
The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis: State of the Art 2025


Abstract
1. Introduction
2. An Overview on Muscle Tissue Biology
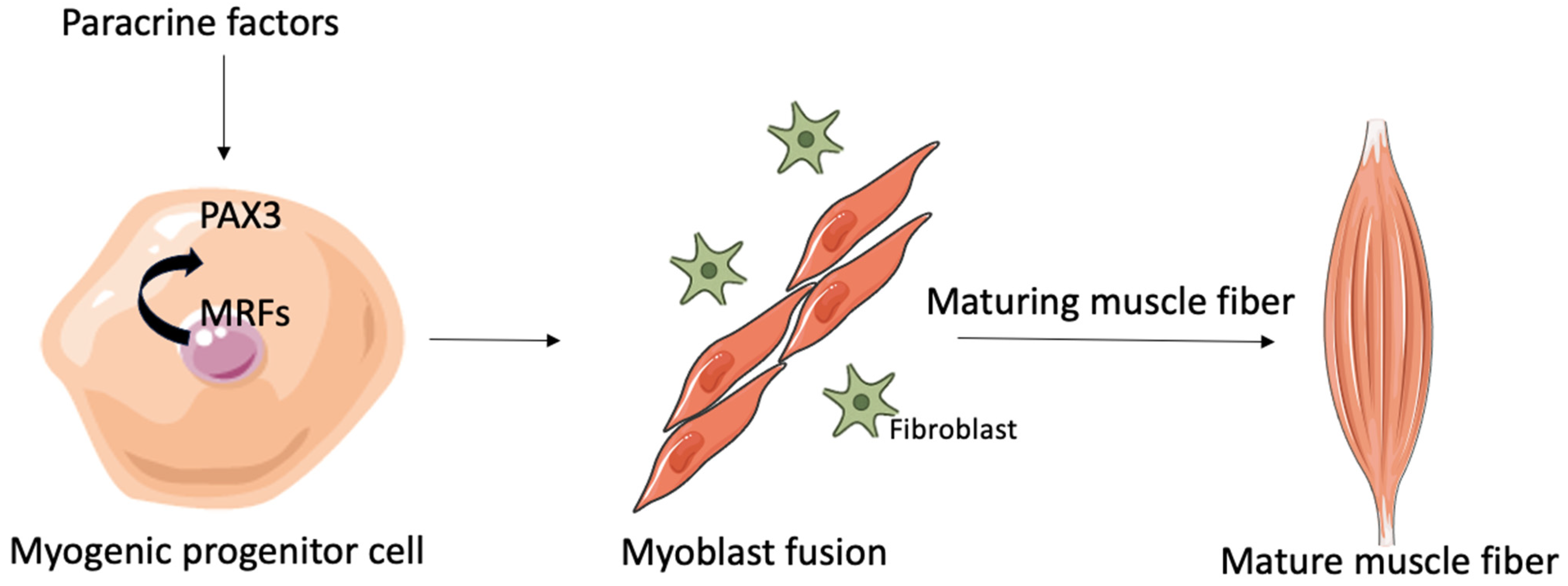
2.1. Myogenesis and Regeneration of Tissue
2.2. Myoblast Fusion
3. A Brief Overview of the Genetic Basis of Pathogenesis in ALS
4. Muscle Pathophysiology in ALS: Mechanisms and Implications
4.1. Mitochondrial Dysfunction
4.2. Protein Degradation
4.3. Muscle Denervation and Reinnervation
4.4. Neuromuscular Junction (NMJ)
5. Altered Regenerative Response of Skeletal Muscle in ALS
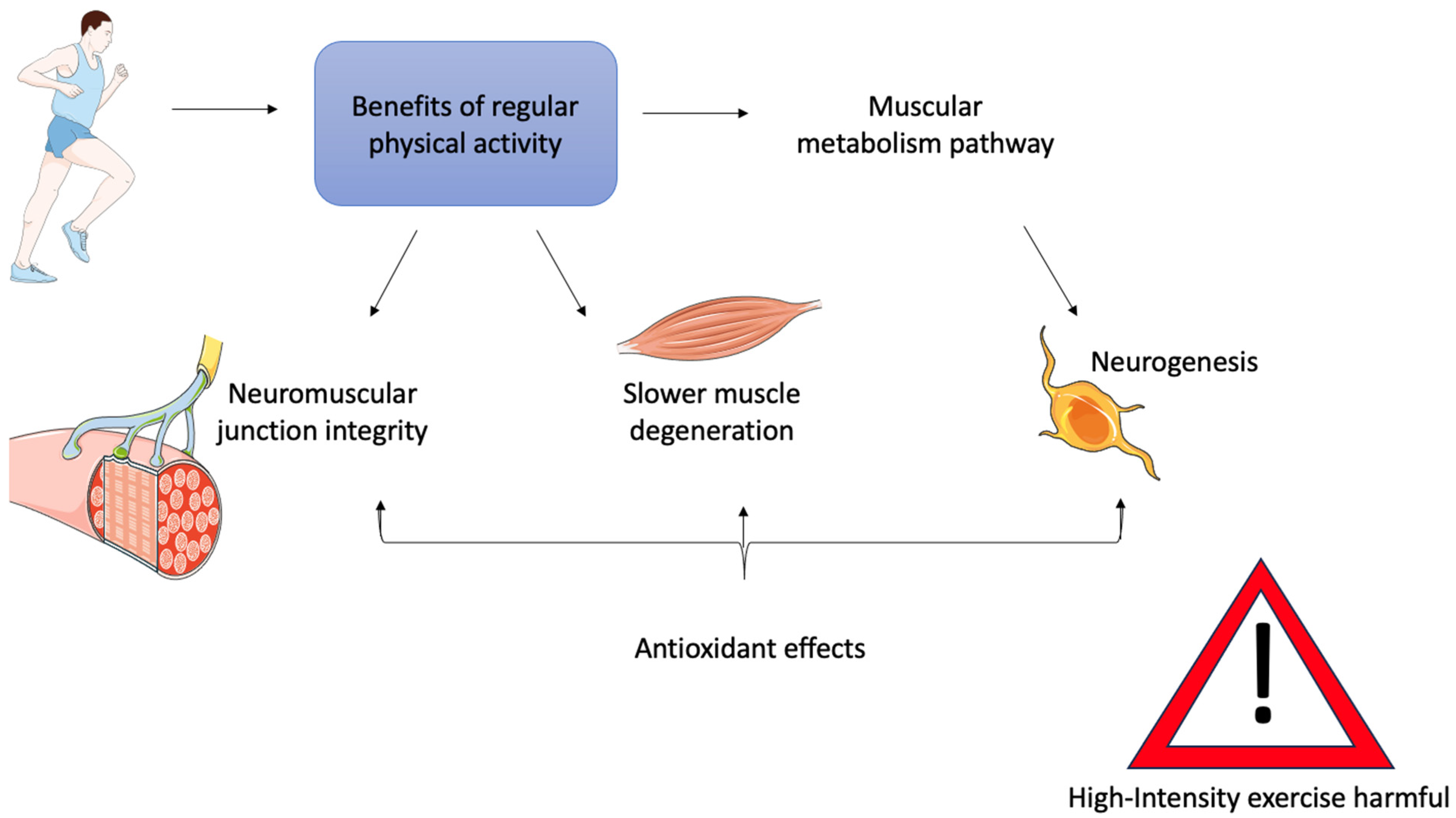
6. Physical Activity as a Therapy for ALS Muscle
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 13, 1688–1700. [Google Scholar]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [PubMed]
- Li, C.; Hou, Y.; Wei, Q.; Lin, J.; Jiang, Z.; Jiang, Q.; Yang, T.; Xiao, Y.; Huang, J.; Cheng, Y.; et al. Mutation screening of SPTLC1 and SPTLC2 in amyotrophic lateral sclerosis. Hum. Genom. 2023, 17, 28. [Google Scholar]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [PubMed]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar]
- Scaramozza, A.; Marchese, V.; Papa, V.; Salaroli, R.; Sorarù, G.; Angelini, C.; Cenacchi, G. Skeletal muscle satellite cells in amyotrophic lateral sclerosis. Ultrastruct. Pathol. 2014, 38, 295–302. [Google Scholar]
- Manzano, R.; Toivonen, J.M.; Calvo, A.C.; Oliván, S.; Zaragoza, P.; Rodellar, C.; Montarras, D.; Osta, R. Altered in vitro proliferation of mouse SOD1-G93A skeletal muscle satellite cells. Neurodegener. Dis. 2013, 11, 153–164. [Google Scholar]
- Tokutake, Y.; Yamada, K.; Ohata, M.; Obayashi, Y.; Tsuchiya, M.; Yonekura, S. ALS-Linked P56S-VAPB Mutation Impairs the Formation of Multinuclear Myotube in C2C12 Cells. Int. J. Mol. Sci. 2015, 16, 18628–18641. [Google Scholar]
- Han, S.M.; El Oussini, H.; Scekic-Zahirovic, J.; Vibbert, J.; Cottee, P.; Prasain, J.K.; Bellen, H.J.; Dupuis, L.; Miller, M.A. VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in caenorhabditis elegans. PLoS Genet. 2013, 9, e1003738. [Google Scholar]
- Doppler, K.; Mittelbronn, M.; Bornemann, A. Myogenesis in human denervated muscle biopsies. Muscle Nerve 2008, 37, 79–83. [Google Scholar] [PubMed]
- Pradat, P.F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombès, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 264–271. [Google Scholar]
- Rothstein, J.D. Edaravone: A New Drug Approved for ALS. Cell 2017, 171, 725. [Google Scholar] [PubMed]
- Cho, H.; Shukla, S. Role of Edaravone as a Treatment Option for Patients with Amyotrophic Lateral Sclerosis. Pharmaceuticals 2020, 14, 29. [Google Scholar] [CrossRef]
- Hinchcliffe, M.; Smith, A. Riluzole: Real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 61–70. [Google Scholar] [PubMed]
- Duranti, E.; Villa, C. From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders. Biology 2024, 13, 719. [Google Scholar] [CrossRef]
