for cryo-EM study of G protein-coupled receptors. Nat. Commun. 2022, 13, 4366. [Google Scholar] [CrossRef]
- Bocquet, N.; Kohler, J.; Hug, M.N.; Kusznir, E.A.; Rufer, A.C.; Dawson, R.J.; Hennig, M.; Ruf, A.; Huber, W.; Huber, S. Real-time monitoring of binding events on a thermostabilized human A2A receptor embedded in a lipid bilayer by surface plasmon resonance. Biochim. Biophys. Acta 2015, 1848, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Takamuku, Y.; Asakawa, T.; Inai, M.; Hino, T.; Iwata, S.; Kan, T.; Murata, T. An efficient screening method for purifying and crystallizing membrane proteins using modified clear-native PAGE. Anal. Biochem. 2018, 548, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Lebon, G. Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol. 2017, 8, 898. [Google Scholar] [CrossRef]
- Cui, M.; Zhou, Q.; Xu, Y.; Weng, Y.; Yao, D.; Zhao, S.; Song, G. Crystal structure of a constitutive active mutant of adenosine A2A receptor. IUCrJ 2022, 9, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K.; Wagner, R.; Reinhart, C.; Desmyter, A.; Cherouati, N.; Magnin, T.; Zeder-Lutz, G.; Courtot, M.; Prual, C.; André, N.; et al. Structural genomics on membrane proteins: Comparison of more than 100 GPCRs in 3 expression systems. J. Struct. Funct. Genom. 2006, 7, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.D.; Dikiy, I.; Chapman, K.; Rödström, K.E.; Aramini, J.; LeVine, M.V.; Khelashvili, G.; Rasmussen, S.G.; Gardner, K.H.; Rosenbaum, D.M. Ligand modulation of sidechain dynamics in a wild-type human GPCR. eLife 2017, 6, e28505. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.-G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef]
- Jespers, W.; Verdon, G.; Azuaje, J.; Majellaro, M.; Keränen, H.; García-Mera, X.; Congreve, M.; Deflorian, F.; de Graaf, C.; Zhukov, A.; et al. X-ray Crystallography and Free Energy Calculations Reveal the Binding Mechanism of A2A Adenosine Receptor Antagonists. Angew. Chem. Int. Ed Engl. 2020, 59, 16536–16543. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Thompson, A.A.; Liu, W.; Roth, C.B.; Griffith, M.T.; Katritch, V.; Kunken, J.; Xu, F.; Cherezov, V.; Hanson, M.A.; et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 2012, 20, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
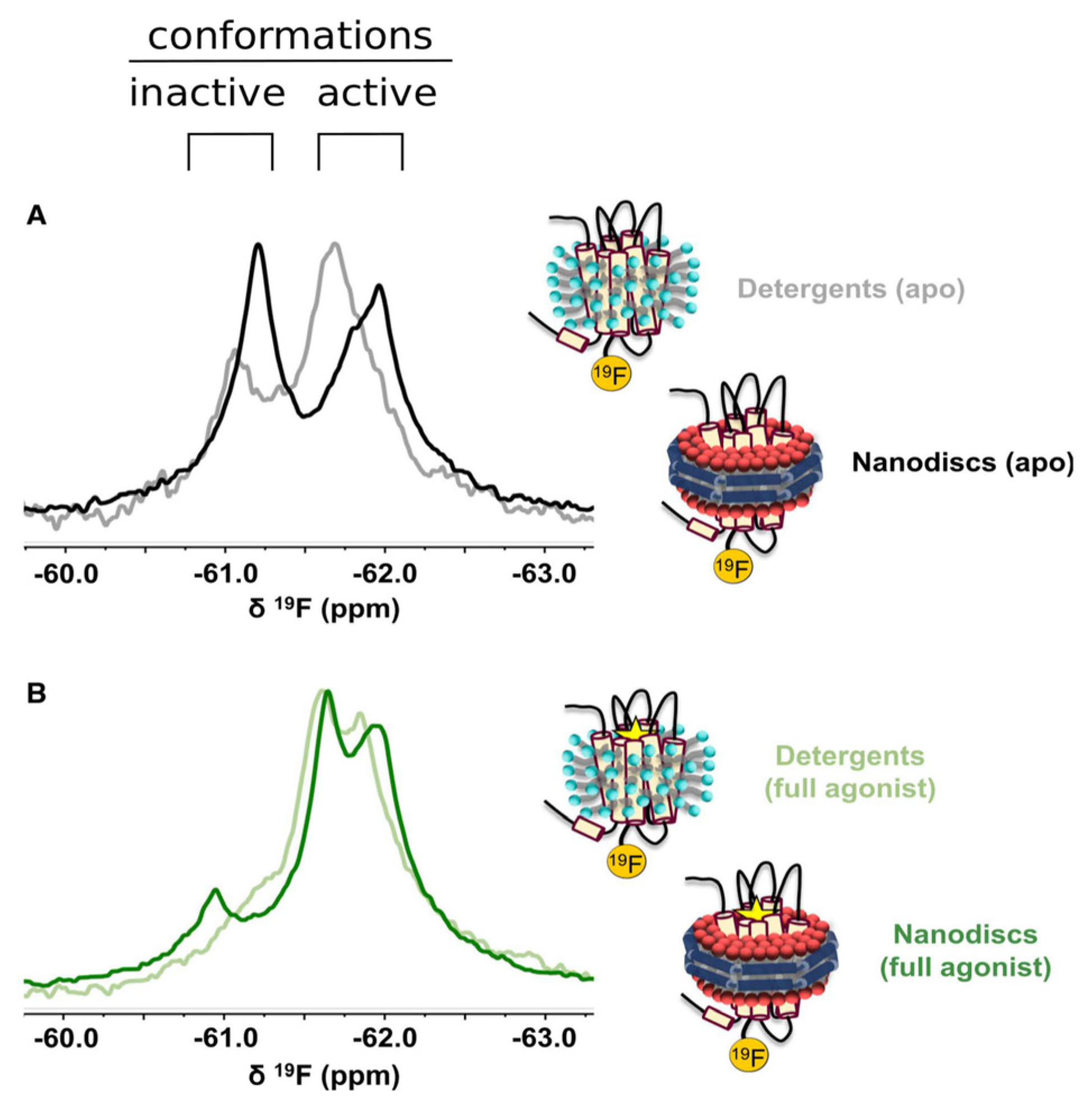
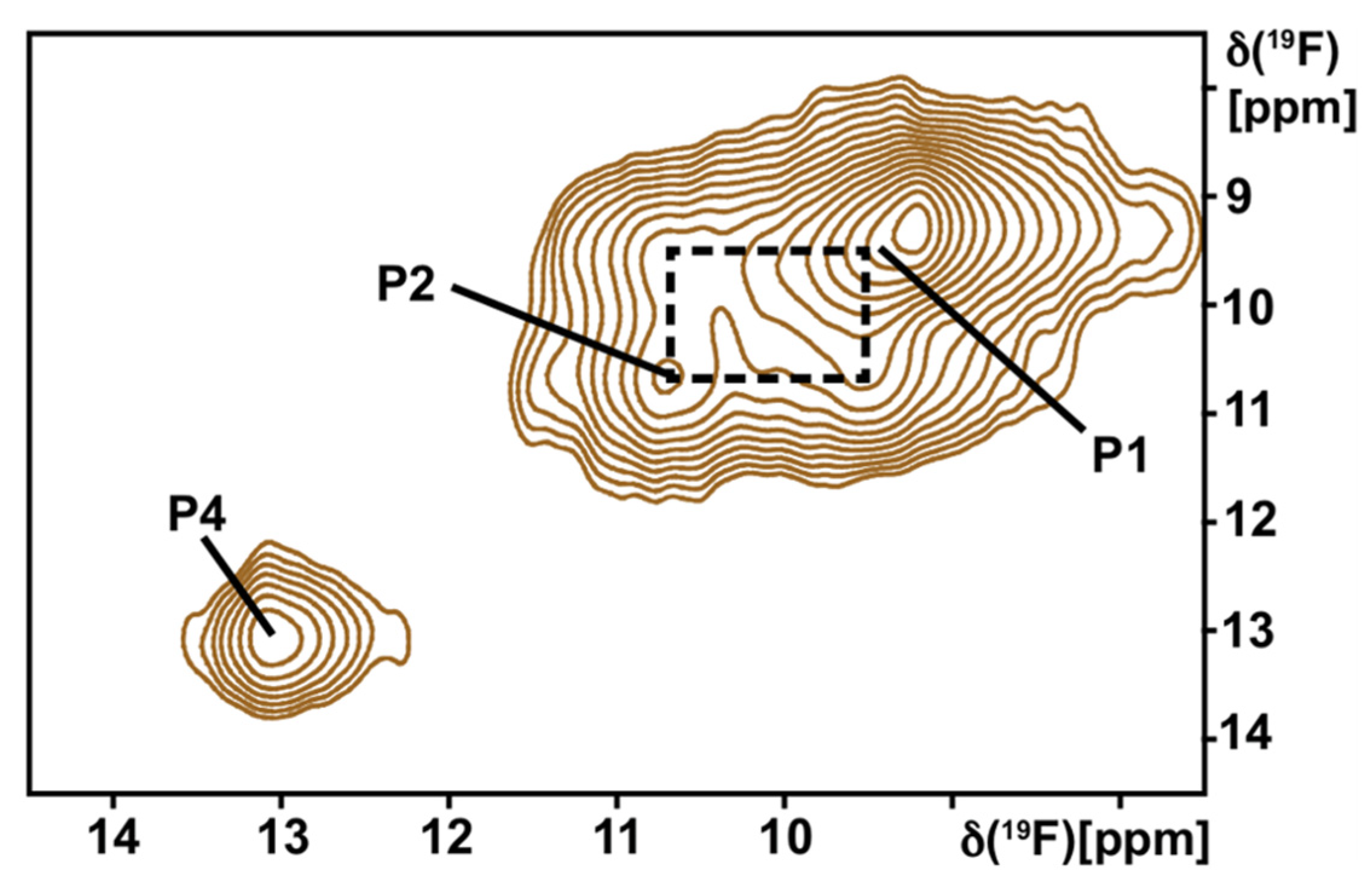
- Sušac, L.; Eddy, M.T.; Didenko, T.; Stevens, R.C.; Wüthrich, K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. USA 2018, 115, 12733–12738. [Google Scholar] [CrossRef]
- Huang, S.K.; Almurad, O.; Pejana, R.J.; Morrison, Z.A.; Pandey, A.; Picard, L.-P.; Nitz, M.; Sljoka, A.; Prosser, R.S. Allosteric modulation of the adenosine A2A receptor by cholesterol. eLife 2022, 11, e73901. [Google Scholar] [CrossRef]
- Thakur, N.; Wei, S.; Ray, A.P.; Lamichhane, R.; Eddy, M.T. Production of human A2AAR in lipid nanodiscs for 19F-NMR and single-molecule fluorescence spectroscopy. STAR Protoc. 2022, 3, 101535. [Google Scholar] [CrossRef] [PubMed]
- Choy, B.C.; Cater, R.J.; Mancia, F.; Pryor, E.E. A 10-year meta-analysis of membrane protein structural biology: Detergents, membrane mimetics, and structure determination techniques. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183533. [Google Scholar] [CrossRef]
- Lecas, L.; Hartmann, L.; Caro, L.; Mohamed-Bouteben, S.; Raingeval, C.; Krimm, I.; Wagner, R.; Dugas, V.; Demesmay, C. Miniaturized weak affinity chromatography for ligand identification of nanodiscs-embedded G-protein coupled receptors. Anal. Chim. Acta 2020, 1113, 26–35. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Geiger, J.; Ishchenko, A.; Han, G.W.; Barty, A.; White, T.A.; Gati, C.; Batyuk, A.; Hunter, M.S.; Aquila, A.; et al. Harnessing the power of an X-ray laser for serial crystallography of membrane proteins crystallized in lipidic cubic phase. IUCrJ 2020, 7, 976–984. [Google Scholar] [CrossRef]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef]
- Reis, R.I.; Moraes, I. Probing Membrane Protein Assembly into Nanodiscs by In Situ Dynamic Light Scattering: A2A Receptor as a Case Study. Biology 2020, 9, 400. [Google Scholar] [CrossRef]
- Batyuk, A.; Galli, L.; Ishchenko, A.; Han, G.W.; Gati, C.; Popov, P.A.; Lee, M.-Y.; Stauch, B.; White, T.A.; Barty, A.; et al. Native phasing of x-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv. 2016, 2, e1600292. [Google Scholar] [CrossRef] [PubMed]
- Dikiy, I.; Clark, L.D.; Gardner, K.H.; Rosenbaum, D.M. Isotopic Labeling of Eukaryotic Membrane Proteins for NMR Studies of Interactions and Dynamics. Methods Enzymol. 2019, 614, 37–65. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, M.A.; Naranjo, A.N.; Lazarova, T.; Robinson, A.S. Analysis of Adenosine A 2 a Receptor Stability: Effects of Ligands and Disulfide Bonds. Biochemistry 2010, 49, 9181–9189. [Google Scholar] [CrossRef] [PubMed]
- Ashok, Y.; Nanekar, R.; Jaakola, V.-P. Defining thermostability of membrane proteins by western blotting. Protein Eng. Des. Sel. 2015, 28, 539–542. [Google Scholar] [CrossRef]
- Zhang, X.; Stevens, R.C.; Xu, F. The importance of ligands for G protein-coupled receptor stability. Trends Biochem. Sci. 2015, 40, 79–87. [Google Scholar] [CrossRef]
- Thompson, A.A.; Liu, J.J.; Chun, E.; Wacker, D.; Wu, H.; Cherezov, V.; Stevens, R.C. GPCR stabilization using the bicelle-like architecture of mixed sterol-detergent micelles. Methods 2011, 55, 310–317. [Google Scholar] [CrossRef]
- O’Malley, M.A.; Helgeson, M.E.; Wagner, N.J.; Robinson, A.S. The morphology and composition of cholesterol-rich micellar nanostructures determine transmembrane protein (GPCR) activity. Biophys. J. 2011, 100, L11–L13. [Google Scholar] [CrossRef]
- Vagenende, V.; Yap, M.G.S.; Trout, B.L. Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 2009, 48, 11084–11096. [Google Scholar] [CrossRef]
- Wittmann, H.-J.; Seifert, R.; Strasser, A. Mathematical analysis of the sodium sensitivity of the human histamine H3 receptor. Silico Pharmacol. 2014, 2, 1. [Google Scholar] [CrossRef]
- Ross, P.; Weihofen, W.; Siu, F.; Xie, A.; Katakia, H.; Wright, S.K.; Hunt, I.; Brown, R.K.; Freire, E. Isothermal chemical denaturation to determine binding affinity of small molecules to G-protein coupled receptors. Anal. Biochem. 2015, 473, 41–45. [Google Scholar] [CrossRef]
- Igonet, S.; Raingeval, C.; Cecon, E.; Pučić-Baković, M.; Lauc, G.; Cala, O.; Baranowski, M.; Perez, J.; Jockers, R.; Krimm, I.; et al. Enabling STD-NMR fragment screening using stabilized native GPCR: A case study of adenosine receptor. Sci. Rep. 2018, 8, 8142. [Google Scholar] [CrossRef]
- Ukena, D.; Jacobson, K.A.; Kirk, K.L.; Daly, J.W. A [3H]amine congener of 1,3-dipropyl-8-phenylxanthine. FEBS Lett. 1986, 199, 269–274. [Google Scholar] [CrossRef]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomès, R.; García, A.E.; Ernst, O.P.; et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.D.Q.; Vigers, M.; Sefah, E.; Seppälä, S.; Hoover, J.P.; Schonenbach, N.S.; Mertz, B.; O’Malley, M.A.; Han, S. Homo-oligomerization of the human adenosine A2A receptor is driven by the intrinsically disordered C-terminus. eLife 2021, 10, e66662. [Google Scholar] [CrossRef]
- Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Nietlispach, D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 2020, 25, 4729. [Google Scholar] [CrossRef]
