1. Introduction
Microarray technology is a cutting-edge approach to gene expression analysis, widely used in fields such as basic biology and medical science. Over the past two decades, the number of gene expression studies employing microarrays has grown rapidly. Two primary microarray technologies are available for expression analysis: spotted cDNA arrays and oligonucleotide arrays. While our research primarily focuses on the statistical analysis of data from spotted cDNA microarrays, the methods discussed here can be adapted for data generated via Affymetrix chips. In microarray experiments, RNA is first extracted from the subject cells. Some of its molecules are then replaced with counterparts containing a fluorescent dye, producing labeled transcripts known as targets. In cDNA microarrays, both targets and probes are cDNA molecules, whereas, in oligonucleotide arrays, the targets remain cDNA molecules, but the probes consist of carefully selected small cDNA segments known as oligos.
A cDNA microarray is commonly referred to as a two-channel array. In this technology, both an experimental sample and a reference sample are labeled with fluorescent dyes—Cyanine 3 (Cy3, green) and Cyanine 5 (Cy5, red)—on a single chip, with the experimental sample typically labeled with Cy5 [
1]. Each chip contains thousands of spots, each corresponding to a specific gene. A laser microscope scanner measures the fluorescence intensity at each spot, providing a quantitative assessment of gene expression. Colored spots indicate genes expressed in one or both samples, while gray areas represent genes not expressed in either sample. Although microarray data capture thousands of genes, only a subset of differentially expressed genes (DEGs) may be biologically significant, particularly in diseases such as cancer. Therefore, identifying these DEGs is a crucial step in microarray analysis. Numerous methods have been proposed in the literature to detect differentially expressed genes.
The fold-change method was one of the earliest techniques used in microarray data analysis [
2,
3,
4]. This simple approach identifies differentially expressed genes (DEGs) by applying a predefined threshold on fold change. However, it is prone to bias, particularly when the data are not properly normalized [
5]. Moreover, since it does not account for statistical variation, the method is considered unreliable. To address these limitations, later approaches incorporated the probabilistic modeling of red (
R) and green (
G) intensities for DEG detection. These methods established classification rules based on distributional assumptions of
. For example, ref. [
6] proposed a data-driven threshold selection for the ratio
, assuming normality and a constant coefficient of variation. However, a key drawback of this approach is that it overlooks the valuable information contained in the product
.
To avoid this problem, ref. [
7] suggested a hierarchical model (Gamma–Gamma–Bernoulli) to capture DEGs based on the posterior odds of change (the odds are functions of
and
). This method assumes that
R and
G are independent. Mav and Chaganty [
8] have shown that the
R and
G are positively correlated. They have built a bivariate distribution with gamma marginals and a positive correlation between
R and
G to incorporate this dependence. They also incorporated a latent Bernoulli variable. They used the EM algorithm to calculate the posterior probabilities of differential expression. The DEGs are the ones with higher posterior probabilities.
A Bayesian approach to testing multiple hypotheses in microarray experiments was proposed by Maria et al. (2020) [
9], pointing to the use of copula functions to model dependence. Their method demonstrated that incorporating dependency structures significantly improves classification accuracy when identifying differentially expressed genes. The term “copula” was first used by Sklar in 1959 [
10]; its meaning is that it “ties” the marginal uniform distributions to create a joint distribution function. An excellent introduction to copulas is the book by Nelsen [
11]. Copula functions are useful for constructing bivariate or, in general, multivariate distributions with given marginal distributions. Since their introduction, the literature and applications of copulas have grown rapidly. Some classic books on the topic include [
11,
12]. More comprehensive coverage of copula models and their applications is given in [
13].
Over the past decade, extensive research has explored the application of copulas in genomics, particularly for analyzing gene expression data. For instance, Chaba (2017) [
14] proposed a semi-parametric copula-based approach to differential gene expression analysis. Likewise, Ray et al. (2020) [
15] introduced a copula-based model that focuses on detecting differential co-expression, rather than differential expression, emphasizing the significance of dependency structures in gene networks.
Although copulas are increasingly used in gene expression studies, existing approaches have not been specifically developed to detect differentially expressed genes in microarray experiments. To address this gap, we introduce a copula-based framework particularly for DEG detection, utilizing copulas to model dependencies between gene intensities while offering a probabilistic basis for differential expression analysis. In this article, we extend the work done by Mav and Chaganty [
8], replacing the joint probability distribution of red and green intensities with a Gaussian copula-based joint distribution. The DEGs can be identified by calculating the Bayes estimate of the differential expression under this model.
The outline of the article is as follows.
Section 2 describes the challenges in analyzing microarray data and the need for sophisticated statistical models to accurately capture the dependence structure between gene expressions. The theoretical foundation of the copula model, including the formulation of the model and the specification of prior distributions for the parameters, is explained in
Section 3.