- Lynch, K. Optimizing pharmacologic treatment for ALS to improve outcomes and quality of life. Am. J. Manag. Care 2023, 29, S112–S119. [Google Scholar]
- Periasamy, M.; Herrera, J.L.; Reis, F.C.G. Skeletal Muscle Thermogenesis and Its Role in Whole Body Energy Metabolism. Diabetes Metab. J. 2017, 41, 327–336. [Google Scholar]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar]
- Fukada, S.I. The roles of muscle stem cells in muscle injury, atrophy and hypertrophy. J. Biochem. 2018, 163, 353–358. [Google Scholar]
- Lepper, C.; Partridge, T.A.; Fan, C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 2011, 138, 3639–3646. [Google Scholar] [PubMed]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [PubMed]
- Barthélémy, F.; Wein, N. Personalized gene and cell therapy for Duchenne Muscular Dystrophy. Neuromuscul. Disord. 2018, 28, 803–824. [Google Scholar]
- Yamamoto, M.; Legendre, N.P.; Biswas, A.A.; Lawton, A.; Yamamoto, S.; Tajbakhsh, S.; Kardon, G.; Goldhamer, D.J. Loss of MyoD and Myf5 in Skeletal Muscle Stem Cells Results in Altered Myogenic Programming and Failed Regeneration. Stem Cell Rep. 2018, 10, 956–969. [Google Scholar]
- McCarthy, J.J.; Mula, J.; Miyazaki, M.; Erfani, R.; Garrison, K.; Farooqui, A.B.; Srikuea, R.; Lawson, B.A.; Grimes, B.; Keller, C.; et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development 2011, 138, 3657–3666. [Google Scholar]
- Kaczmarek, A.; Kaczmarek, M.; Ciałowicz, M.; Clemente, F.M.; Wolański, P.; Badicu, G.; Murawska-Ciałowicz, E. The Role of Satellite Cells in Skeletal Muscle Regeneration-The Effect of Exercise and Age. Biology 2021, 10, 1056. [Google Scholar]
- Fochi, S.; Giuriato, G.; De Simone, T.; Gomez-Lira, M.; Tamburin, S.; Del Piccolo, L.; Schena, F.; Venturelli, M.; Romanelli, M.G. Regulation of microRNAs in Satellite Cell Renewal, Muscle Function, Sarcopenia and the Role of Exercise. Int. J. Mol. Sci. 2020, 21, 6732. [Google Scholar]
- Sheveleva, O.N.; Payushina, O.V.; Butorina, N.N.; Domaratskaya, E.I. The Myogenic Potential of Mesenchymal Stromal Cells and Their Effect on Skeletal Muscle Regeneration. Biol. Bull. Russ. Acad. Sci. 2020, 47, 455–465. [Google Scholar]
- Forcina, L.; Cosentino, M.; Musarò, A. Mechanisms Regulating Muscle Regeneration: Insights into the Interrelated and Time-Dependent Phases of Tissue Healing. Cells 2020, 9, 1297. [Google Scholar] [CrossRef]
- Chal, J.; Pourquié, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar]
- Yusuf, F.; Brand-Saberi, B. Myogenesis and muscle regeneration. Histochem. Cell Biol. 2012, 138, 187–199. [Google Scholar] [PubMed]
- Doyonnas, R.; LaBarge, M.A.; Sacco, A.; Charlton, C.; Blau, H.M. Hematopoietic contribution to skeletal muscle regeneration by myelomonocytic precursors. Proc. Natl. Acad. Sci. USA 2004, 101, 13507–13512. [Google Scholar]
- Mourkioti, F.; Rosenthal, N. IGF-1, inflammation and stem cells: Interactions during muscle regeneration. Trends Immunol. 2005, 26, 535–542. [Google Scholar] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar]
