
Genetic Diversity Among Independent Isolates of the Dolichocephalovirinae Subfamily


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Culture Preparation
2.2. Bacteriophage Isolation and Cultivation
2.3. Host Range Determination
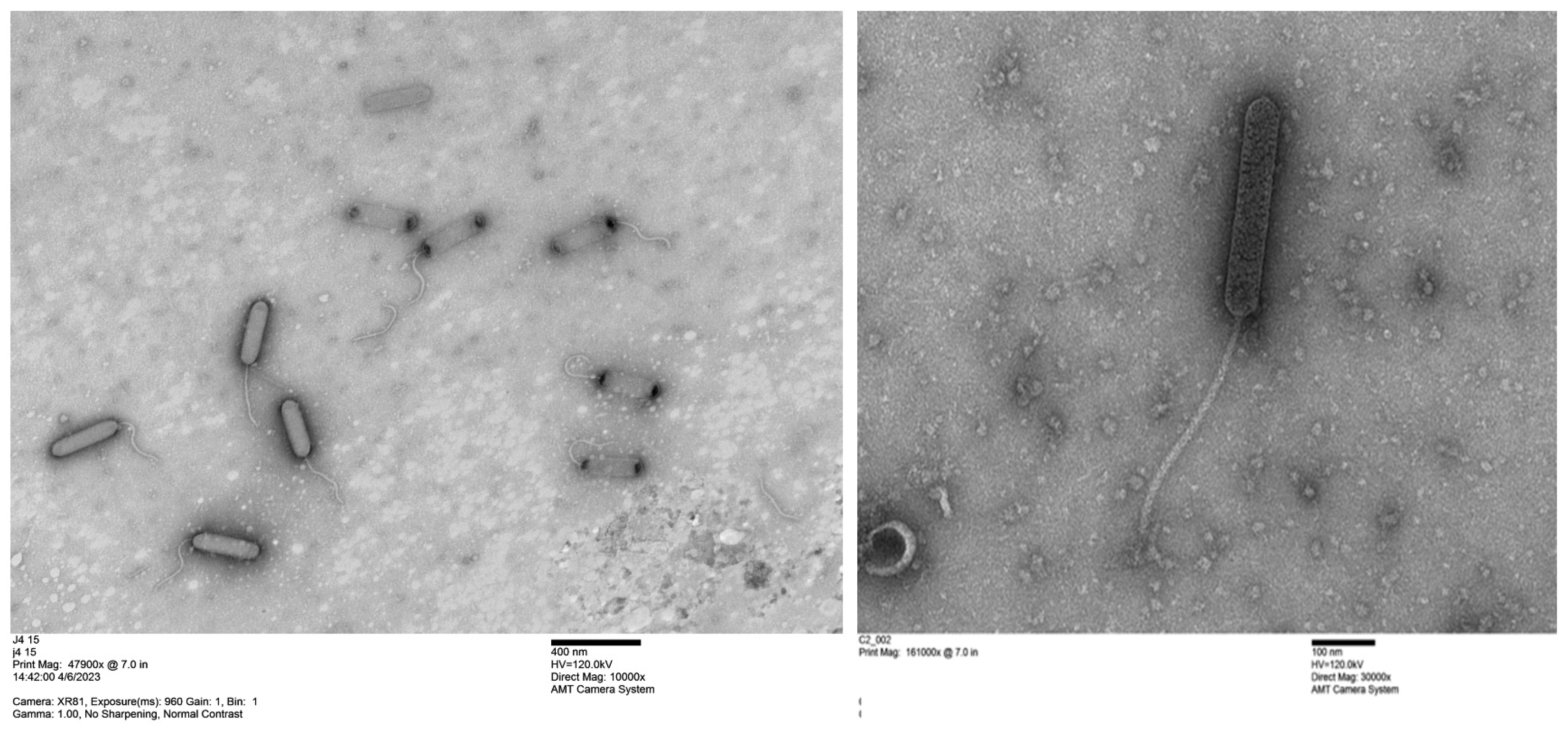
2.4. Transmission Electron Microscope Analysis of Purified Phages with Uranyl Acetate Staining
2.5. Phage Genome Sequencing and Annotation
3. Results and Discussion
3.1. Isolation and Characterization of Ten Bacteriophages from Rocky Branch Creek
3.2. Characterization of Bacteriophage Host Ranges
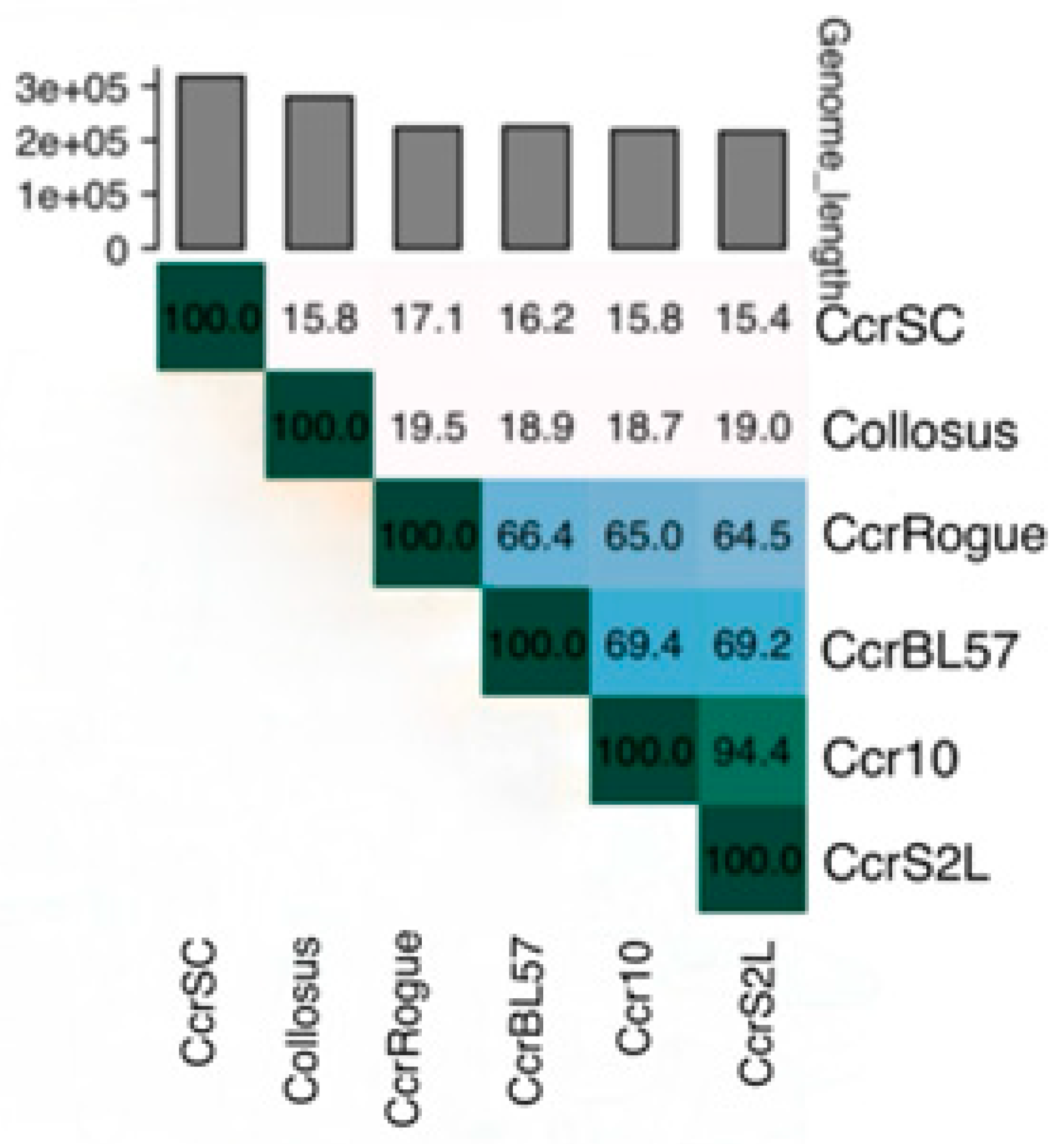
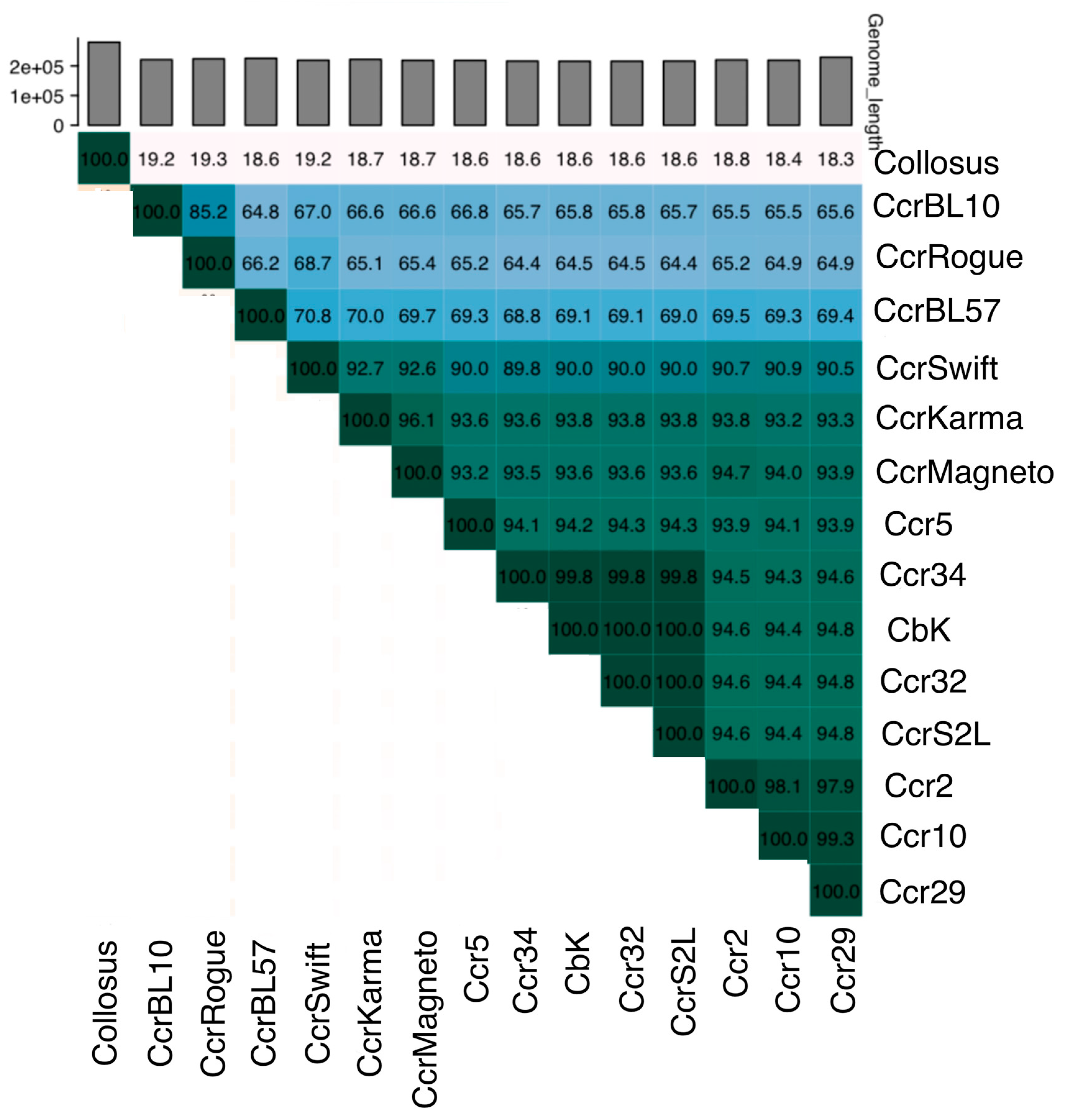
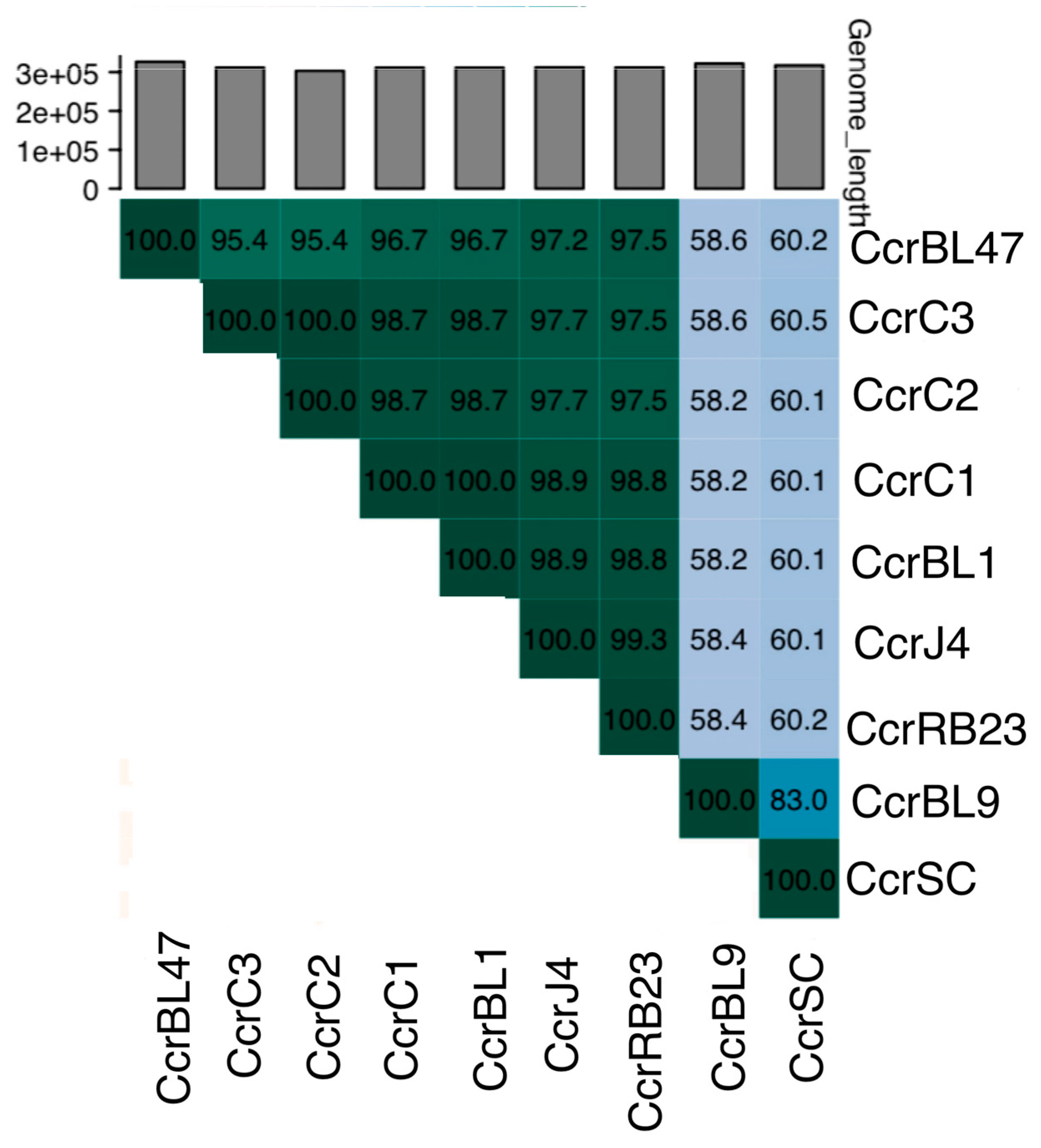
3.3. Genetic Diversity and Taxonomic Classification of Dolichocephalovirinae Phages
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tubman, E. Bacteriophages: Methods and Protocols; Springer: New York, NY, USA, 2024; Volume 2738, pp. 1–431. [Google Scholar] [CrossRef]
- Kosznik-Kwasnicka, K.; Topka, G.; Mantej, J.; Grabowski, L.; Necel, A.; Wegrzyn, G.; Wegrzyn, A. Propagation, purification, and characterization of bacteriophages for phage therapy. In Bacteriophages: Methods and Protocols; Springer: New York, NY, USA, 2024; Volume 2738, pp. 357–400. [Google Scholar]