In
Section 4, we present the methodology for estimating the parameters of the copula model using maximum likelihood estimation and Bayesian inferential procedures. The section also details the numerical optimization routines employed.
Section 4 provides an in-depth discussion of the estimation process, including the steps involved in maximizing the log-likelihood function and implementing the quasi-Newton algorithm.
Section 5 focuses on identifying differentially expressed genes using the estimated copula model parameters and explaining the statistical tests and criteria used. In
Section 6, we explore the relationship between the copula parameter and the correlation coefficient, providing insights into how the copula model captures dependencies between gene expression levels.
Section 7 presents the results of simulation studies conducted to evaluate the performance of the copula-based Bayesian model in identifying differentially expressed genes and comparing it with other existing methods.
Section 8 contains the application of the copula-based Bayesian model to real datasets of gene expression levels in
Escherichia coli, demonstrating the practical utility of the model. Finally, by comparing the log-likelihood values, we show that this Gaussian copula model improves over the models given in [
7,
8].
2. Motivation
Escherichia coli (
E. coli) is a bacteria that generally lives in the intestines of people and animals. The motivating data for this article is the experiment designed to study gene expression levels in
E. coli, described in [
16]. The
E. coli genome consists of approximately 4.6 million base pairs (Mbp), but it is suspected of encoding only about forty-two hundred genes. To study differential gene expressions in
E. coli, ref. [
16] used two traditional treatments that affect gene expression levels. The first treatment was induction with isopropyl-
-D-thiogalactopyranoside (IPTG), which tests the methods since only a few gene transcripts are expected to change. Secondly, heat shock treatment allows global regulatory effects to be observed. A single colony of
E. coli K-12 was divided into five samples for the experiments.
IPTG treatment was performed independently on two samples (IPTG-A and IPTG-B), while one sample (control) was untreated. Heat shock induction was carried out by treating the culture to a 50 °C shaking water bath for seven minutes on the remaining two samples (Heat Shock A and Heat Shock B). Following the hybridization of the samples on E. coli microarrays, the signal intensities for each spot were determined using the ScanAlyze software 1.0.3. The average fluorescence intensity for each spot was measured, and the background was chosen as the median pixel intensity in a square surrounding each spot. The red and green signal intensities were recalculated and normalized after background subtraction.
Newton et al. [
7] proposed a Bayesian hierarchical model with a latent variable to identify differentially expressed genes. In their approach, the marginal distributions of red and green intensities were modeled as gamma distributions with common shape parameters but different scale parameters. However, they assumed that the red and green intensities for the same gene were independent. To address this limitation, Mav and Chaganty [
8] introduced a bivariate distribution with gamma marginals that incorporated a positive correlation between the red and green intensities. In this article, we extend their approach by replacing the bivariate distribution with a bivariate Gaussian copula, constructing a joint distribution with gamma marginals. The performance of our proposed model is evaluated through log-likelihood analysis using
E. coli data.
3. Copula-Based Bayesian Model for Expression Level
The typical objective when analyzing data from microarray experiments is to identify differentially expressed genes. This section will propose a copula-based Bayesian model that can filter these genes. Consider a microarray consisting of
n genes. Let
and
denote the red and green intensities of gene
j, respectively. The concepts based on the red and green intensity ratio have been widely used in the literature to identify differentially expressed genes. Some of those were discussed briefly in
Section 1. To filter the differentially expressed genes, we will use the ratio of expected expression levels, which are given by
, where
E stands for the expected value.