- Shirakawa, T.; Toyono, T.; Inoue, A.; Matsubara, T.; Kawamoto, T.; Kokabu, S. Factors Regulating or Regulated by Myogenic Regulatory Factors in Skeletal Muscle Stem Cells. Cells 2022, 11, 1493. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.S. Function of the Myogenic Regulatory Factors Myf5, MyoD, Myogenin and MRF4 in Skeletal Muscle, Satellite Cells and Regenerative Myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar]
- Romagnoli, C.; Iantomasi, T.; Brandi, M.L. Available In Vitro Models for Human Satellite Cells from Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 13221. [Google Scholar]
- Lehka, L.; Rędowicz, M.J. Mechanisms regulating myoblast fusion: A multilevel interplay. Semin. Cell Dev. Biol. 2020, 104, 81–92. [Google Scholar]
- Feng, L.T.; Chen, Z.N.; Bian, H. Skeletal Muscle: Molecular Structure, Myogenesis, Biological Functions, and Diseases. MedComm 2024, 5, e649. [Google Scholar]
- Pizza, F.X.; Buckley, K.H. Regenerating Myofibers after an Acute Muscle Injury: What Do We Really Know about Them? Int. J. Mol. Sci. 2023, 24, 12545. [Google Scholar]
- Millay, D.P. Regulation of the myoblast fusion reaction for muscle development, regeneration, and adaptations. Exp. Cell Res. 2022, 415, 113134. [Google Scholar] [PubMed]
- Deleu, M.; Crowet, J.M.; Nasir, M.N.; Lins, L. Complementary biophysical tools to investigate lipid specificity in the interaction between bioactive molecules and the plasma membrane: A review. Biochim. Biophys. Acta 2014, 1838, 3171–3190. [Google Scholar] [PubMed]
- Mukai, A.; Kurisaki, T.; Sato, S.B.; Kobayashi, T.; Kondoh, G.; Hashimoto, N. Dynamic clustering and dispersion of lipid rafts contribute to fusion competence of myogenic cells. Exp. Cell Res. 2009, 315, 3052–3063. [Google Scholar]
- Schwander, M.; Leu, M.; Stumm, M.; Dorchies, O.M.; Ruegg, U.T.; Schittny, J.; Müller, U. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev. Cell 2003, 4, 673–685. [Google Scholar]
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [PubMed]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar]
- Boillée, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar]
- Zhou, W.; Xu, R. Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis. Front. Neurosci. 2023, 17, 1189470. [Google Scholar]
- Ticozzi, N.; Tiloca, C.; Morelli, C.; Colombrita, C.; Poletti, B.; Doretti, A.; Maderna, L.; Messina, S.; Ratti, A.; Silani, V. Genetics of familial Amyotrophic lateral sclerosis. Arch. Ital. Biol. 2011, 149, 65–82. [Google Scholar] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [PubMed]
- Khosravi, B.; Hartmann, H.; May, S.; Möhl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2017, 26, 790–800. [Google Scholar] [PubMed]
- Eck, R.J.; Kraemer, B.C.; Liachko, N.F. Regulation of TDP-43 Phosphorylation in Aging and Disease. Geroscience 2021, 43, 1605–1614. [Google Scholar]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar]
- Chook, Y.M.; Süel, K.E. Nuclear import by karyopherin-βs: Recognition and inhibition. Biochim. Biophys. Acta 2011, 1813, 1593–1606. [Google Scholar]