- Laanto, E. Overcoming bacteriophage resistance in phage therapy. In Bacteriophages: Methods and Protocols; Springer: New York, NY, USA, 2024; Volume 2738, pp. 401–410. [Google Scholar]
- Rout, A.K.; Dixit, S.; Tripathy, P.S.; Rout, S.S.; Parida, S.N.; Parida, P.K.; Sarkar, D.J.; Das, B.K.; Singh, A.K.; Behera, B.K. Metagenomic Landscape of Sediments of River Ganga Reveals Microbial Diversity, Potential Plastic and Xenobiotic Degradation Enzymes. J. Hazard. Mater. 2024, 471, 134377. [Google Scholar] [CrossRef]
- Wang, S.; Nie, W.; Gu, Q.; Wang, X.; Yang, D.; Li, H.; Wang, P.; Liao, W.; Huang, J.; Yuan, Q.; et al. Spread of antibiotic resistance genes in drinking water reservoirs: Insights from a deep metagenomic study using a curated database. Water Res 2024, 1, 121572. [Google Scholar] [CrossRef]
- Das, B.K.; Behera, B.K.; Chakraborty, H.J.; Paria, P.; Gangopadhyay, A.; Rout, A.K.; Nayak, K.K.; Parida, P.K.; Rai, A. Metagenomic Study Focusing on Antibiotic Resistance Genes from the Sediments of River Yamuna. Gene 2020, 758, 144951. [Google Scholar] [CrossRef]
- O’Neil, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. The Review on Antimicrobial Resistance 2014. Available online: https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (accessed on 4 October 2024).
- Wittebole, X.; De Roock, S.; Opal, S.M. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef]