As explained in
Section 2, this study was motivated by
E. coli data. Gamma distribution has been widely used in statistical analysis in microarray experiments [
17,
18,
19], not only for its analytical convenience but also for its ability to be interpreted in a deeper way. The family of gamma distributions is supported on the positive line and provides a rich class of distributions as special cases [
17]. In addition, Chen et al. in 1997 [
6] presented an intriguing argument that, while expected expression levels vary from gene to gene in the microarray, the measurements are connected via a constant coefficient of variation (
) and
. Having a common shape parameter,
, ensures that the variability structure remains consistent across genes and makes it convenient to model the variability in gene expression. Taking into account all of the above facts, we assume
and
are gamma distributions with a common shape parameter,
, but different scale parameters,
and
for
. The probability density function of
is given by
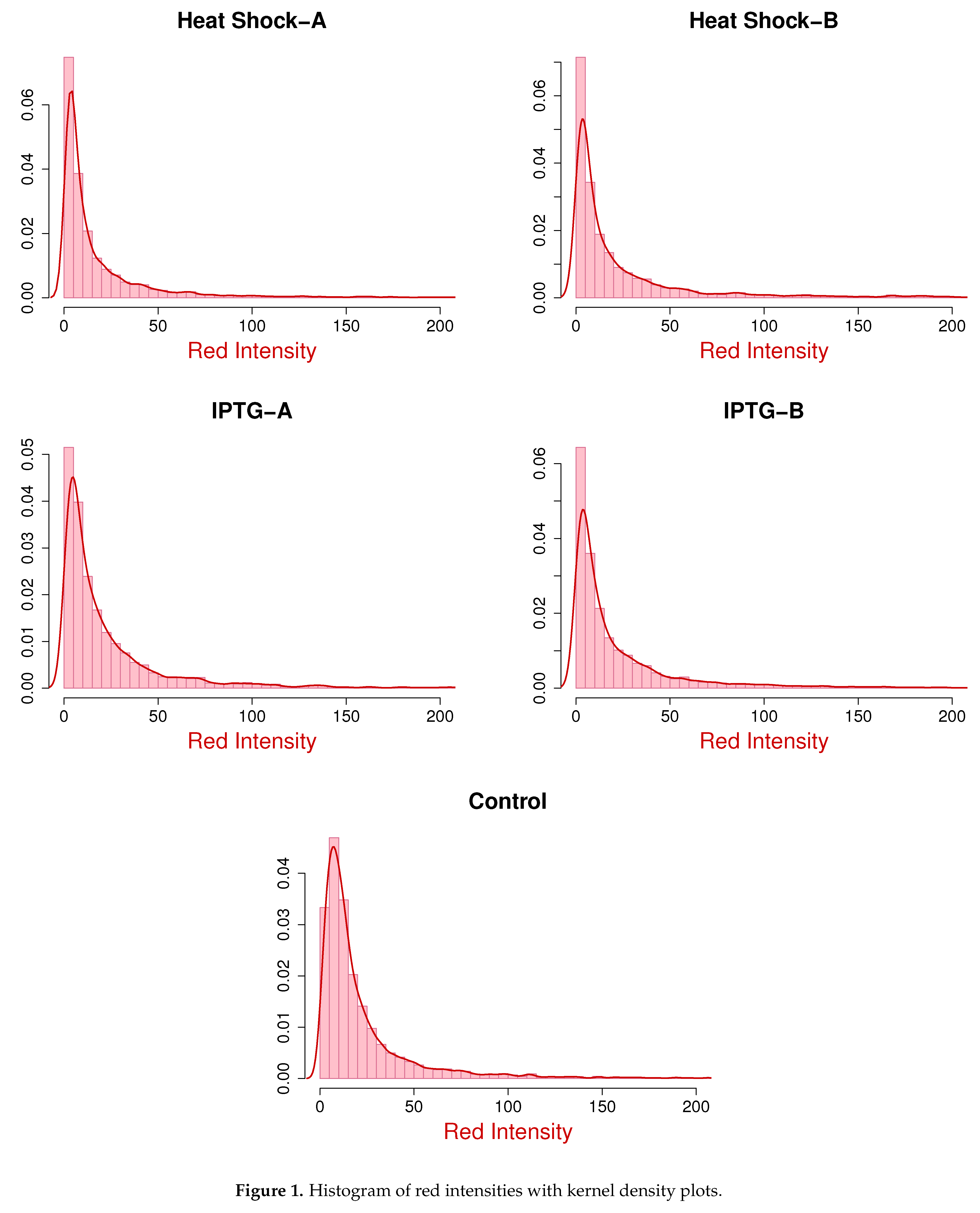
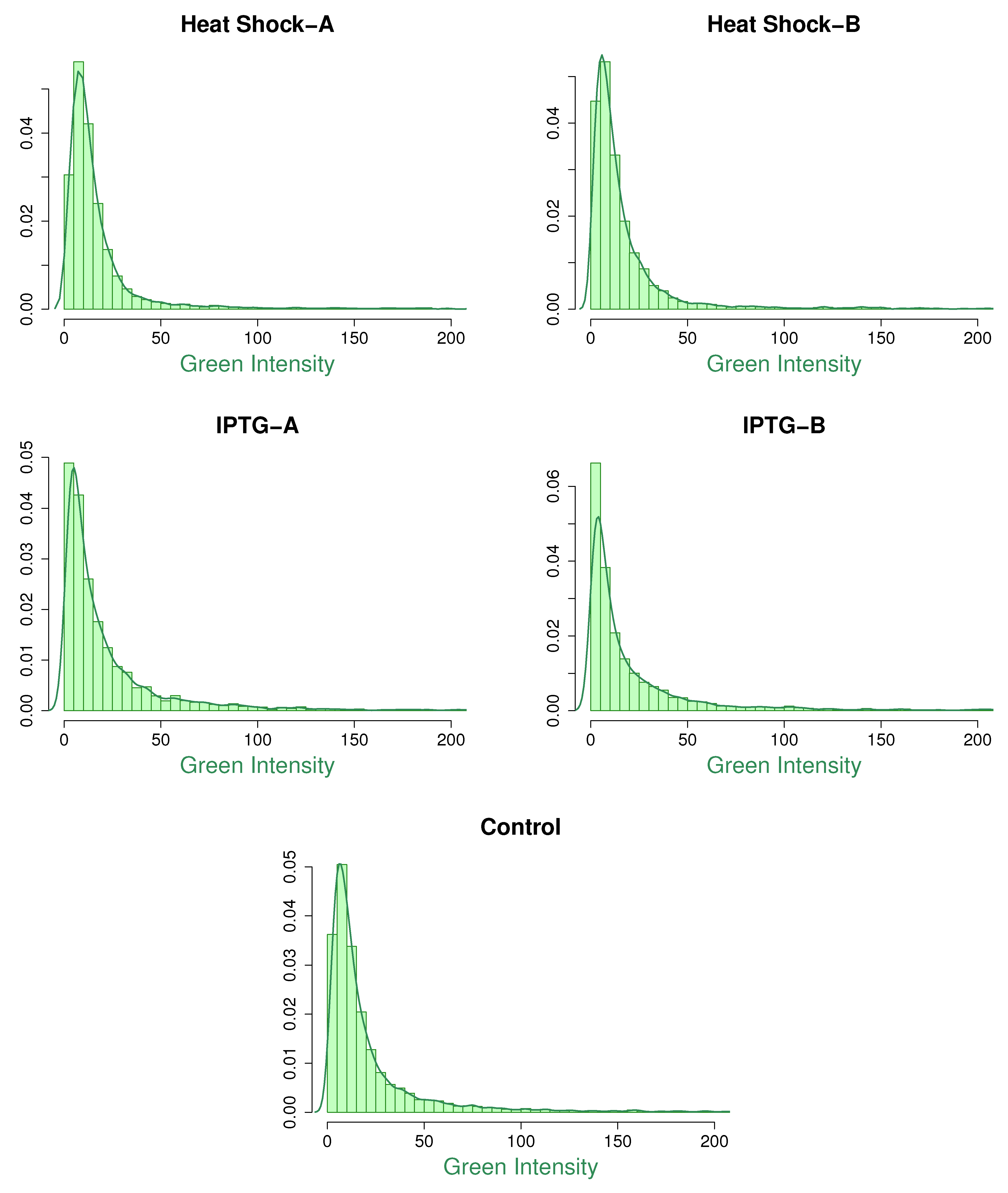
Figure 1 and
Figure 2 show the histograms of red and green intensities along with nonparametric kernel density plots for the five microarray experiments. The positively skewed shape of the density curves suggests that the assumption of gamma marginals is reasonable. Note that
for
. Therefore, the ratio of expected expression levels
for
. To model the dependence between the two intensities, we assume the joint distribution of
is given by the bivariate Gaussian copula and can be written as
where
and
is the cumulative distribution function of a gamma distribution with parameters
. Note that the Gaussian copula density is
where
, and
is the parameter of the copula density. To simplify the notation moving forward, we write