- Serebryany, E.; Zhu, G.A.; Yan, E.C.Y. Artificial membrane-like environments for in vitro studies of purified G-protein coupled receptors. Biochim. Biophys. Acta 2012, 1818, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Overduin, M.; Trieber, C.; Prosser, R.S.; Picard, L.-P.; Sheff, J.G. Structures and Dynamics of Native-State Transmembrane Protein Targets and Bound Lipids. Membranes 2021, 11, 451. [Google Scholar] [CrossRef]
- Park, S.H.; Das, B.B.; Casagrande, F.; Tian, Y.; Nothnagel, H.J.; Chu, M.; Kiefer, H.; Maier, K.; de Angelis, A.A.; Marassi, F.M.; et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 2012, 491, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Duc, N.M.; Du, Y.; Zhang, C.; Lee, S.Y.; Thorsen, T.S.; Kobilka, B.K.; Chung, K.Y. Effective application of bicelles for conformational analysis of G protein-coupled receptors by hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 808–817. [Google Scholar] [CrossRef]
- Schmidt, P.; Bender, B.J.; Kaiser, A.; Gulati, K.; Scheidt, H.A.; Hamm, H.E.; Meiler, J.; Beck-Sickinger, A.G.; Huster, D. Improved in Vitro Folding of the Y2 G Protein-Coupled Receptor into Bicelles. Front. Mol. Biosci. 2017, 4, 100. [Google Scholar] [CrossRef]
- Brea, R.J.; Cole, C.M.; Lyda, B.R.; Ye, L.; Prosser, R.S.; Sunahara, R.K.; Devaraj, N.K. In Situ Reconstitution of the Adenosine A2A Receptor in Spontaneously Formed Synthetic Liposomes. J. Am. Chem. Soc. 2017, 139, 3607–3610. [Google Scholar] [CrossRef]
- Bayburt, T.H.; Grinkova, Y.V.; Sligar, S.G. Self-Assembly of Discoidal Phospholipid Bilayer Nanoparticles with Membrane Scaffold Proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar] [CrossRef]
- Ritchie, T.K.; Grinkova, Y.V.; Bayburt, T.H.; Denisov, I.G.; Zolnerciks, J.K.; Atkins, W.M.; Sligar, S.G. Chapter 11—Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009, 464, 211–231. [Google Scholar] [CrossRef]
- Lavington, S.; Watts, A. Lipid nanoparticle technologies for the study of G protein-coupled receptors in lipid environments. Biophys. Rev. 2020, 12, 1287–1302. [Google Scholar] [CrossRef]
- Segala, E.; Errey, J.C.; Fiez-Vandal, C.; Zhukov, A.; Cooke, R.M. Biosensor-based affinities and binding kinetics of small molecule antagonists to the adenosine A2A receptor reconstituted in HDL like particles. FEBS Lett. 2015, 589, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lu, Y.; Wu, D.; Peng, Y.; Loa-Kum-Cheung, W.; Peng, C.; Quinn, R.J.; Shui, W.; Liu, Z.-J. Ligand identification of the adenosine A2A receptor in self-assembled nanodiscs by affinity mass spectrometry. Anal. Methods 2017, 9, 5851–5858. [Google Scholar] [CrossRef]
- Fredriksson, K.; Lottmann, P.; Hinz, S.; Onila, I.; Shymanets, A.; Harteneck, C.; Müller, C.E.; Griesinger, C.; Exner, T.E. Nanodiscs for INPHARMA NMR Characterization of GPCRs: Ligand Binding to the Human A2A Adenosine Receptor. Angew. Chem. Int. Ed Engl. 2017, 56, 5750–5754. [Google Scholar] [CrossRef]
- Mary, S.; Damian, M.; Rahmeh, R.; Mouillac, B.; Marie, J.; Granier, S.; Banères, J.-L. Amphipols in G protein-coupled receptor pharmacology: What are they good for? J. Membr. Biol. 2014, 247, 853–860. [Google Scholar] [CrossRef]
- Zoonens, M.; Popot, J.-L. Amphipols for each season. J. Membr. Biol. 2014, 247, 759–796. [Google Scholar] [CrossRef] [PubMed]
- Overduin, M.; Klumperman, B. Advancing membrane biology with poly(styrene-co-maleic acid)-based native nanodiscs. Eur. Polym. J. 2019, 110, 63–68. [Google Scholar] [CrossRef]
- Grime, R.L.; Goulding, J.; Uddin, R.; Stoddart, L.A.; Hill, S.J.; Poyner, D.R.; Briddon, S.J.; Wheatley, M. Single molecule binding of a ligand to a G-protein-coupled receptor in real time using fluorescence correlation spectroscopy, rendered possible by nano-encapsulation in styrene maleic acid lipid particles. Nanoscale 2020, 12, 11518–11525. [Google Scholar] [CrossRef]
- Routledge, S.J.; Jamshad, M.; Little, H.A.; Lin, Y.-P.; Simms, J.; Thakker, A.; Spickett, C.M.; Bill, R.M.; Dafforn, T.R.; Poyner, D.R.; et al. Ligand-induced conformational changes in a SMALP-encapsulated GPCR. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183235. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.T.; Hoi, K.K.; Robinson, C.V. Interfacing Membrane Mimetics with Mass Spectrometry. Acc. Chem. Res. 2016, 49, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Keener, J.E.; Zhang, G.; Marty, M.T. Native Mass Spectrometry of Membrane Proteins. Anal. Chem. 2021, 93, 583–597. [Google Scholar] [CrossRef]
- Young, G.; Hundt, N.; Cole, D.; Fineberg, A.; Andrecka, J.; Tyler, A.; Olerinyova, A.; Ansari, A.; Marklund, E.G.; Collier, M.P.; et al. Quantitative mass imaging of single biological macromolecules. Science 2018, 360, 423–427. [Google Scholar] [CrossRef]
- Olerinyova, A.; Sonn-Segev, A.; Gault, J.; Eichmann, C.; Schimpf, J.; Kopf, A.H.; Rudden, L.S.P.; Ashkinadze, D.; Bomba, R.; Frey, L.; et al. Mass Photometry of Membrane Proteins. Chem 2021, 7, 224–236. [Google Scholar] [CrossRef]
- Schlick, K.H.; Lange, C.K.; Gillispie, G.D.; Cloninger, M.J. Characterization of protein aggregation via intrinsic fluorescence lifetime. J. Am. Chem. Soc. 2009, 131, 16608–16609. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Zhang, M.; Bertheleme, N.; Kara, E.; Strange, P.G.; Byrne, B. Radioligand binding analysis as a tool for quality control of GPCR production for structural characterization: Adenosine A2aR as a template for study. Curr. Protoc. Protein Sci. 2012, 29, 29.3. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ghosh, S.; Jana, S.; Robertson, N.; Tate, C.G.; Vaidehi, N. How Do Branched Detergents Stabilize GPCRs in Micelles? Biochemistry 2020, 59, 2125–2134. [Google Scholar] [CrossRef]
- Holdgate, G.; Embrey, K.; Milbradt, A.; Davies, G. Biophysical methods in early drug discovery. ADMET DMPK 2019, 7, 222–241. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, W.J.C.; Heitman, L.H.; Rosenkilde, M.M. Perspective: Implications of Ligand-Receptor Binding Kinetics for Therapeutic Targeting of G Protein-Coupled Receptors. ACS Pharmacol. Transl. Sci. 2020, 3, 179–189. [Google Scholar] [CrossRef]
- Copeland, R.A. The drug-target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef]
- Guo, D.; Heitman, L.H.; IJzerman, A.P. Kinetic Aspects of the Interaction between Ligand and G Protein-Coupled Receptor: The Case of the Adenosine Receptors. Chem. Rev. 2017, 117, 38–66. [Google Scholar] [CrossRef] [PubMed]
- IJzerman, A.P.; Guo, D. Drug-Target Association Kinetics in Drug Discovery. Trends Biochem. Sci. 2019, 44, 861–871. [Google Scholar] [CrossRef]
- Rich, R.L.; Myszka, D.G. Advances in surface plasmon resonance biosensor analysis. Curr. Opin. Biotechnol. 2000, 11, 54–61. [Google Scholar] [CrossRef]
- Locatelli-Hoops, S.; Yeliseev, A.A.; Gawrisch, K.; Gorshkova, I. Surface plasmon resonance applied to G protein-coupled receptors. Biomed. Spectrosc. Imaging 2013, 2, 155–181. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Surface plasmon resonance spectroscopy for characterisation of membrane protein-ligand interactions and its potential for drug discovery. Biochim. Biophys. Acta 2014, 1838, 43–55. [Google Scholar] [CrossRef]
- Aristotelous, T.; Hopkins, A.L.; Navratilova, I. Surface plasmon resonance analysis of seven-transmembrane receptors. Methods Enzymol. 2015, 556, 499–525. [Google Scholar] [CrossRef]
- Olaru, A.; Bala, C.; Jaffrezic-Renault, N.; Aboul-Enein, H.Y. Surface plasmon resonance (SPR) biosensors in pharmaceutical analysis. Crit. Rev. Anal. Chem. 2015, 45, 97–105. [Google Scholar] [CrossRef]