- Lanson, N.A., Jr.; Pandey, U.B. FUS-related proteinopathies: Lessons from animal models. Brain Res. 2012, 1462, 44–60. [Google Scholar]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: Translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar]
- Gendron, T.F.; Petrucelli, L. Disease Mechanisms of C9ORF72 Repeat Expansions. Cold Spring Harb. Perspect. Med. 2018, 8, a024224. [Google Scholar] [PubMed]
- Jiang, L.; Ngo, S.T. Altered TDP-43 Structure and Function: Key Insights into Aberrant RNA, Mitochondrial, and Cellular and Systemic Metabolism in Amyotrophic Lateral Sclerosis. Metabolites 2022, 12, 709. [Google Scholar] [CrossRef]
- Peggion, C.; Massimino, M.L.; Biancotto, G.; Angeletti, R.; Reggiani, C.; Sorgato, M.C.; Bertoli, A.; Stella, R. Absolute Quantification of Myosin Heavy Chain Isoforms by Selected Reaction Monitoring Can Underscore Skeletal Muscle Changes in a Mouse Model of Amyotrophic Lateral Sclerosis. Anal. Bioanal. Chem. 2017, 409, 2143–2153. [Google Scholar] [PubMed]
- Li, Q.; Xu, L.; Duan, H.; Yang, H.; Luo, Y.B. Common and Key Differential Pathogenic Pathways in Desminopathy and Titinopathy. Int. J. Med. Sci. 2024, 21, 2040–2051. [Google Scholar]
- Pikatza-Menoio, O.; Elicegui, A.; Bengoetxea, X.; Naldaiz-Gastesi, N.; López de Munain, A.; Gerenu, G.; Gil-Bea, F.J.; Alonso-Martín, S. The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis. J. Pers. Med. 2021, 11, 671. [Google Scholar] [PubMed]
- Yashooa, R.K.; Duranti, E.; Conconi, D.; Lavitrano, M.; Mustafa, S.A.; Villa, C. Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases. Int. J. Mol. Sci. 2025, 26, 626. [Google Scholar]
- Venditti, P.; Di Meo, S. The Role of Reactive Oxygen Species in the Life Cycle of the Mitochondrion. Int. J. Mol. Sci. 2020, 21, 2173. [Google Scholar]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123 Pt 7, 1339–1348. [Google Scholar]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [PubMed]
- Al-Sarraj, S.; King, A.; Cleveland, M.; Pradat, P.F.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar]
- Biedasek, K.; Andres, J.; Mai, K.; Adams, S.; Spuler, S.; Fielitz, J.; Spranger, J. Skeletal muscle 11beta-HSD1 controls glucocorticoid-induced proteolysis and expression of E3 ubiquitin ligases atrogin-1 and MuRF-1. PLoS ONE 2011, 6, e16674. [Google Scholar]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar]
- Chen, W.; Guo, L.; Li, M.; Wei, C.; Li, S.; Xu, R. The pathogenesis of amyotrophic lateral sclerosis: Mitochondrial dysfunction, protein misfolding and epigenetics. Brain Res. 2022, 1786, 147904. [Google Scholar]
- Shefner, J.M.; Musaro, A.; Ngo, S.T.; Lunetta, C.; Steyn, F.J.; Robitaille, R.; De Carvalho, M.; Rutkove, S.; Ludolph, A.C.; Dupuis, L. Skeletal muscle in amyotrophic lateral sclerosis. Brain 2023, 146, 4425–4436. [Google Scholar]