- Ely, B.; Hils, M.; Clarke, A.; Albert, M.; Holness, N.; Lenski, J.; Mohammadi, T. New genera and species of Caulobacter and Brevundimonas bacteriophages provide insights into phage genome evolution. Viruses 2024, 16, 641. [Google Scholar] [CrossRef]
- Barrows, J.M.; Goley, E.D. Synchronized swarmers and sticky stalks: Caulobacter crescentus as a model for bacterial cell biology. J. Bacteriol. 2023, 205, e00384-22. [Google Scholar] [CrossRef]
- Chong, T.N.; Shapiro, L. Bacterial cell differentiation enables population level survival strategies. mBio 2024, 15, e00758-24. [Google Scholar] [CrossRef]
- Hallgren, J.; Jonas, K. Nutritional control of bacterial DNA replication. Curr. Opin. Microbiol. 2024, 77, 102403. [Google Scholar] [CrossRef]
- Fröhlich, K.S.; Gomariz, M.V. RNA-controlled regulation in Caulobacter crescentus. Curr. Opin. Microbiol. 2021, 60, 1–7. [Google Scholar] [CrossRef]
- Ely, B.; Berrios, L.; Thomas, Q. S2B, a temperate bacteriophage that infects Caulobacter crescentus strain CB15. Curr. Microbiol. 2022, 79, 98. [Google Scholar]
- Gill, J.J.; Berry, J.D.; Russell, W.K.; Lessor, L.; Escobar-Garcia, D.A.; Hernandez, D.; Kane, A.; Keene, J.; Maddox, M.; Martin, R.; et al. The Caulobacter crescentus phage phiCbK: Genomics of a canonical phage. BMC Genom. 2012, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Ash, K.T.; Drake, K.M.; Gibbs, W.S.; Ely, B. Genomic diversity of type B3 bacteriophages of Caulobacter crescentus. Curr. Microbiol. 