- Giannetti, A.M. From experimental design to validated hits a comprehensive walk-through of fragment lead identification using surface plasmon resonance. Methods Enzymol. 2011, 493, 169–218. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.L.; Errey, J.; Marshall, F.; Myszka, D.G. Biacore analysis with stabilized G-protein-coupled receptors. Anal. Biochem. 2011, 409, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.A.; Hopkins, A.L.; Navratilova, I. Fragment screening by SPR and advanced application to GPCRs. Prog. Biophys. Mol. Biol. 2014, 116, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.; Jazayeri, A.; Errey, J.; Baig, A.; Hurrell, E.; Zhukov, A.; Langmead, C.J.; Weir, M.; Marshall, F.H. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology 2011, 60, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Rich, R.L.; Myszka, D.G.; Figaroa, F.; Siegal, G.; Marshall, F.H. Fragment screening of stabilized G-protein-coupled receptors using biophysical methods. Methods Enzymol. 2011, 493, 115–136. [Google Scholar] [CrossRef]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef]
- Zhukov, A.; Andrews, S.P.; Errey, J.C.; Robertson, N.; Tehan, B.; Mason, J.S.; Marshall, F.H.; Weir, M.; Congreve, M. Biophysical mapping of the adenosine A2A receptor. J. Med. Chem. 2011, 54, 4312–4323. [Google Scholar] [CrossRef]
- Roos, H.; Karlsson, R.; Nilshans, H.; Persson, A. Thermodynamic analysis of protein interactions with biosensor technology. J. Mol. Recognit. 1998, 11, 204–210. [Google Scholar] [CrossRef]
- Babazada, H.; Alekberli, T.; Hajieva, P.; Farajov, E. Biosensor-based kinetic and thermodynamic characterization of opioids interaction with human μ-opioid receptor. Eur. J. Pharm. Sci. 2019, 138, 105017. [Google Scholar] [CrossRef]
- Deganutti, G.; Zhukov, A.; Deflorian, F.; Federico, S.; Spalluto, G.; Cooke, R.M.; Moro, S.; Mason, J.S.; Bortolato, A. Impact of protein-ligand solvation and desolvation on transition state thermodynamic properties of adenosine A2A ligand binding kinetics. Silico Pharmacol. 2017, 5, 16. [Google Scholar] [CrossRef]
- Coyle, J.; Walser, R. Applied Biophysical Methods in Fragment-Based Drug Discovery. SLAS Discov. 2020, 25, 471–490. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, S.; Zhang, B.; Dai, A.; Cai, X.; Ma, M.; Gao, Z.-G.; Yang, D.; Stevens, R.C.; Jacobson, K.A.; et al. Accelerating the Throughput of Affinity Mass Spectrometry-Based Ligand Screening toward a G Protein-Coupled Receptor. Anal. Chem. 2019, 91, 8162–8169. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Robinson, S.; Berizzi, A.; Thompson, L.E.J.; Bird, L.; Culurgioni, S.; Varzandeh, S.; Rawlins, P.B.; Olsen, R.H.J.; Navratilova, I.H. Surface Plasmon Resonance Screening to Identify Active and Selective Adenosine Receptor Binding Fragments. ACS Med. Chem. Lett. 2022, 13, 1172–1181. [Google Scholar] [CrossRef]
- Koretz, K.S.; McGraw, C.E.; Stradley, S.; Elbaradei, A.; Malmstadt, N.; Robinson, A.S. Characterization of binding kinetics of A2AR to Gαs protein by surface plasmon resonance. Biophys. J. 2021, 120, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Damian, L. Isothermal titration calorimetry for studying protein-ligand interactions. Methods Mol. Biol. 2013, 1008, 103–118. [Google Scholar] [CrossRef]
- Pierce, M.M.; Raman, C.S.; Nall, B.T. Isothermal titration calorimetry of protein-protein interactions. Methods 1999, 19, 213–221. [Google Scholar] [CrossRef]
- Draczkowski, P.; Matosiuk, D.; Jozwiak, K. Isothermal titration calorimetry in membrane protein research. J. Pharm. Biomed. Anal. 2014, 87, 313–325. [Google Scholar] [CrossRef]
- Rajarathnam, K.; Rösgen, J. Isothermal titration calorimetry of membrane proteins-progress and challenges. Biochim. Biophys. Acta 2014, 1838, 69–77. [Google Scholar] [CrossRef]
- Magalhaes, A.C.; Dunn, H.; Ferguson, S.S. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 2012, 165, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Klaasse, E.C.; IJzerman, A.P.; de Grip, W.J.; Beukers, M.W. Internalization and desensitization of adenosine receptors. Purinergic Signal. 2008, 4, 21–37. [Google Scholar] [CrossRef]
- Burgueño, J.; Blake, D.J.; Benson, M.A.; Tinsley, C.L.; Esapa, C.T.; Canela, E.I.; Penela, P.; Mallol, J.; Mayor, F.; Lluis, C.; et al. The adenosine A2A receptor interacts with the actin-binding protein alpha-actinin. J. Biol. Chem. 2003, 278, 37545–37552. [Google Scholar] [CrossRef]
- Woods, A.S.; Marcellino, D.; Jackson, S.N.; Franco, R.; Ferré, S.; Agnati, L.F.; Fuxe, K. How calmodulin interacts with the adenosine A2A and the dopamine D2 receptors. J. Proteome Res. 2008, 7, 3428–3434. [Google Scholar] [CrossRef]
- Milojevic, T.; Reiterer, V.; Stefan, E.; Korkhov, V.M.; Dorostkar, M.M.; Ducza, E.; Ogris, E.; Boehm, S.; Freissmuth, M.; Nanoff, C. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol. Pharmacol. 2006, 69, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Canela, L.; Luján, R.; Lluís, C.; Burgueño, J.; Mallol, J.; Canela, E.I.; Franco, R.; Ciruela, F. The neuronal Ca2+-binding protein 2 (NECAB2) interacts with the adenosine A2A receptor and modulates the cell surface expression and function of the receptor. Mol. Cell. Neurosci. 2007, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-N.; Cheng, H.-C.; Chou, J.-L.; Lee, S.-Y.; Lin, Y.-W.; Lai, H.-L.; Chen, H.-M.; Chern, Y. Rescue of p53 blockage by the A2A adenosine receptor via a novel interacting protein, translin-associated protein X. Mol. Pharmacol. 2006, 70, 454–466. [Google Scholar] [CrossRef]
- Gsandtner, I.; Charalambous, C.; Stefan, E.; Ogris, E.; Freissmuth, M.; Zezula, J. Heterotrimeric G protein-independent signaling of a G protein-coupled receptor. Direct binding of ARNO/cytohesin-2 to the carboxyl terminus of the A2A adenosine receptor is necessary for sustained activation of the ERK/MAP kinase pathway. J. Biol. Chem. 2005, 280, 31898–31905. [Google Scholar] [CrossRef]
- Navarro, G.; Hradsky, J.; Lluís, C.; Casadó, V.; McCormick, P.J.; Kreutz, M.R.; Mikhaylova, M. NCS-1 associates with adenosine A2A receptors and modulates receptor function. Front. Mol. Neurosci. 2012, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Piirainen, H.; Hellman, M.; Tossavainen, H.; Permi, P.; Kursula, P.; Jaakola, V.-P. Human adenosine A2A receptor binds calmodulin with high affinity in a calcium-dependent manner. Biophys. J. 2015, 108, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Piirainen, H.; Taura, J.; Kursula, P.; Ciruela, F.; Jaakola, V.-P. Calcium modulates calmodulin/α-actinin 1 interaction with and agonist-dependent internalization of the adenosine A2A receptor. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 674–686. [Google Scholar] [CrossRef]
- Quast, R.B.; Margeat, E. Studying GPCR conformational dynamics by single molecule fluorescence. Mol. Cell. Endocrinol. 2019, 493, 110469. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Fürstenberg, A.; Huber, T. Labeling and Single-Molecule Methods To Monitor G Protein-Coupled Receptor Dynamics. Chem. Rev. 2017, 117, 186–245. [Google Scholar] [CrossRef]
- Hoffmann, C.; Gaietta, G.; Bünemann, M.; Adams, S.R.; Oberdorff-Maass, S.; Behr, B.; Vilardaga, J.-P.; Tsien, R.Y.; Ellisman, M.H.; Lohse, M.J. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat. Methods 2005, 2, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Kindon, N.D.; Otun, O.; Harwood, C.R.; Patera, F.; Veprintsev, D.B.; Woolard, J.; Briddon, S.J.; Franks, H.A.; Hill, S.J.; et al. Ligand-directed covalent labelling of a GPCR with a fluorescent tag in live cells. Commun. Biol. 2020, 3, 722. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Thakur, N.; Ray, A.P.; Jin, B.; Obeng, S.; McCurdy, C.R.; McMahon, L.R.; Gutiérrez-de-Terán, H.; Eddy, M.T.; Lamichhane, R. Slow conformational dynamics of the human A2A adenosine receptor are temporally ordered. Structure 2022, 30, 329–337.e5. [Google Scholar] [CrossRef]