- Chan, Y.; Alix, J.J.P.; Neuwirth, C.; Barkhaus, P.E.; Castro, J.; Jenkins, T.M.; McDermott, C.J.; Shaw, P.J.; de Carvalho, M.; Nandedkar, S.; et al. Reinnervation as measured by the motor unit size index is associated with preservation of muscle strength in amyotrophic lateral sclerosis, but not all muscles reinnervate. Muscle Nerve 2022, 65, 203–210. [Google Scholar]
- Ding, Q.; Kesavan, K.; Lee, K.M.; Wimberger, E.; Robertson, T.; Gill, M.; Power, D.; Chang, J.; Fard, A.T.; Mar, J.C.; et al. Impaired signaling for neuromuscular synaptic maintenance is a feature of Motor Neuron Disease. Acta Neuropathol. Commun. 2022, 10, 61. [Google Scholar] [PubMed]
- Quessada, C.; Bouscary, A.; René, F.; Valle, C.; Ferri, A.; Ngo, S.T.; Loeffler, J.P. Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells 2021, 10, 1449. [Google Scholar] [CrossRef]
- Anderson, G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut Microbiome, and Muscle Interactions via the Mitochondrial Melatonergic Pathway, with Disruption by Glyphosate-Based Herbicides. Int. J. Mol. Sci. 2022, 24, 587. [Google Scholar] [PubMed]
- Dupuis, L.; Loeffler, J.P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: Insights from transgenic models. Curr. Opin. Pharmacol. 2009, 9, 341–346. [Google Scholar]
- Lloyd, E.M.; Pinniger, G.J.; Murphy, R.M.; Grounds, M.D. Slow or fast: Implications of myofibre type and associated differences for manifestation of neuromuscular disorders. Acta Physiol. 2023, 238, e14012. [Google Scholar]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar]
- Liu, W.; Chakkalakal, J.V. The Composition, Development, and Regeneration of Neuromuscular Junctions. Curr. Top. Dev. Biol. 2018, 126, 99–124. [Google Scholar]
- Cunningham, M.E.; Meehan, G.R.; Robinson, S.; Yao, D.; McGonigal, R.; Willison, H.J. Perisynaptic Schwann cells phagocytose nerve terminal debris in a mouse model of Guillain-Barré syndrome. J. Peripher. Nerv. Syst. 2020, 25, 143–151. [Google Scholar]
- Martineau, É.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. eLife 2018, 7, e41973. [Google Scholar]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar]
- Harrison, J.M.; Rafuse, V.F. Muscle fiber-type specific terminal Schwann cell pathology leads to sprouting deficits following partial denervation in SOD1(G93A) mice. Neurobiol. Dis. 2020, 145, 105052. [Google Scholar]
- Liu, W.; Klose, A.; Forman, S.; Paris, N.D.; Wei-LaPierre, L.; Cortés-Lopéz, M.; Tan, A.; Flaherty, M.; Miura, P.; Dirksen, R.T.; et al. Loss of adult skeletal muscle stem cells drives age-related neuromuscular junction degeneration. eLife 2017, 6, e26464. [Google Scholar]
- Picchiarelli, G.; Demestre, M.; Zuko, A.; Been, M.; Higelin, J.; Dieterlé, S.; Goy, M.A.; Mallik, M.; Sellier, C.; Scekic-Zahirovic, J.; et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 2019, 22, 1793–1805. [Google Scholar] [PubMed]
- Castellanos-Montiel, M.J.; Durcan, T.M. TDP-43 Dysregulation and Neuromuscular Junction Disruption in Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2022, 11, 56. [Google Scholar]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Investig. 2010, 120, 11–19. [Google Scholar] [PubMed]