2017, 74, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Dillard, R.S.; Storms, R.E.; De Masi, L.; Hampton, C.; Panis, G.; Viollier, P.H.; Wright, E.R. Analysis of phage-pilus interactions in Caulobacter crescentus. Microsc. Microanal. 2016, 22 (Suppl. S3), 202. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.C.; Viollier, P.H.; Ely, B.; Poindexter, J.S.; Georgieva, M.; Jensen, G.J.; Wright, E.R. A novel mechanism for bacteriophage adsorption to the motile bacterium Caulobacter crescentus. Proc. Nat. Acad. Sci. USA 2011, 108, 9963–9968. [Google Scholar] [CrossRef]
- Johnson, R.C.; Ely, B. Analysis of non-motile mutants of the dimorphic bacterium Caulobacter crescentus. J. Bacteriol. 1979, 137, 627–634. [Google Scholar] [CrossRef]
- Wilson, K.; Ely, B. Analyses of four new Caulobacter PhiCbKviruses indicate independent lineages. J. Gen. Virol. 2019, 100, 321–331. [Google Scholar] [CrossRef]
- Agabian-Keshishian, N.; Shapiro, L. Stalked bacteria: Properties of deoxyribonucleic acid bacteriophage PhiCbK. J. Virol. 1970, 5, 795–800. [Google Scholar] [CrossRef]