- Fernandes, D.D.; Neale, C.; Gomes, G.-N.W.; Li, Y.; Malik, A.; Pandey, A.; Orazietti, A.P.; Wang, X.; Ye, L.; Scott Prosser, R.; et al. Ligand modulation of the conformational dynamics of the A2A adenosine receptor revealed by single-molecule fluorescence. Sci. Rep. 2021, 11, 5910. [Google Scholar] [CrossRef] [PubMed]
- Zemella, A.; Richter, T.; Thoring, L.; Kubick, S. A Combined Cell-Free Protein Synthesis and Fluorescence-Based Approach to Investigate GPCR Binding Properties. Methods Mol. Biol. 2019, 1947, 57–77. [Google Scholar] [CrossRef]
- Raingeval, C.; Krimm, I. NMR investigation of protein-ligand interactions for G-protein coupled receptors. Future Med. Chem. 2019, 11, 1811–1825. [Google Scholar] [CrossRef]
- Stauch, B.; Orts, J.; Carlomagno, T. The description of protein internal motions aids selection of ligand binding poses by the INPHARMA method. J. Biomol. NMR 2012, 54, 245–256. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Hoi, K.K.; Liko, I.; Hedger, G.; Horrell, M.R.; Song, W.; Wu, D.; Heine, P.; Warne, T.; Lee, Y.; et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 2018, 559, 423–427. [Google Scholar] [CrossRef]
- Agasid, M.T.; Sørensen, L.; Urner, L.H.; Yan, J.; Robinson, C.V. The Effects of Sodium Ions on Ligand Binding and Conformational States of G Protein-Coupled Receptors-Insights from Mass Spectrometry. J. Am. Chem. Soc. 2021, 143, 4085–4089. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.H.J.; English, J.G. Advancements in G protein-coupled receptor biosensors to study GPCR-G protein coupling. Br. J. Pharmacol. 2022. [Google Scholar] [CrossRef]
- Zhou, Y.; Meng, J.; Xu, C.; Liu, J. Multiple GPCR Functional Assays Based on Resonance Energy Transfer Sensors. Front. Cell Dev. Biol. 2021, 9, 611443. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. Principles: Extending the utility of [35S]GTPγS binding assays. Trends Pharmacol. Sci. 2003, 24, 87–90. [Google Scholar] [CrossRef]
- McEwen, D.P.; Gee, K.R.; Kang, H.C.; Neubig, R.R. Fluorescent BODIPY-GTP analogs: Real-time measurement of nucleotide binding to G proteins. Anal. Biochem. 2001, 291, 109–117. [Google Scholar] [CrossRef]
- Ravyn, V.; Bostwick, J.R. Functional coupling of the Galpha(olf) variant XLGalpha(olf) with the human adenosine A2A receptor. J. Recept. Signal Transduct. Res. 2006, 26, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Beckner, R.L.; Zoubak, L.; Hines, K.G.; Gawrisch, K.; Yeliseev, A.A. Probing thermostability of detergent-solubilized CB2 receptor by parallel G protein-activation and ligand-binding assays. J. Biol. Chem. 2020, 295, 181–190. [Google Scholar] [CrossRef]
- Arcemisbéhère, L.; Sen, T.; Boudier, L.; Balestre, M.-N.; Gaibelet, G.; Detouillon, E.; Orcel, H.; Mendre, C.; Rahmeh, R.; Granier, S.; et al. Leukotriene BLT2 receptor monomers activate the G(i2) GTP-binding protein more efficiently than dimers. J. Biol. Chem. 2010, 285, 6337–6347. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Ghirlando, R.; White, J.F.; Gvozdenovic-Jeremic, J.; Northup, J.K.; Grisshammer, R. Modulation of the interaction between neurotensin receptor NTS1 and Gq protein by lipid. J. Mol. Biol. 2012, 417, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Damian, M.; Marie, J.; Leyris, J.-P.; Fehrentz, J.-A.; Verdié, P.; Martinez, J.; Banères, J.-L.; Mary, S. High constitutive activity is an intrinsic feature of ghrelin receptor protein: A study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J. Biol. Chem. 2012, 287, 3630–3641. [Google Scholar] [CrossRef]
- Gregorio, G.G.; Masureel, M.; Hilger, D.; Terry, D.S.; Juette, M.; Zhao, H.; Zhou, Z.; Perez-Aguilar, J.M.; Hauge, M.; Mathiasen, S.; et al. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 2017, 547, 68–73. [Google Scholar] [CrossRef]
- Damian, M.; Louet, M.; Gomes, A.A.S.; M’Kadmi, C.; Denoyelle, S.; Cantel, S.; Mary, S.; Bisch, P.M.; Fehrentz, J.-A.; Catoire, L.J.; et al. Allosteric modulation of ghrelin receptor signaling by lipids. Nat. Commun. 2021, 12, 3938. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Vishnivetskiy, S.A.; McLean, M.A.; Morizumi, T.; Huang, C.-C.; Tesmer, J.J.G.; Ernst, O.P.; Sligar, S.G.; Gurevich, V.V. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J. Biol. Chem. 2011, 286, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Baidya, M.; Dwivedi-Agnihotri, H.; Shukla, A.K. Site-directed labeling of β-arrestin with monobromobimane for measuring their interaction with G protein-coupled receptors. Methods Enzymol. 2020, 633, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.E.; Farrens, D.L. Arrestin can act as a regulator of rhodopsin photochemistry. Vis. Res. 2006, 46, 4532–4546. [Google Scholar] [CrossRef] [PubMed]
- Rahmeh, R.; Damian, M.; Cottet, M.; Orcel, H.; Mendre, C.; Durroux, T.; Sharma, K.S.; Durand, G.; Pucci, B.; Trinquet, E.; et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 6733–6738. [Google Scholar] [CrossRef]
- Kumari, P.; Srivastava, A.; Banerjee, R.; Ghosh, E.; Gupta, P.; Ranjan, R.; Chen, X.; Gupta, B.; Gupta, C.; Jaiman, D.; et al. Functional competence of a partially engaged GPCR-β-arrestin complex. Nat. Commun. 2016, 7, 13416. [Google Scholar] [CrossRef]
- Staus, D.P.; Wingler, L.M.; Choi, M.; Pani, B.; Manglik, A.; Kruse, A.C.; Lefkowitz, R.J. Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in β-arrestin coupling. Proc. Natl. Acad. Sci. USA 2018, 115, 3834–3839. [Google Scholar] [CrossRef]
- Prosser, R.S.; Ye, L.; Pandey, A.; Orazietti, A. Activation processes in ligand-activated G protein-coupled receptors: A case study of the adenosine A2A receptor. Bioessays 2017, 39, 1700072. [Google Scholar] [CrossRef] [PubMed]
- Welihinda, A.A.; Kaur, M.; Greene, K.; Zhai, Y.; Amento, E.P. The adenosine metabolite inosine is a functional agonist of the adenosine A2A receptor with a unique signaling bias. Cell. Signal. 2016, 28, 552–560. [Google Scholar] [CrossRef]
- Kolb, P.; Kenakin, T.; Alexander, S.P.H.; Bermudez, M.; Bohn, L.M.; Breinholt, C.S.; Bouvier, M.; Hill, S.J.; Kostenis, E.; Martemyanov, K.A.; et al. Community guidelines for GPCR ligand bias: IUPHAR review 32. Br. J. Pharmacol. 2022, 179, 3651–3674. [Google Scholar] [CrossRef]
- Benleulmi-Chaachoua, A.; Chen, L.; Sokolina, K.; Wong, V.; Jurisica, I.; Emerit, M.B.; Darmon, M.; Espin, A.; Stagljar, I.; Tafelmeyer, P.; et al. Protein interactome mining defines melatonin MT1 receptors as integral component of presynaptic protein complexes of neurons. J. Pineal Res. 2016, 60, 95–108. [Google Scholar] [CrossRef]
- Overton, M.C.; Blumer, K.J. The extracellular N-terminal domain and transmembrane domains 1 and 2 mediate oligomerization of a yeast G protein-coupled receptor. J. Biol. Chem. 2002, 277, 41463–41472. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, G.; Jaiswal, P.; Dey, S.; Sengupta, J.; Mukherjee, S. Cloning, Expression, Purification and Characterization of Oligomeric States of the Native 5HT2A G-Protein-Coupled Receptor. Protein Pept. Lett. 2018, 25, 390–397. [Google Scholar] [CrossRef]
- Fernández-Dueñas, V.; Bonaventura, J.; Aso, E.; Luján, R.; Ferré, S.; Ciruela, F. Overcoming the Challenges of Detecting GPCR Oligomerization in the Brain. Curr. Neuropharmacol. 2022, 20, 1035–1045. [Google Scholar] [CrossRef]