in place of
.
The joint distribution in (
2) consists of
unknown parameters. Since there are too many unknown parameters, we adopt the empirical Bayes approach to make the model parsimonious. This requires specification of prior distributions for the gene-specific parameters
and
. We assume independent gamma distributions with parameters
and
as the prior distributions for
.
The prior density
is
Multiplying (
2) and (
4), we get the joint density of
and
as
where
is the vector of model parameters. Recall that this model has gene-specific parameters,
, for
. The marginal density of
is
Here,
is the cumulative distribution function of gamma with parameters
and
for
and
.
The double integral in Equation (
6) does not simplify because of the presence of the Gaussian copula function
in the integrand. A numerical computation of the double integral (
6) is also challenging. To compute the double integral, we could use the R libraries, such as the
cubature by [
20] or
pracma by [
21]. We were unsuccessful with these packages and encountered numerous errors with the functions embedded in these packages when evaluating the double integral iteratively. To overcome the computational problems, we have developed our own R code to evaluate the double integral and obtain the marginal density of
.
6. Relation Between Copula Parameter and the Correlation Coefficient
It is a well-known fact that the correlation coefficient of two random variables is the magnitude and the direction of the linear relationship between those two random variables. However, it fails to capture nonlinear dependence, which the copula function captures, specifically the dependence in the tail region for non-normal variables. In this section, we derive the relationship between the linear correlation coefficient between and and the Gaussian copula parameter .