- Johnson, R.; Wood, N.; Ely, B. Isolation and characterization of bacteriophages for Caulobacter crescentus. J. Gen. Virol. 1977, 37, 323–335. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Mohammadi, T.; Ely, B. The isolation and characterization of novel Caulobacter and Non-Caulobacter lysogenic bacteria from soil and the discovery of broad-host-range phages infecting multiple genera. Microorganisms 2024, 12, 1894. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the rapid annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2013, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 10, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Bau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Ely, B. Genomic GC content drifts downward in most bacterial genomes. PLoS ONE 2021, 16, e0244163. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage Name | CcrS2L | CcrC1 | CcrC2 | CcrC3 | CcrJ4 | CcrBL1 | Ccr BL47 | Ccr RB23 | Ccr BL57 |
|---|---|---|---|---|---|---|---|---|---|
| Date collected | January 2018 | January 2018 | January 2018 | January 2018 | June 2018 | January 2016 | January 2016 | January 2019 | January 2016 |
| Plaque size (mm) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Genome size (kbp) | 216 | 312 | 303 | 312 | 313 | 312 | 327 | 312 | 226 |
| GC content (%) | 66.2 | 64.2 | 64.2 | 64.1 | 64.2 | 64.2 | 64.1 | 64.2 | 66.3 |
| tRNA genes | 24 | 25 | 25 | 25 | 25 | 25 | 25 | 25 | 25 |
| # of genes | 347 | 542 | 519 | 530 | 563 | 539 | 581 | 543 | 384 |