- Mansoor, S.; Kayık, G.; Durdagi, S.; Sensoy, O. Mechanistic insight into the impact of a bivalent ligand on the structure and dynamics of a GPCR oligomer. Comput. Struct. Biotechnol. J. 2022, 20, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Canals, M.; Burgueño, J.; Marcellino, D.; Cabello, N.; Canela, E.I.; Mallol, J.; Agnati, L.; Ferré, S.; Bouvier, M.; Fuxe, K.; et al. Homodimerization of adenosine A2A receptors: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Neurochem. 2004, 88, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Franco, R.; Cordomí, A.; Del Llinas Torrent, C.; Lillo, A.; Serrano-Marín, J.; Navarro, G.; Pardo, L. Structure and function of adenosine receptor heteromers. Cell. Mol. Life Sci. 2021, 78, 3957–3968. [Google Scholar] [CrossRef] [PubMed]
- Carriba, P.; Ortiz, O.; Patkar, K.; Justinova, Z.; Stroik, J.; Themann, A.; Müller, C.; Woods, A.S.; Hope, B.T.; Ciruela, F.; et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology 2007, 32, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Hillion, J.; Canals, M.; Torvinen, M.; Casado, V.; Scott, R.; Terasmaa, A.; Hansson, A.; Watson, S.; Olah, M.E.; Mallol, J.; et al. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J. Biol. Chem. 2002, 277, 18091–18097. [Google Scholar] [CrossRef] [PubMed]
- Canals, M.; Marcellino, D.; Fanelli, F.; Ciruela, F.; de Benedetti, P.; Goldberg, S.R.; Neve, K.; Fuxe, K.; Agnati, L.F.; Woods, A.S.; et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 2003, 278, 46741–46749. [Google Scholar] [CrossRef]
- Kamiya, T.; Saitoh, O.; Yoshioka, K.; Nakata, H. Oligomerization of adenosine A2A and dopamine D2 receptors in living cells. Biochem. Biophys. Res. Commun. 2003, 306, 544–549. [Google Scholar] [CrossRef]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A receptor and dopamine D3 receptor interactions: Evidence of functional A2A/D3 heteromeric complexes. Mol. Pharmacol. 2005, 67, 400–407. [Google Scholar] [CrossRef]
- Cabello, N.; Gandía, J.; Bertarelli, D.C.G.; Watanabe, M.; Lluís, C.; Franco, R.; Ferré, S.; Luján, R.; Ciruela, F. Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J. Neurochem. 2009, 109, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Trifilieff, P.; Rives, M.-L.; Urizar, E.; Piskorowski, R.A.; Vishwasrao, H.D.; Castrillon, J.; Schmauss, C.; Slättman, M.; Gullberg, M.; Javitch, J.A. Detection of antigen interactions ex vivo by proximity ligation assay: Endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. Biotechniques 2011, 51, 111–118. [Google Scholar] [CrossRef]
- Fernández-Dueñas, V.; Gómez-Soler, M.; Valle-León, M.; Watanabe, M.; Ferrer, I.; Ciruela, F. Revealing Adenosine A2A-Dopamine D2 Receptor Heteromers in Parkinson’s Disease Post-Mortem Brain through a New AlphaScreen-Based Assay. Int. J. Mol. Sci. 2019, 20, 3600. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Antonelli, T.; Tomasini, M.C.; Borelli, A.C.; Agnati, L.F.; Tanganelli, S.; Fuxe, K.; Ferraro, L. Adenosine A2A-D2 receptor-receptor interactions in putative heteromers in the regulation of the striato-pallidal gaba pathway: Possible relevance for parkinson’s disease and its treatment. Curr. Protein Pept. Sci. 2014, 15, 673–680. [Google Scholar] [CrossRef]
- Ferré, S.; Bonaventura, J.; Tomasi, D.; Navarro, G.; Moreno, E.; Cortés, A.; Lluís, C.; Casadó, V.; Volkow, N.D. Allosteric mechanisms within the adenosine A2A-dopamine D2 receptor heterotetramer. Neuropharmacology 2016, 104, 154–160. [Google Scholar] [CrossRef]
- Romero-Fernandez, W.; Taura, J.J.; Crans, R.A.J.; Lopez-Cano, M.; Fores-Pons, R.; Narváez, M.; Carlsson, J.; Ciruela, F.; Fuxe, K.; Borroto-Escuela, D.O. The mGlu5 Receptor Protomer-Mediated Dopamine D2 Receptor Trans-Inhibition Is Dependent on the Adenosine A2A Receptor Protomer: Implications for Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 5955–5969. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.A.; Denisov, I.G.; Sligar, S.G. Nanodiscs as a new tool to examine lipid-protein interactions. Methods Mol. Biol. 2013, 974, 415–433. [Google Scholar] [CrossRef] [PubMed]
- White, J.F.; Grodnitzky, J.; Louis, J.M.; Trinh, L.B.; Shiloach, J.; Gutierrez, J.; Northup, J.K.; Grisshammer, R. Dimerization of the class A G protein-coupled neurotensin receptor NTS1 alters G protein interaction. Proc. Natl. Acad. Sci. USA 2007, 104, 12199–12204. [Google Scholar] [CrossRef] [PubMed]
- Bayburt, T.H.; Leitz, A.J.; Xie, G.; Oprian, D.D.; Sligar, S.G. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J. Biol. Chem. 2007, 282, 14875–14881. [Google Scholar] [CrossRef]
- El Moustaine, D.; Granier, S.; Doumazane, E.; Scholler, P.; Rahmeh, R.; Bron, P.; Mouillac, B.; Banères, J.-L.; Rondard, P.; Pin, J.-P. Distinct roles of metabotropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proc. Natl. Acad. Sci. USA 2012, 109, 16342–16347. [Google Scholar] [CrossRef]
- Mary, S.; Fehrentz, J.-A.; Damian, M.; Gaibelet, G.; Orcel, H.; Verdié, P.; Mouillac, B.; Martinez, J.; Marie, J.; Banères, J.-L. Heterodimerization with Its splice variant blocks the ghrelin receptor 1a in a non-signaling conformation: A study with a purified heterodimer assembled into lipid discs. J. Biol. Chem. 2013, 288, 24656–24665. [Google Scholar] [CrossRef]
- Mesnier, D.; Banères, J.-L. Cooperative conformational changes in a G-protein-coupled receptor dimer, the leukotriene B(4) receptor BLT1. J. Biol. Chem. 2004, 279, 49664–49670. [Google Scholar] [CrossRef]
- Damian, M.; Pons, V.; Renault, P.; M’Kadmi, C.; Delort, B.; Hartmann, L.; Kaya, A.I.; Louet, M.; Gagne, D.; Ben Haj Salah, K.; et al. GHSR-D2R heteromerization modulates dopamine signaling through an effect on G protein conformation. Proc. Natl. Acad. Sci. USA 2018, 115, 4501–4506. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Sinha, A.; DeWitt, M.; Farrens, D.L. Monomeric rhodopsin is the minimal functional unit required for arrestin binding. J. Mol. Biol. 2010, 399, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Piirainen, H.; Ashok, Y.; Nanekar, R.T.; Jaakola, V.-P. Structural features of adenosine receptors: From crystal to function. Biochim. Biophys. Acta 2011, 1808, 1233–1244. [Google Scholar] [CrossRef]
- Langelaan, D.N.; Ngweniform, P.; Rainey, J.K. Biophysical characterization of G-protein coupled receptor-peptide ligand binding. Biochem. Cell Biol. 2011, 89, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Cianférani, S. Towards integrative structural mass spectrometry: Benefits from hybrid approaches. Methods 2015, 89, 4–12. [Google Scholar] [CrossRef]
- Dafun, A.S.; Marcoux, J. Structural mass spectrometry of membrane proteins. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140813. [Google Scholar] [CrossRef] [PubMed]
- Khanal, A.; Pan, Y.; Brown, L.S.; Konermann, L. Pulsed hydrogen/deuterium exchange mass spectrometry for time-resolved membrane protein folding studies. J. Mass Spectrom. 2012, 47, 1620–1626. [Google Scholar] [CrossRef]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef]
- Möller, I.R.; Slivacka, M.; Hausner, J.; Nielsen, A.K.; Pospíšilová, E.; Merkle, P.S.; Lišková, R.; Polák, M.; Loland, C.J.; Kádek, A.; et al. Improving the Sequence Coverage of Integral Membrane Proteins during Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. Anal. Chem. 2019, 91, 10970–10978. [Google Scholar] [CrossRef]
- Trabjerg, E.; Nazari, Z.E.; Rand, K.D. Conformational analysis of complex protein states by hydrogen/deuterium exchange mass spectrometry (HDX-MS): Challenges and emerging solutions. TrAC Trends Anal. Chem. 2018, 106, 125–138. [Google Scholar] [CrossRef]
- Martens, C.; Politis, A. A glimpse into the molecular mechanism of integral membrane proteins through hydrogen-deuterium exchange mass spectrometry. Protein Sci. 2020, 29, 1285–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rempel, D.L.; Zhang, J.; Sharma, A.K.; Mirica, L.M.; Gross, M.L. Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 14604–14609. [Google Scholar] [CrossRef]