Case 1. Suppose that
is distributed as gamma
for
, and the joint distribution is given by the bivariate Gaussian copula with parameter
. Note that the marginal mean and variance of
are
and
, respectively. The joint probability density function of
is given by
where
, for
and
, and
is the cumulative distribution function. Therefore, the expected value of
is
If
is the correlation coefficient between
and
, then we have
This can be numerically computed for different values of
,
, and
.
Case 2. Suppose that
is distributed as gamma
, and
is also distributed as gamma
for
Then, the joint probability distribution of
is
We can show that the marginal probability density function of
is the beta distribution of the second type (
) with parameters
.
The marginal mean and the variance of
are
Note that
and
are functions of
. Assuming the joint distribution of
is determined by the Gaussian copula with parameter
, Equation (
6) gives the marginal density of
. The expected value of the product of
is given by
Equation (
12) has a double integral with respect to
and
, and another additional double integral with respect to
and
. The function
adaptIntegrate in the R package
cubature is useful to evaluate this multidimensional integral numerically. We have developed an R function that uses
adaptIntegrate to calculate (
12). The relationship between the copula parameter
and
, in this case, is given by
The above-derived relationship between
and
is applied in the simulation study in
Section 7, demonstrating its practical relevance. Furthermore, in
Table 1, the correlation coefficient
and its estimate
were reasonably close for all sample sizes, validating the robustness of the proposed approach.
7. Simulation Studies
In this section, we perform simulations to gauge the performance of our Gaussian copula model. The data are simulated for two sets of values of with three sample sizes, . The data simulation steps are as follows.
Fix a value for .
Generate n pairs of bivariate normal random variables from a standard bivariate normal distribution (BVN) with the correlation parameter .
Calculate for where is the cumulative distribution function of the normal standard.
Generate from a gamma distribution with parameters for and .
Calculate , where is the cumulative distribution function of a gamma distribution with parameters .
For our first simulation, we have fixed the parameter values as
and
. We simulated samples of sizes
n = 100, 500, and 3000 with these parameter values. The parameter estimation results are given in
Table 1. In
Table 1,
is the correlation coefficient calculated from simulated data, and
is the correlation coefficient calculated after substituting the estimated values of
in Equation (
13). The parameter estimates are closer to the actual parameter values for large sample sizes, and the standard errors get smaller as the sample size increases. The correlation coefficient,
, and its estimate,
, were reasonably close for all sample sizes.
For our second simulation, we fixed the parameter values as
and
, and as before, we took three sample sizes, 100, 500, and 3000.
Table 2 consists of the parameter estimation results for this second simulation.
For , the estimate is an overestimate of , and is a terrible underestimate of . Otherwise, the results are consistent with the first simulation. All the parameter estimates are closer to their true values for a larger sample size, . This is good news because, in practice, n, which represents the number of genes, is in thousands.
8. Analysis of E. coli Microarray Data
In this section, we apply the Gaussian copula-based Bayesian model developed in
Section 3 to some actual data obtained from microarray experiments on
E. coli. These data consist of observations from five microarrays. There are two IPTG-treated samples labeled IPTG-A and IPTG-B, as well as two heat shock samples labeled Heat Shock A and Heat Shock B, and the fifth is a control (untreated). We described these data earlier in
Section 2. There are 4253, 4083, 4141, 4208, and 4071 genes in the control, IPTG-A, IPTG-B, Heat Shock A, and Heat Shock-B samples, respectively.