| Host | CcrS2L | CcrC1 | CcrC2 | CcrC3 | CcrJ4 | Ccr BL1 | Ccr BL49 | Ccr RB23 | Ccr BL57 |
|---|---|---|---|---|---|---|---|---|---|
| CB15 | + | + | + | + | + | + | + | + | + |
| CB13 | + | + | + | + | + | + | + | + | + |
| CB2 | + | + | + | + | + | + | + | + | + |
| CBR1 | + | + | + | + | + | − | + | + | + |
| TK0059 | + | − | − | − | − | − | + | + | |
| RBW14 | + | + | + | + | - | + | + | + | |
| RBW21 | − | + | + | - | - | - | + | + | − |
| RBW22 | + | + | + | + | + | + | + | + | + |
| RBW23 | + | + | + | + | + | + | − | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ely, B.; Thomas, Q.; Mohammadi, T. Genetic Diversity Among Independent Isolates of the Dolichocephalovirinae Subfamily. Bacteria 2025, 4, 8. https://doi.org/10.3390/bacteria4010008
Ely B, Thomas Q, Mohammadi T. Genetic Diversity Among Independent Isolates of the Dolichocephalovirinae Subfamily. Bacteria. 2025; 4(1):8. https://doi.org/10.3390/bacteria4010008
Chicago/Turabian StyleEly, Bert, Quill Thomas, and Tannaz Mohammadi. 2025. "Genetic Diversity Among Independent Isolates of the Dolichocephalovirinae Subfamily" Bacteria 4, no. 1: 8. https://doi.org/10.3390/bacteria4010008
APA StyleEly, B., Thomas, Q., & Mohammadi, T. (2025). Genetic Diversity Among Independent Isolates of the Dolichocephalovirinae Subfamily. Bacteria, 4(1), 8. https://doi.org/10.3390/bacteria4010008