- Du, Y.; Duc, N.M.; Rasmussen, S.G.F.; Hilger, D.; Kubiak, X.; Wang, L.; Bohon, J.; Kim, H.R.; Wegrecki, M.; Asuru, A.; et al. Assembly of a GPCR-G Protein Complex. Cell 2019, 177, 1232–1242.e11. [Google Scholar] [CrossRef]
- Konermann, L.; Tong, X.; Pan, Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J. Mass Spectrom. 2008, 43, 1021–1036. [Google Scholar] [CrossRef]
- Wang, L.; Chance, M.R. Protein Footprinting Comes of Age: Mass Spectrometry for Biophysical Structure Assessment. Mol. Cell. Proteom. 2017, 16, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, C.; Piotrowski, C.; Aebersold, R.; Amaral, B.C.; Andrews, P.; Bernfur, K.; Borchers, C.; Brodie, N.I.; Bruce, J.E.; Cao, Y.; et al. First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal. Chem. 2019, 91, 6953–6961. [Google Scholar] [CrossRef]
- Steigenberger, B.; Albanese, P.; Heck, A.J.R.; Scheltema, R.A. To Cleave or Not To Cleave in XL-MS? J. Am. Soc. Mass Spectrom. 2020, 31, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Ma, Z.; Tong, J.; Tang, Y.; Li, S.; Qin, S.; Lou, R.; Zhao, S.; Lei, X.; Shui, W. Evaluation of chemical cross-linkers for in-depth structural analysis of G protein-coupled receptors through cross-linking mass spectrometry. Anal. Chim. Acta 2020, 1102, 53–62. [Google Scholar] [CrossRef]
- Jones, A.X.; Cao, Y.; Tang, Y.-L.; Wang, J.-H.; Ding, Y.-H.; Tan, H.; Chen, Z.-L.; Fang, R.-Q.; Yin, J.; Chen, R.-C.; et al. Improving mass spectrometry analysis of protein structures with arginine-selective chemical cross-linkers. Nat. Commun. 2019, 10, 3911. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Yao, X.-J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef]
- Magnani, F.; Serrano-Vega, M.J.; Shibata, Y.; Abdul-Hussein, S.; Lebon, G.; Miller-Gallacher, J.; Singhal, A.; Strege, A.; Thomas, J.A.; Tate, C.G. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016, 11, 1554–1571. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Rubio, P.; Lieberman, D.I.; Jacobson, K.A. A3 adenosine receptor activation mechanisms: Molecular dynamics analysis of inactive, active, and fully active states. J. Comput. Aided Mol. Des. 2019, 33, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A.; Cooke, R.M.; Marshall, F.H.; Doré, A.S. Towards high throughput GPCR crystallography: In Meso soaking of Adenosine A2A Receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef]
- García-Nafría, J.; Lee, Y.; Bai, X.; Carpenter, B.; Tate, C.G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 2018, 7, e35946. [Google Scholar] [CrossRef]
- Amelia, T.; van Veldhoven, J.P.D.; Falsini, M.; Liu, R.; Heitman, L.H.; van Westen, G.J.P.; Segala, E.; Verdon, G.; Cheng, R.K.Y.; Cooke, R.M.; et al. Crystal Structure and Subsequent Ligand Design of a Nonriboside Partial Agonist Bound to the Adenosine A2A Receptor. J. Med. Chem. 2021, 64, 3827–3842. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Heydenreich, F.M.; Flock, T.; Miljus, T.; Balaji, S.; Bouvier, M.; Veprintsev, D.B.; Tate, C.G.; et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 2016, 536, 484–487. [Google Scholar] [CrossRef]
- Shiriaeva, A.; Park, D.; Kim, G.; Lee, Y.; Hou, X.; Jarhad, D.B.; Kim, G.; Yu, J.; Hyun, Y.E.; Kim, W.; et al. GPCR Agonist-to-Antagonist Conversion: Enabling the Design of Nucleoside Functional Switches for the A2A Adenosine Receptor. J. Med. Chem. 2022, 65, 11648–11657. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Bachhawat, P.; Chu, M.L.-H.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef]
- Martin-Garcia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.; Olieric, N.; Cheng, R.; Brünle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef]
- Martin-Garcia, J.M.; Zhu, L.; Mendez, D.; Lee, M.-Y.; Chun, E.; Li, C.; Hu, H.; Subramanian, G.; Kissick, D.; Ogata, C.; et al. High-viscosity injector-based pink-beam serial crystallography of microcrystals at a synchrotron radiation source. IUCrJ 2019, 6, 412–425. [Google Scholar] [CrossRef]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, Y.; Tono, K.; Tanaka, T.; Yamanaka, Y.; Nakane, T.; Mori, C.; Terakado Kimura, K.; Fujiwara, T.; Sugahara, M.; Tanaka, R.; et al. High-viscosity sample-injection device for serial femtosecond crystallography at atmospheric pressure. J. Appl. Crystallogr. 2019, 52, 1280–1288. [Google Scholar] [CrossRef]
- Ihara, K.; Hato, M.; Nakane, T.; Yamashita, K.; Kimura-Someya, T.; Hosaka, T.; Ishizuka-Katsura, Y.; Tanaka, R.; Tanaka, T.; Sugahara, M.; et al. Isoprenoid-chained lipid EROCOC17+4: A new matrix for membrane protein crystallization and a crystal delivery medium in serial femtosecond crystallography. Sci. Rep. 2020, 10, 19305. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Structure determination of GPCRs: Cryo-EM compared with X-ray crystallography. Biochem. Soc. Trans. 2021, 49, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Vénien-Bryan, C.; Li, Z.; Vuillard, L.; Boutin, J.A. Cryo-electron microscopy and X-ray crystallography: Complementary approaches to structural biology and drug discovery. Acta Crystallogr. F Struct. Biol. Commun. 2017, 73, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Boutin, J.A.; Li, Z.; Vuillard, L.; Vénien-Bryan, C. La cryo-microscopie, une alternative à la cristallographie aux rayons X? Med. Sci. 2016, 32, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G.; Battisti, A.J.; Plevka, P. Future prospects. Adv. Protein Chem. Struct. Biol. 2011, 82, 101–121. [Google Scholar] [CrossRef]
- Blundell, T.L.; Chaplin, A.K. The resolution revolution in X-ray diffraction, Cryo-EM and other Technologies. Prog. Biophys. Mol. Biol. 2021, 160, 2–4. [Google Scholar] [CrossRef]
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Khoshouei, M.; Radjainia, M.; Zhang, Y.; Glukhova, A.; Tarrasch, J.; Thal, D.M.; Furness, S.G.B.; Christopoulos, G.; Coudrat, T.; et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 2017, 546, 118–123. [Google Scholar] [CrossRef]
- Martynowycz, M.W.; Shiriaeva, A.; Ge, X.; Hattne, J.; Nannenga, B.L.; Cherezov, V.; Gonen, T. MicroED structure of the human adenosine receptor determined from a single nanocrystal in LCP. Proc. Natl. Acad. Sci. USA 2021, 118, e2106041118. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef]
- Kitevski-LeBlanc, J.L.; Prosser, R.S. Current applications of 19F NMR to studies of protein structure and dynamics. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 62, 1–33. [Google Scholar] [CrossRef]
- Picard, L.-P.; Prosser, R.S. Advances in the study of GPCRs by 19F NMR. Curr. Opin. Struct. Biol. 2021, 69, 169–176. [Google Scholar] [CrossRef]
- Liu, D.; Wüthrich, K. Ring current shifts in (19)F-NMR of membrane proteins. J. Biomol. NMR 2016, 65, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hagn, F.; Etzkorn, M.; Raschle, T.; Wagner, G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135, 1919–1925. [Google Scholar] [CrossRef]
- Pozza, A.; Giraud, F.; Cece, Q.; Casiraghi, M.; Point, E.; Damian, M.; Le Bon, C.; Moncoq, K.; Banères, J.-L.; Lescop, E.; et al. Exploration of the dynamic interplay between lipids and membrane proteins by hydrostatic pressure. Nat. Commun. 2022, 13, 1780. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Damian, M.; Lescop, E.; Point, E.; Moncoq, K.; Morellet, N.; Levy, D.; Marie, J.; Guittet, E.; Banères, J.-L.; et al. Functional Modulation of a G Protein-Coupled Receptor Conformational Landscape in a Lipid Bilayer. J. Am. Chem. Soc. 2016, 138, 11170–11175. [Google Scholar] [CrossRef]