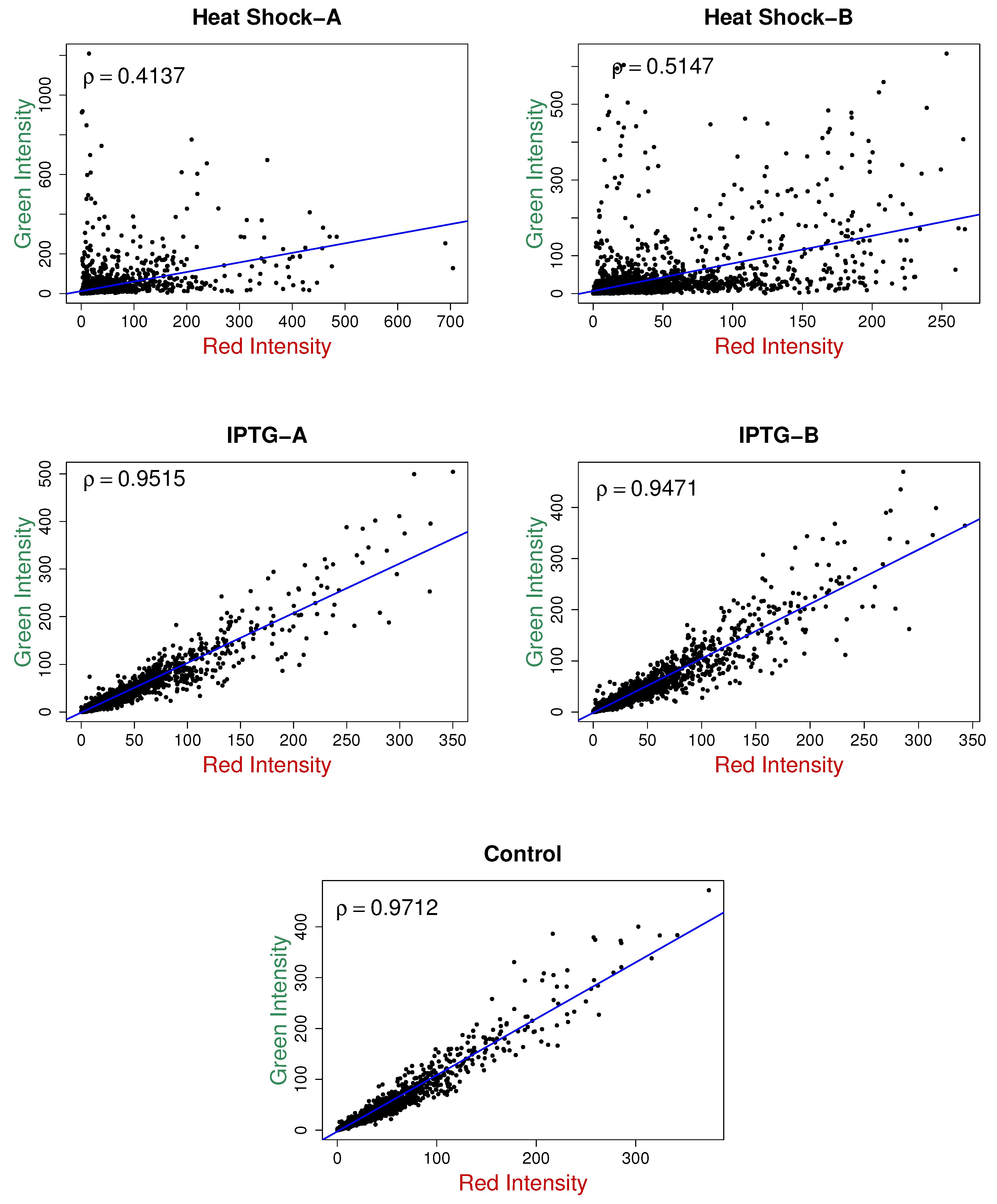
The scatter plots for the red and green intensities for the five samples are shown in
Figure 3, along with the sample correlation coefficients. There is a high positive correlation between red and green intensities in all five samples. The positively skewed shape of the density curves suggests the assumption of gamma marginals is reasonable. Thus, following [
7], as a parsimonious model, we assume the marginal distributions of the red and green intensities as gamma with standard shape parameters but different scale parameters.
Table 3 contains the parameter estimates and their standard errors for the Gaussian copula-based Bayesian models for the five microarray samples. The standard errors are small because the sample sizes are large—more than 4000 in all cases. This suggests that the parameter estimates are pretty accurate.
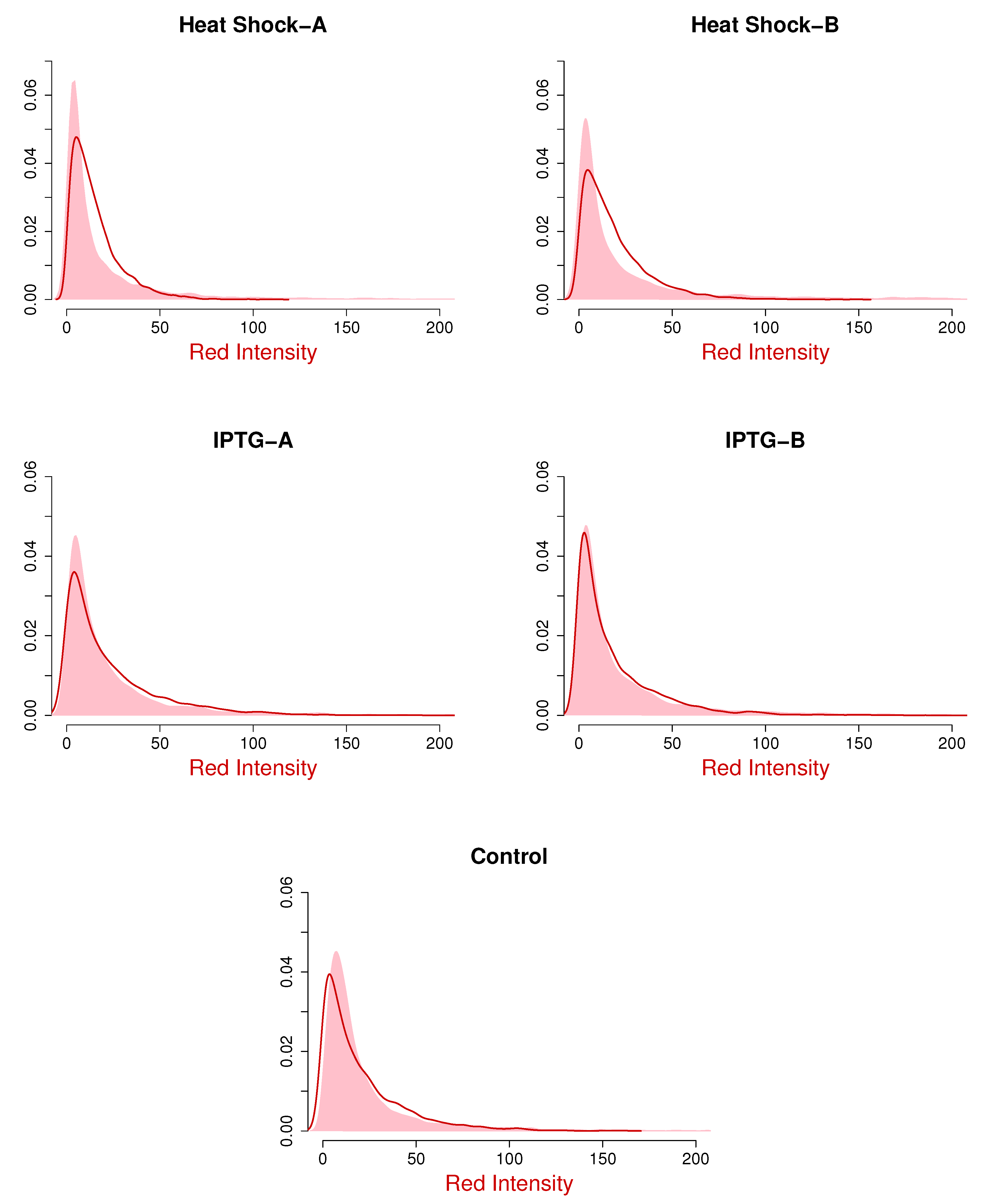
The empirical density plots, along with the fitted density plots, are shown in
Figure 4 and
Figure 5 for the red and green intensities, respectively. The solid curves in
Figure 4 and
Figure 5 are the fitted gamma marginals, and the shaded curves are the empirical plots. Note that the fitted marginals are gamma densities with the estimated parameter values in
Table 3. These figures show that the fitted marginals are very good for the IPTG and control samples, but there is some improvement for the heat shock samples, especially the red intensities.
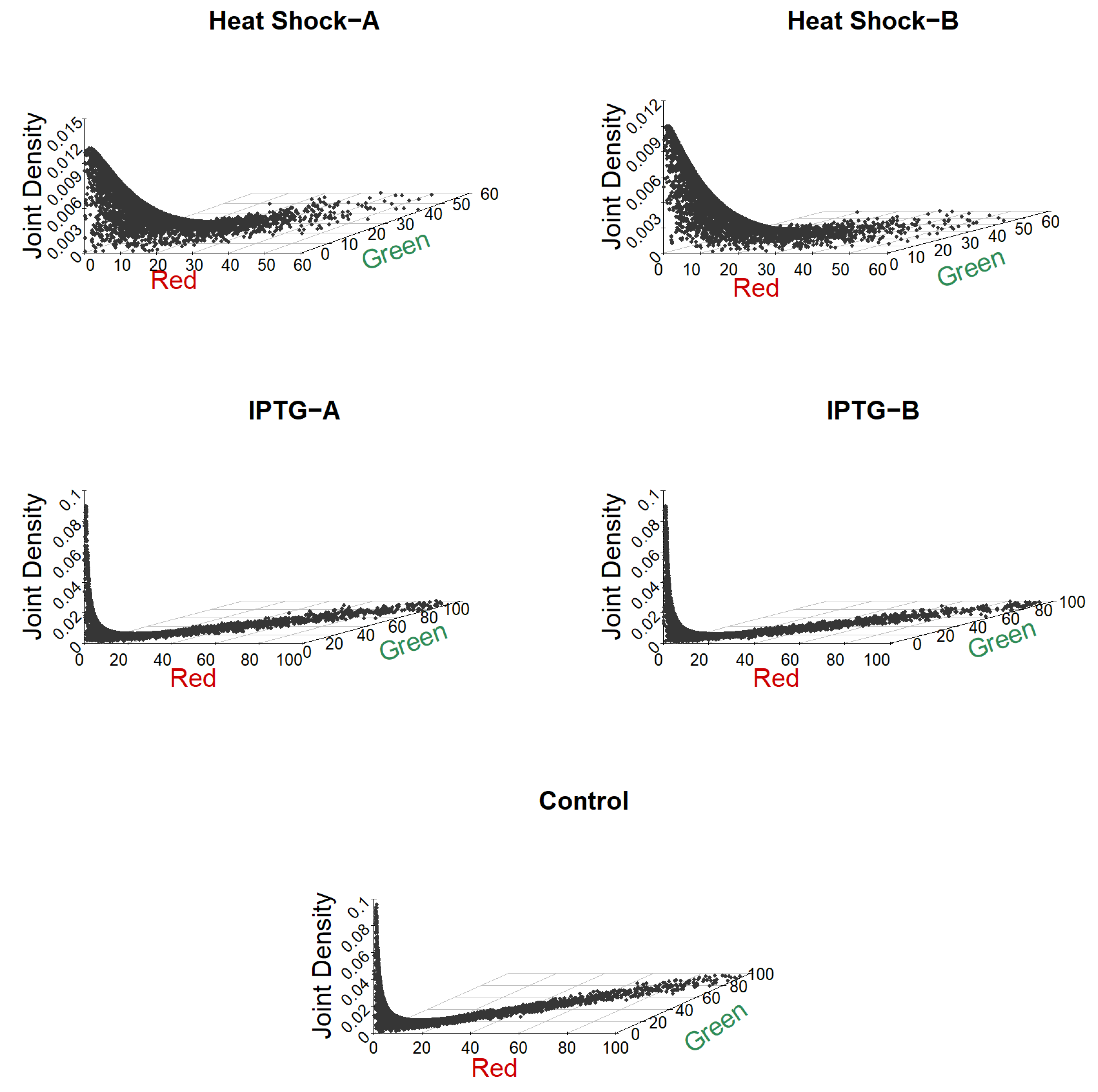
Figure 6 shows the fitted bivariate density plots obtained using the parameter estimates in