- Le Grand, F.; Rudnicki, M. Satellite and stem cells in muscle growth and repair. Development 2007, 134, 3953–3957. [Google Scholar]
- Buckingham, M. Skeletal muscle progenitor cells and the role of Pax genes. Comptes Rendus Biol. 2007, 330, 530–533. [Google Scholar]
- Manzano, R.; Toivonen, J.M.; Oliván, S.; Calvo, A.C.; Moreno-Igoa, M.; Muñoz, M.J.; Zaragoza, P.; García-Redondo, A.; Osta, R. Altered expression of myogenic regulatory factors in the mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 2011, 8, 386–396. [Google Scholar]
- Krivickas, L.S.; Yang, J.I.; Kim, S.K.; Frontera, W.R. Skeletal muscle fiber function and rate of disease progression in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 636–643. [Google Scholar]
- Hegedus, J.; Putman, C.T.; Gordon, T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007, 28, 154–164. [Google Scholar]
- Manzano, R.; Toivonen, J.M.; Calvo, A.C.; Oliván, S.; Zaragoza, P.; Muñoz, M.J.; Montarras, D.; Osta, R. Quantity and activation of myofiber-associated satellite cells in a mouse model of amyotrophic lateral sclerosis. Stem Cell Rev. Rep. 2012, 8, 279–287. [Google Scholar] [PubMed]
- Manzano, R.; Toivonen, J.M.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; López-Royo, T.; Molina, N.; Zaragoza, P.; Calvo, A.C.; Osta, R. What skeletal muscle has to say in amyotrophic lateral sclerosis: Implications for therapy. Br. J. Pharmacol. 2021, 178, 1279–1297. [Google Scholar]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2016, 2016, 5930621. [Google Scholar]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [PubMed]
- Gillon, A.; Nielsen, K.; Steel, C.; Cornwall, J.; Sheard, P. Exercise attenuates age-associated changes in motoneuron number, nucleocytoplasmic transport proteins and neuromuscular health. Geroscience 2018, 40, 177–192. [Google Scholar] [PubMed]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar]
- Garbugino, L.; Golini, E.; Giuliani, A.; Mandillo, S. Prolonged Voluntary Running Negatively Affects Survival and Disease Prognosis of Male SOD1G93A Low-Copy Transgenic Mice. Front. Behav. Neurosci. 2018, 12, 275. [Google Scholar]
- Nicolis di Robilant, V.; Scardigli, R.; Strimpakos, G.; Tirone, F.; Middei, S.; Scopa, C.; De Bardi, M.; Battistini, L.; Saraulli, D.; Farioli Vecchioli, S. Running-Activated Neural Stem Cells Enhance Subventricular Neurogenesis and Improve Olfactory Behavior in p21 Knockout Mice. Mol. Neurobiol. 2019, 56, 7534–7556. [Google Scholar]
- Sailani, M.R.; Halling, J.F.; Møller, H.D.; Lee, H.; Plomgaard, P.; Pilegaard, H.; Snyder, M.P.; Regenberg, B. Lifelong physical activity is associated with promoter hypomethylation of genes involved in metabolism, myogenesis, contractile properties and oxidative stress resistance in aged human skeletal muscle. Sci. Rep. 2019, 9, 3272. [Google Scholar]