- Nygaard, R.; Frimurer, T.M.; Holst, B.; Rosenkilde, M.M.; Schwartz, T.W. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol. Sci. 2009, 30, 249–259. [Google Scholar] [CrossRef]
- Eddy, M.T.; Gao, Z.-G.; Mannes, P.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Extrinsic Tryptophans as NMR Probes of Allosteric Coupling in Membrane Proteins: Application to the A2A Adenosine Receptor. J. Am. Chem. Soc. 2018, 140, 8228–8235. [Google Scholar] [CrossRef]
- Eddy, M.T.; Martin, B.T.; Wüthrich, K. A2A Adenosine Receptor Partial Agonism Related to Structural Rearrangements in an Activation Microswitch. Structure 2021, 29, 170–176.e3. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Damian, M.; Lescop, E.; Banères, J.-L.; Catoire, L.J. Illuminating the Energy Landscape of GPCRs: The Key Contribution of Solution-State NMR Associated with Escherichia coli as an Expression Host. Biochemistry 2018, 57, 2297–2307. [Google Scholar] [CrossRef]
- Sekhar, A.; Kay, L.E. NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc. Natl. Acad. Sci. USA 2013, 110, 12867–12874. [Google Scholar] [CrossRef]
- Gunkel, M.; Schöneberg, J.; Alkhaldi, W.; Irsen, S.; Noé, F.; Kaupp, U.B.; Al-Amoudi, A. Higher-order architecture of rhodopsin in intact photoreceptors and its implication for phototransduction kinetics. Structure 2015, 23, 628–638. [Google Scholar] [CrossRef]
- Fotiadis, D.; Liang, Y.; Filipek, S.; Saperstein, D.A.; Engel, A.; Palczewski, K. Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature 2003, 421, 127–128. [Google Scholar] [CrossRef]
- Sapra, K.T.; Spoerri, P.M.; Engel, A.; Alsteens, D.; Müller, D.J. Seeing and sensing single G protein-coupled receptors by atomic force microscopy. Curr. Opin. Cell Biol. 2019, 57, 25–32. [Google Scholar] [CrossRef] [PubMed]
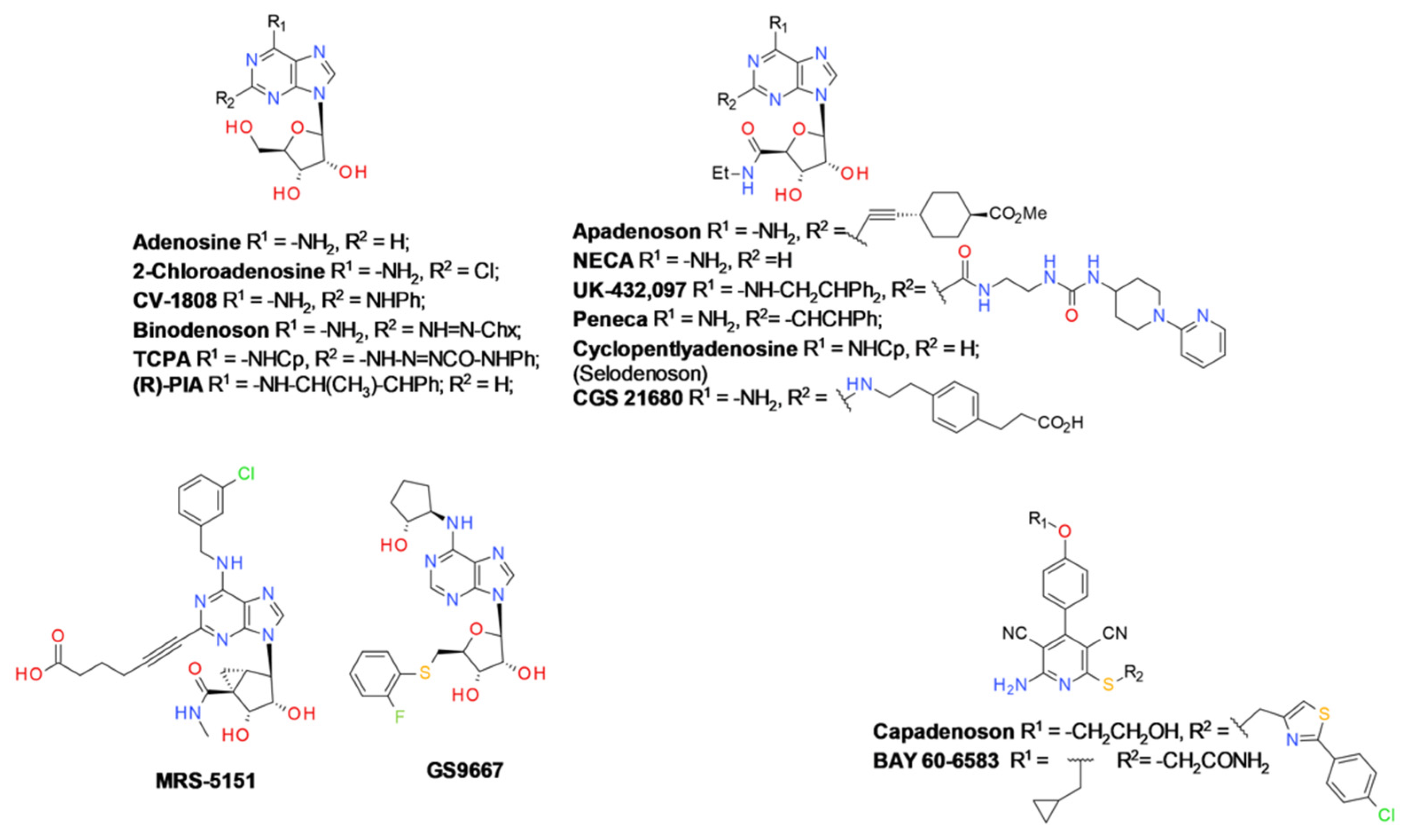
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.-G. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef]
- Melman, A.; Gao, Z.-G.; Kumar, D.; Wan, T.C.; Gizewski, E.; Auchampach, J.A.; Jacobson, K.A. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg. Med. Chem. Lett. 2008, 18, 2813–2819. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Jacobson, K.A. Molecular modeling of a PAMAM-CGS21680 dendrimer bound to an A2A adenosine receptor homodimer. Bioorg. Med. Chem. Lett. 2008, 18, 4312–4315. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Lambertucci, C.; Buccioni, M.; Martí Navia, A.; Marucci, G.; Spinaci, A.; Volpini, R. Non-Nucleoside Agonists of the Adenosine Receptors: An Overview. Pharmaceuticals 2019, 12, 150. [Google Scholar] [CrossRef] [PubMed]
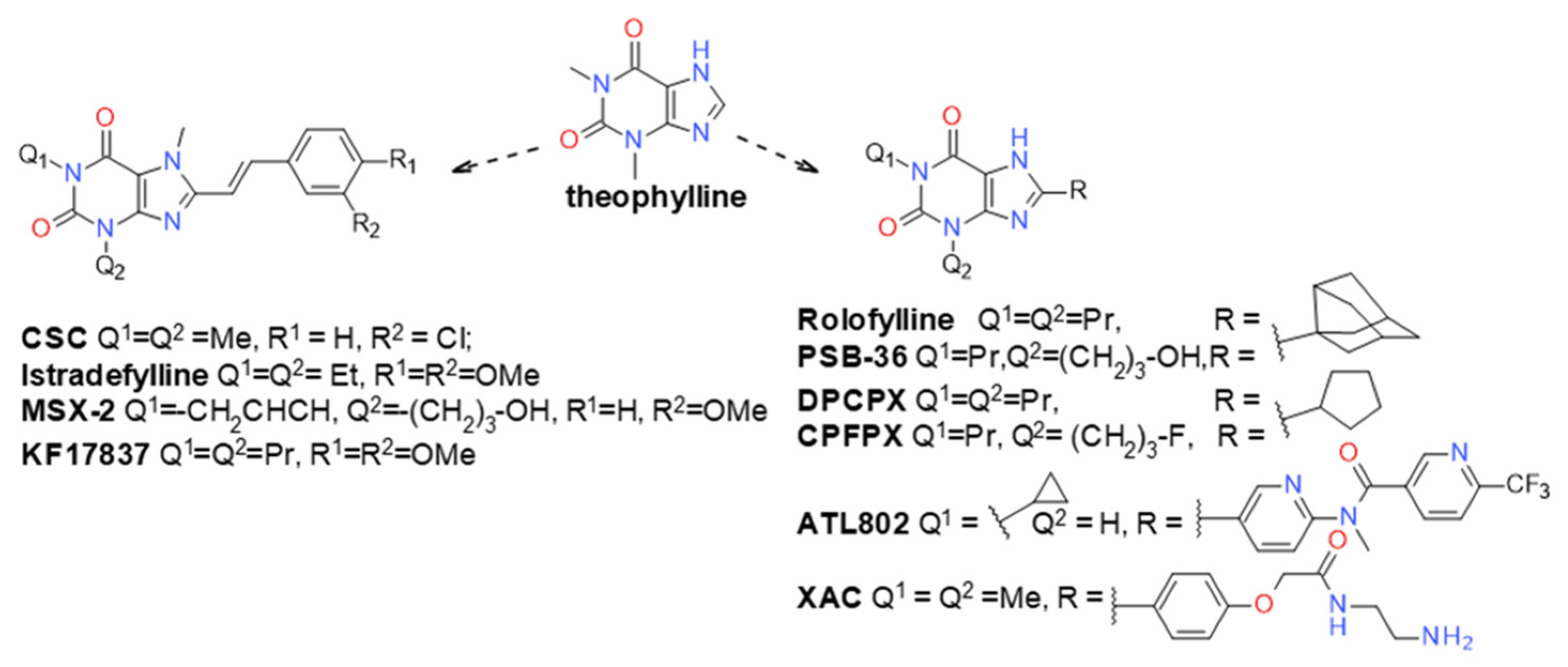
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 200, 151–199. [Google Scholar] [CrossRef]
- Ukena, D.; Padgett, W.L.; Hong, O.; Daly, J.W.; Daly, D.T.; Olsson, R.A. N6-substituted 9-methyladenines: A new class of adenosine receptor antagonists. FEBS Lett. 1987, 215, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Khanapur, S.; Huizing, A.P.; Zijlma, R.; Schepers, M.; Dierckx, R.A.J.O.; van Waarde, A.; de Vries, E.F.J.; Elsinga, P.H. Synthesis and preclinical evaluation of 2-(2-furanyl)-7-2-4-4-(2-11Cmethoxyethoxy)phenyl-1-piperazinylethyl7H-pyrazolo4,3-e1,2,4triazolo1,5-cpyrimidine-5-amine (11CPreladenant) as a PET tracer for the imaging of cerebral adenosine A2A receptors. J. Med. Chem. 2014, 57, 9204–9210. [Google Scholar] [CrossRef]
- Shinkre, B.A.; Kumar, T.S.; Gao, Z.-G.; Deflorian, F.; Jacobson, K.A.; Trenkle, W.C. Synthesis and evaluation of 1,2,4-triazolo1,5-cpyrimidine derivatives as A2A receptor-selective antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 5690–5694. [Google Scholar] [CrossRef] [PubMed]
- Todde, S.; Moresco, R.M.; Simonelli, P.; Baraldi, P.G.; Cacciari, B.; Spalluto, G.; Varani, K.; Monopoli, A.; Matarrese, M.; Carpinelli, A.; et al. Design, radiosynthesis, and biodistribution of a new potent and selective ligand for in vivo imaging of the adenosine A2A receptor system using positron emission tomography. J. Med. Chem. 2000, 43, 4359–4362. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, C.; Ongini, E.; Conti, A.; Monopoli, A.; Negretti, A.; Baraldi, P.G.; Dionisotti, S. The non-xanthine heterocyclic compound SCH 58261 is a new potent and selective A2a adenosine receptor antagonist. J. Pharmacol. Exp. Ther. 1996, 276, 398–404. [Google Scholar] [PubMed]
- Marques, T.R.; Natesan, S.; Rabiner, E.A.; Searle, G.E.; Gunn, R.; Howes, O.D.; Kapur, S. Adenosine A2A receptor in schizophrenia: An in vivo brain PET imaging study. Psychopharmacology 2021, 239, 3439–3445. [Google Scholar] [CrossRef]
- Boutin, J.A.; Witt-Enderby, P.A.; Sotriffer, C.; Zlotos, D.P. Melatonin receptor ligands: A pharmaco-chemical perspective. J. Pineal Res. 2020, 69, e12672. [Google Scholar] [CrossRef]
- Bojarski, A.J. Pharmacophore models for metabotropic 5-HT receptor ligands. Curr. Top. Med. Chem. 2006, 6, 2005–2026. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Jacobson, K.A. Allosteric modulation and functional selectivity of G protein-coupled receptors. Drug Discov. Today Technol. 2013, 10, e237–e243. [Google Scholar] [CrossRef]
- Lütjens, R.; Rocher, J.-P. Recent advances in drug discovery of GPCR allosteric modulators for neurodegenerative disorders. Curr. Opin. Pharmacol. 2017, 32, 91–95. [Google Scholar] [CrossRef]
- van der Westhuizen, E.T.; Valant, C.; Sexton, P.M.; Christopoulos, A. Endogenous allosteric modulators of G protein-coupled receptors. J. Pharmacol. Exp. Ther. 2015, 353, 246–260. [Google Scholar] [CrossRef]
- Madsen, J.J.; Ye, L.; Frimurer, T.M.; Olsen, O.H. Mechanistic basis of GPCR activation explored by ensemble refinement of crystallographic structures. Protein Sci. 2022, 31, e4456. [Google Scholar] [CrossRef]
- Swaminath, G.; Lee, T.W.; Kobilka, B. Identification of an allosteric binding site for Zn2+ on the beta2 adrenergic receptor. J. Biol. Chem. 2003, 278, 352–356. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, H.; Yang, D.; Zhong, L.; Xin, Y.; Zhao, S.; Wang, M.-W.; Zhou, Q.; Shui, W. Affinity Mass Spectrometry-Based Fragment Screening Identified a New Negative Allosteric Modulator of the Adenosine A2A Receptor Targeting the Sodium Ion Pocket. ACS Chem. Biol. 2021, 16, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Korkutata, M.; Saitoh, T.; Cherasse, Y.; Ioka, S.; Duo, F.; Qin, R.; Murakoshi, N.; Fujii, S.; Zhou, X.; Sugiyama, F.; et al. Enhancing endogenous adenosine A2A receptor signaling induces slow-wave sleep without affecting body temperature and cardiovascular function. Neuropharmacology 2019, 144, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Welihinda, A.A.; Amento, E.P. Positive allosteric modulation of the adenosine A2a receptor attenuates inflammation. J. Inflamm. 2014, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Welihinda, A.A.; Kaur, M.; Raveendran, K.S.; Amento, E.P. Enhancement of inosine-mediated A2AR signaling through positive allosteric modulation. Cell. Signal. 2018, 42, 227–235. [Google Scholar] [CrossRef]
- Salovich, J.M.; Sheffler, D.J.; Vinson, P.N.; Lamsal, A.; Utley, T.J.; Blobaum, A.L.; Bridges, T.M.; Le, U.; Jones, C.K.; Wood, M.R.; et al. Probe Reports from the NIH Molecular Libraries Program: Discovery of a Novel Structural Class of M4 Positive Allosteric Modulators: Characterization of ML293, N-(4-Methoxy-7-Methylbenzodthiazol-2-yl)isonicotinamide, with CNS Exposure in Rats; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010.