Table 3. In these plots, the 45° line indicates equal red and green intensities, and the points that fall on this line correspond to genes that are not differentially expressed. We can see that most of the control group’s genes are not differentially expressed using this criteria. For the IPTG samples, a few points lie away from the 45° line, indicating the presence of differentially expressed genes in these samples. Finally, for the two heat shock samples, a large number of points are away from the 45° line, indicating that there is a large number of differentially expressed genes in these samples.
Table 4 displays the observed correlation (
) and correlation coefficient (
) calculated from the estimated copula parameter as in
Table 3 using Equation (
13). Except for Heat Shock B, for all the other four samples, the values of
and
are very close, indicating that our copula model was fairly successful in quantifying the dependence between the two intensities.
We calculated
using Equation (
8), the Bayes estimate
of
which is a measure of differential expression of the
jth gene.
Table 5 displays the top twenty down-regulated (
is small) genes, and
Table 6 lists the top twenty up-regulated (
is large) genes for all five samples.
Plots of ordered
values for the five samples are displayed in
Figure 7. These plots are S-shaped; the left tails contain the down-regulated genes, whereas the right tails contain the up-regulated genes. In their paper, ref. [
16] have listed the genes significantly affected by heat shock and IPTG treatments. According to their findings, the control sample has none of the differentially expressed genes, the IPTG samples have few, and the heat-shock samples have many differentially expressed genes. Therefore, by considering the number of differentially expressed genes and the plots of
of five microarrays,
is a good candidate cut-off value to filter up-regulated genes, while
is for down-regulated genes. The horizontal lines in
Figure 7 indicate the possible cut-off values to separate the normal genes from the two extremes.
The total number of differentially expressed genes for each microarray is listed in
Table 7, along with the total number of differentially expressed genes filtered with the bivariate gamma model proposed by [
8].
As expected, none of the genes are identified as differentially expressed in the control sample, and very few in IPTG-A and IPTG-B. Many genes were filtered as up- or down-regulated from both the Heat Shock A and Heat Shock B models. The number of genes filtered from the Gaussian copula model is somewhat smaller than that from the bivariate gamma model.
The best model cannot be determined by looking at this total number of genes. Therefore, the log-likelihoods for the two models under each microarray are compared and shown in
Table 8. The log-likelihood values under our model are higher than those of the bivariate gamma model, which was proposed by [
8] for each microarray. Hence, we conclude that the Gaussian copula-based Bayesian model achieves an improvement over the model given in [
8]. Further, our method’s filtered differentially expressed genes of heat-shock samples are well matched with the genes listed in [
16]. Recall that, in [
16], the control sample had no differentially expressed genes, and the IPTG samples had few, consistent with our findings.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}