- Tseng, C.; Sinha, K.; Pan, H.; Cui, Y.; Guo, P.; Lin, C.Y.; Yang, F.; Deng, Z.; Eltzschig, H.K.; Lu, A.; et al. Markers of Accelerated Skeletal Muscle Regenerative Response in Murphy Roths Large Mice: Characteristics of Muscle Progenitor Cells and Circulating Factors. Stem Cells 2019, 37, 357–367. [Google Scholar] [PubMed]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [PubMed]
- Zhao, H.; He, Z.; Yun, H.; Wang, R.; Liu, C. A Meta-Analysis of the Effects of Different Exercise Modes on Inflammatory Response in the Elderly. Int. J. Environ. Res. Public Health 2022, 19, 10451. [Google Scholar]
- Simioni, C.; Zauli, G.; Martelli, A.M.; Vitale, M.; Sacchetti, G.; Gonelli, A.; Neri, L.M. Oxidative stress: Role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget 2018, 9, 17181–17198. [Google Scholar]
- Rebelo-Marques, A.; De Sousa Lages, A.; Andrade, R.; Ribeiro, C.F.; Mota-Pinto, A.; Carrilho, F.; Espregueira-Mendes, J. Aging Hallmarks: The Benefits of Physical Exercise. Front. Endocrinol. 2018, 9, 258. [Google Scholar]
- Wyckelsma, V.L.; Levinger, I.; McKenna, M.J.; Formosa, L.E.; Ryan, M.T.; Petersen, A.C.; Anderson, M.J.; Murphy, R.M. Preservation of skeletal muscle mitochondrial content in older adults: Relationship between mitochondria, fibre type and high-intensity exercise training. J. Physiol. 2017, 595, 3345–3359. [Google Scholar]
- Ruiz-Iglesias, P.; Estruel-Amades, S.; Camps-Bossacoma, M.; Massot-Cladera, M.; Franch, À.; Pérez-Cano, F.J.; Castell, M. Influence of Hesperidin on Systemic Immunity of Rats Following an Intensive Training and Exhausting Exercise. Nutrients 2020, 12, 1291. [Google Scholar] [CrossRef] [PubMed]
- Lanuza, M.A.; Just-Borràs, L.; Hurtado, E.; Cilleros-Mañé, V.; Tomàs, M.; Garcia, N.; Tomàs, J. The Impact of Kinases in Amyotrophic Lateral Sclerosis at the Neuromuscular Synapse: Insights into BDNF/TrkB and PKC Signaling. Cells 2019, 8, 1578. [Google Scholar]
- Deforges, S.; Branchu, J.; Biondi, O.; Grondard, C.; Pariset, C.; Lécolle, S.; Lopes, P.; Vidal, P.P.; Chanoine, C.; Charbonnier, F. Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2009, 587, 3561–3572. [Google Scholar]
- Flis, D.J.; Dzik, K.; Kaczor, J.J.; Cieminski, K.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 233. [Google Scholar]
- Pradhan, J.; Noakes, P.G.; Bellingham, M.C. The Role of Altered BDNF/TrkB Signaling in Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2019, 13, 368. [Google Scholar]
- Park, D.; Kwak, S.G.; Park, J.S.; Choo, Y.J.; Chang, M.C. Can Therapeutic Exercise Slow Down Progressive Functional Decline in Patients With Amyotrophic Lateral Sclerosis? A Meta-Analysis. Front. Neurol. 2020, 11, 853. [Google Scholar]
- Musarò, A.; Dobrowolny, G.; Cambieri, C.; Onesti, E.; Ceccanti, M.; Frasca, V.; Pisano, A.; Cerbelli, B.; Lepore, E.; Ruffolo, G.; et al. Neuromuscular magnetic stimulation counteracts muscle decline in ALS patients: Results of a randomized, double-blind, controlled study. Sci. Rep. 2019, 9, 2837. [Google Scholar]