- Jones, C.K.; Bubser, M.; Thompson, A.D.; Dickerson, J.W.; Turle-Lorenzo, N.; Amalric, M.; Blobaum, A.L.; Bridges, T.M.; Morrison, R.D.; Jadhav, S.; et al. The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with L-DOPA or an adenosine 2A antagonist in preclinical rodent models of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2012, 340, 404–421. [Google Scholar] [CrossRef] [PubMed]
- Pinhal-Enfield, G.; Ramanathan, M.; Hasko, G.; Vogel, S.N.; Salzman, A.L.; Boons, G.-J.; Leibovich, S.J. An Angiogenic Switch in Macrophages Involving Synergy between Toll-Like Receptors 2, 4, 7, and 9 and Adenosine A2A Receptors. Am. J. Pathol. 2003, 163, 711–721. [Google Scholar] [CrossRef]
- Jung, S.-M.; Peyton, L.; Essa, H.; Choi, D.-S. Adenosine receptors: Emerging non-opioids targets for pain medications. Neurobiol. Pain 2022, 11, 100087. [Google Scholar] [CrossRef]
- Federico, S.; Lassiani, L.; Spalluto, G. Chemical Probes for the Adenosine Receptors. Pharmaceuticals 2019, 12, 168. [Google Scholar] [CrossRef]
- Soave, M.; Briddon, S.J.; Hill, S.J.; Stoddart, L.A. Fluorescent ligands: Bringing light to emerging GPCR paradigms. Br. J. Pharmacol. 2020, 177, 978–991. [Google Scholar] [CrossRef]
- Kozma, E.; Jayasekara, P.S.; Squarcialupi, L.; Paoletta, S.; Moro, S.; Federico, S.; Spalluto, G.; Jacobson, K.A. Fluorescent ligands for adenosine receptors. Bioorg. Med. Chem. Lett. 2013, 23, 26–36. [Google Scholar] [CrossRef]
- McCabe, R.T.; Skolnick, P.; Jacobson, K.A. 2-2-4-2-2-1,3-Dihydro- 1,1-bis (4-hydroxyphenyl)-3-oxo-5-isobenzofuranthioureidylethylaminocarbonylethylphenyl ethylamino-5′-N-ethylcarboxamidoadenosine (FITC-APEC): A Fluorescent Ligand for A2a-Adenosine Receptors. J. Fluoresc. 1992, 2, 217–223. [Google Scholar] [CrossRef]
- Brand, F.; Klutz, A.M.; Jacobson, K.A.; Fredholm, B.B.; Schulte, G. Adenosine A2A receptor dynamics studied with the novel fluorescent agonist Alexa488-APEC. Eur. J. Pharmacol. 2008, 590, 36–42. [Google Scholar] [CrossRef]
- Comeo, E.; Kindon, N.D.; Soave, M.; Stoddart, L.A.; Kilpatrick, L.E.; Scammells, P.J.; Hill, S.J.; Kellam, B. Subtype-Selective Fluorescent Ligands as Pharmacological Research Tools for the Human Adenosine A2A Receptor. J. Med. Chem. 2020, 63, 2656–2672. [Google Scholar] [CrossRef]
- Toti, K.S.; Campbell, R.G.; Lee, H.; Salmaso, V.; Suresh, R.R.; Gao, Z.-G.; Jacobson, K.A. Fluorescent A2A and A3 adenosine receptor antagonists as flow cytometry probes. Purinergic Signal. 2022. [Google Scholar] [CrossRef] [PubMed]
- McNeely, P.M.; Naranjo, A.N.; Forsten-Williams, K.; Robinson, A.S. A2AR Binding Kinetics in the Ligand Depletion Regime. SLAS Discov. 2017, 22, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Rieger, J.M.; Brown, M.L.; Sullivan, G.W.; Linden, J.; Macdonald, T.L. Design, synthesis, and evaluation of novel A2A adenosine receptor agonists. J. Med. Chem. 2001, 44, 531–539. [Google Scholar] [CrossRef]
- Chen, J.-B.; Liu, E.M.; Chern, T.-R.; Yang, C.-W.; Lin, C.-I.; Huang, N.-K.; Lin, Y.-L.; Chern, Y.; Lin, J.-H.; Fang, J.-M. Design and synthesis of novel dual-action compounds targeting the adenosine A2A receptor and adenosine transporter for neuroprotection. ChemMedChem 2011, 6, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Gedeon, N.G.; Jankins, T.C.; Jones, G.B. Novel approaches for targeting the adenosine A2A receptor. Expert Opin. Drug Discov. 2015, 10, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Hosila, F.J. The effect of different extraction sites upon incisor retraction. Am. J. Orthod. 1976, 69, 388–410. [Google Scholar] [CrossRef]
- Jazayeri, A.; Andrews, S.P.; Marshall, F.H. Structurally Enabled Discovery of Adenosine A2A Receptor Antagonists. Chem. Rev. 2017, 117, 21–37. [Google Scholar] [CrossRef]
- Hu, E.; Kunz, R.K.; Rumfelt, S.; Andrews, K.L.; Li, C.; Hitchcock, S.A.; Lindstrom, M.; Treanor, J. Use of structure based design to increase selectivity of pyridyl-cinnoline phosphodiesterase 10A (PDE10A) inhibitors against phosphodiesterase 3 (PDE3). Bioorg. Med. Chem. Lett. 2012, 22, 6938–6942. [Google Scholar] [CrossRef]
- Deganutti, G.; Moro, S. Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example. Molecules 2017, 22, 818. [Google Scholar] [CrossRef] [PubMed]
- McGraw, C.; Yang, L.; Levental, I.; Lyman, E.; Robinson, A.S. Membrane cholesterol depletion reduces downstream signaling activity of the adenosine A2A receptor. Biochim. Biophys. Acta Biomembr. 2019, 1861, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed Engl. 2015, 54, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Pan, A.C.; Dror, R.O.; Mocking, T.; Liu, R.; Heitman, L.H.; Shaw, D.E.; IJzerman, A.P. Molecular Basis of Ligand Dissociation from the Adenosine A2A Receptor. Mol. Pharmacol. 2016, 89, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Mondal, C.; Halder, A.K.; Adhikari, N.; Jha, T. Structural findings of cinnolines as anti-schizophrenic PDE10A inhibitors through comparative chemometric modeling. Mol. Divers. 2014, 18, 655–671. [Google Scholar] [CrossRef]
- Rodríguez, D.; Gao, Z.-G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular docking screening using agonist-bound GPCR structures: Probing the A2A adenosine receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef]
- Novikov, G.V.; Sivozhelezov, V.S.; Shaĭtan, K.V. Investigation of the conformational dynamics of the adenosine A2A receptor by means of molecular dynamics simulation. Biofizika 2013, 58, 618–634. [Google Scholar]
- Kalash, L.; Winfield, I.; Safitri, D.; Bermudez, M.; Carvalho, S.; Glen, R.; Ladds, G.; Bender, A. Structure-based identification of dual ligands at the A2AR and PDE10A with anti-proliferative effects in lung cancer cell-lines. J. Cheminform. 2021, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Laughton, C.A.; Doughty, S.W. Molecular dynamics simulations of the adenosine A2a receptor: Structural stability, sampling, and convergence. J. Chem. Inf. Model. 2013, 53, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ranganathan, A.; IJzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2A adenosine receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef]
- Chen, D.; Errey, J.C.; Heitman, L.H.; Marshall, F.H.; IJzerman, A.P.; Siegal, G. Fragment screening of GPCRs using biophysical methods: Identification of ligands of the adenosine A2A receptor with novel biological activity. ACS Chem. Biol. 2012, 7, 2064–2073. [Google Scholar] [CrossRef]
- Matricon, P.; Ranganathan, A.; Warnick, E.; Gao, Z.-G.; Rudling, A.; Lambertucci, C.; Marucci, G.; Ezzati, A.; Jaiteh, M.; Dal Ben, D.; et al. Fragment optimization for GPCRs by molecular dynamics free energy calculations: Probing druggable subpockets of the A2A adenosine receptor binding site. Sci. Rep. 2017, 7, 6398. [Google Scholar] [CrossRef]
- Tosh, D.K.; Phan, K.; Gao, Z.-G.; Gakh, A.A.; Xu, F.; Deflorian, F.; Abagyan, R.; Stevens, R.C.; Jacobson, K.A.; Katritch, V. Optimization of adenosine 5′-carboxamide derivatives as adenosine receptor agonists using structure-based ligand design and fragment screening. J. Med. Chem. 2012, 55, 4297–4308. [Google Scholar] [CrossRef]
- Zhang, M.; Fan, S.; Zhou, X.; Xie, F.; Li, S.; Zhong, W. Design, synthesis and biological evaluation of 2-hydrazinyladenosine derivatives as A2A adenosine receptor ligands. Eur. J. Med. Chem. 2019, 179, 310–324. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banères, J.-L.; Botzanowski, T.; Boutin, J.A.; Calamini, B.; Castel, J.; Catoire, L.J.; Cianférani, S.; Demesmay, C.; Ferguson, G.; Ferry, G.; et al. Biophysical Dissection of Isolated GPCRs: The Adenosine A2A Receptor under the Bistouries. Receptors 2023, 2, 47-92. https://doi.org/10.3390/receptors2010004
Banères J-L, Botzanowski T, Boutin JA, Calamini B, Castel J, Catoire LJ, Cianférani S, Demesmay C, Ferguson G, Ferry G, et al. Biophysical Dissection of Isolated GPCRs: The Adenosine A2A Receptor under the Bistouries. Receptors. 2023; 2(1):47-92. https://doi.org/10.3390/receptors2010004
Chicago/Turabian StyleBanères, Jean-Louis, Thomas Botzanowski, Jean A. Boutin, Barbara Calamini, Jérôme Castel, Laurent J. Catoire, Sarah Cianférani, Claire Demesmay, Gavin Ferguson, Gilles Ferry, and et al. 2023. "Biophysical Dissection of Isolated GPCRs: The Adenosine A2A Receptor under the Bistouries" Receptors 2, no. 1: 47-92. https://doi.org/10.3390/receptors2010004
APA StyleBanères, J.-L., Botzanowski, T., Boutin, J. A., Calamini, B., Castel, J., Catoire, L. J., Cianférani, S., Demesmay, C., Ferguson, G., Ferry, G., Kniazeff, J., Krimm, I., Langer, T., Lebon, G., Ley, M., Nyerges, M., Schwob, M., Venien-Bryan, C., Wagner, R., ... Zilian-Stohrer, C. (2023). Biophysical Dissection of Isolated GPCRs: The Adenosine A2A Receptor under the Bistouries. Receptors, 2(1), 47-92. https://doi.org/10.3390/receptors2010004