- Lunetta, C.; Lizio, A.; Sansone, V.A.; Cellotto, N.M.; Maestri, E.; Bettinelli, M.; Gatti, V.; Melazzini, M.G.; Meola, G.; Corbo, M. Strictly Monitored Exercise Programs Reduce Motor Deterioration in ALS: Preliminary Results of a Randomized Controlled. Trial. J. Neurol. 2016, 263, 52–60. [Google Scholar] [PubMed]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.; Owegi, M.; Goutman, S.A.; et al. Trial of AMX0035 for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Phase 3 Trial of Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Gene | Common Mutations | Main Pathological Mechanism | Clinical Features |
|---|---|---|---|
| SOD1 | G93A, A4V, H46R, D90A (recessive in Scandinavian population) |
|
|
| TARDBP (TDP-43) | Mutations in the glycine-rich C-terminal region |
|
|
| FUS/TLS | Mutations in C-terminal region of NLS |
|
|
| C9ORF72 | GGGGCC (G4C2) intronic hexanucleotide expansion |
|
|
| Mechanism | Physiological Role | Alterations in ALS | Consequences on Muscle |
|---|---|---|---|
| Neuromuscular innervation | Signal transmission between motor neurons and muscle fibers | Motor neuron degeneration and progressive loss of neuromuscular junctions (NMJs) | Denervation, muscle atrophy |
| Mitochondria | ATP production, redox homeostasis | Structural disorganization, increased ROS, mitochondrial DNA damage | Energy crisis, oxidative stress |
| Proteostasis (UPS) | Degradation of damaged or misfolded proteins | Accumulation of toxic protein aggregates (e.g., mutant SOD1), UPS dysfunction | Cellular toxicity, pathological inclusions |
| Autophagy | Clearance of damaged organelles and protein aggregates | Dysregulated activation or chronic stimulation | Impaired turnover, buildup of dysfunctional components |
| Local inflammation | Controlled immune response to injury | Chronic activation of microglia and astrocytes, increased TNF-α and IL-1β secretion | Hostile environment, inhibition of regeneration |
| Muscle fiber structure | Contraction, adaptation to mechanical stimuli | Reduction in cross-sectional area, selective loss of fast-twitch fibers | Progressive weakness, altered morphology |
| NMJ and synaptic signaling | Stabilization of acetylcholine receptors and nerve-muscle communication | Disruption of pre/post-synaptic elements, loss of nAChR clustering | NMJ instability, functional impairment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duranti, E. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis: State of the Art 2025. Muscles 2025, 4, 22. https://doi.org/10.3390/muscles4030022
Duranti E. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis: State of the Art 2025. Muscles. 2025; 4(3):22. https://doi.org/10.3390/muscles4030022
Chicago/Turabian StyleDuranti, Elisa. 2025. "The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis: State of the Art 2025" Muscles 4, no. 3: 22. https://doi.org/10.3390/muscles4030022
APA StyleDuranti, E. (2025). The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis: State of the Art 2025. Muscles, 4(3), 22. https://doi.org/10.3390/muscles4030022