1. Introduction
Influenza A virus is classified as being part of the family of
Orthomyxoviridae and the genus of Influenza A virus, and it contains a negative-sense, single-stranded RNA genome composed of eight gene segments encoding at least 11 viral proteins [
1,
2]. IAVs are divided into low-pathogenic (LP) and highly pathogenic (HP) pathotypes based on the genetic features and/or disease severity in chickens. To date, 16 hemagglutinin (HA) and 9 neuraminidase (NA) subtypes of IAVs have been isolated from aquatic and domestic birds [
3]. However, only the H5 and H7 subtypes of IAV have been found to possess the potential to mutate into highly pathogenic viruses during circulation in poultry [
4,
5,
6]. All H5 and H7 IA viruses have been classified as notifiable viruses by the World Organization for Animal Health (WOAH) since 2005 [
7]. Highly pathogenic avian influenza viruses (HPIAVs) that originated from low-pathogenic avian influenza viruses (LPIAVs) have mostly been eradicated from poultry via stamping-out programs, except for the Gs/GD lineage of H5, which rose to prominence in 1996. The Gs/GD lineage of H5 viruses has spread and been maintained in poultry and wild aquatic bird reservoirs until today, producing infections in poultry, wild birds, and mammals, including humans in many countries in Asia, Africa, Europe, and North America [
8,
9]. Currently, H5 HPIAVs have been classified into clades 0–9 and different subclades based on phylogenetic analysis of their HA genes [
10]. The clade 2.3.4.4 HA of H5 viruses has evolved into eight subclades, namely, clades 2.3.4.4a to 2.3.4.4h [
11]. Since the H5N1 viruses bearing the clade 2.3.4.4b HA gene emerged in October 2020 in the Netherlands, the viruses have spread to many countries in Europe, Africa, Asia, and North America and are responsible for the loss of millions of domestic poultry [
12,
13,
14,
15]. The same viruses emerged in Canada in the winter of 2021 through migratory wild birds and have spread to all 10 provinces, resulting in a loss of over 7 million domestic birds [
16,
17].
To prevent the extensive circulation of notifiable IAVs in both wild birds and domestic poultry populations, mandated serological surveillance and survey programs have been implemented worldwide. Classical laboratory serologic tools include agar gel immunodiffusion (AGID), the hemagglutination inhibition (HI) test, and the virus neutralization (VN) test. The AGID test is used to detect group-specific antigens of the influenza A virus (i.e., nucleoprotein and matrix proteins). It is a simple and economical assay but it is time-consuming and not suitable for large-scale specimen screening [
18]. The HI assay is considered to be the reference test by WOAH. It is the subtype-specific test for influenza serologic differential diagnosis, but the downsides of this test include antigen selection and titration, nonspecific inhibitor removal, and a long processing time [
19]. The VN test is recommended for the identification of HPIAV using live infectious viruses. Thus, the test needs to be performed in higher-level containment labs and has significant limitations in rapid and high-throughput diagnostics [
19]. Conversely, enzyme-linked immunosorbent assay (ELISA) is widely accepted as an essential method that is superior in regard to throughput, turnaround time, and accuracy for early diagnosis [
20]. Among the 11 proteins encoded by the IAV, haemagglutinin (HA) and nucleoprotein (NP) are particularly targeted for serological diagnosis due to their antigenic abundance [
21]. An indirect ELISA is considered a valuable and inexpensive test routinely used for the detection of antibodies to IAV in birds. However, this technique is not feasible when screening samples from a wide range of bird species. Alternatively, the competitive ELISA (cELISA) is suitable for broader use in multiple animal species as a species-specific enzyme conjugate is not required [
22].
The success of a reliable cELISA requires a highly specific monoclonal antibody (mAb) to recognize a broadly conserved and dominant antigenic epitope. In addition to the availability of two commercial multispecies NP-based cELISA kits (i.e., IDVet, Montpellier, France, and Idexx, Westbrook, ME, USA), a recombinant NP-based cELISA developed in our laboratory is routinely used as a screening tool for IAV serological monitoring owing to the group-specific feature of the NP antigen [
23]. Previously, Yang and colleagues in our laboratory successfully generated a high-affinity, subtype-specific mAb #10 (F37H5N1-56) based on immunizing mice with the inactivated American-H5 (clade Am-nonGs/GD) LPIAV strain, A/Turkey/ON/6213/1966 (H5N1). This mAb has been shown to react with sera raised from a wide range of HP and LP H5 strains of North American and Eurasian origins [
24]. In this study, a cELISA immunoassay based on mAb #10 (F37H5N1-56) and the recombinant H5 HA antigen derived from a Eurasian strain A/Teal/Germany/Wv632/2005 (H5N1) (clade EA-nonGs/GD) was developed, validated and evaluated with high specificity, sensitivity, and low variability. In particular, the developed cELISA is capable of detecting antibodies in serum samples collected from different species of animals that were infected with currently circulating clade 2.3.4.4b H5Nx viruses.
2. Materials and Methods
2.1. Preparation of Viruses
IAVs of the H5 subtype used in this study were acquired from the National Centre for Foreign Animal Disease (NCFAD) viral depository. Allantoic fluid from virus-inoculated embryonated chicken eggs was collected and tested by hemagglutination assay (HA) as previously described [
24]. All procedures were performed under biocontainment level (BSL) 3 conditions.
2.2. Preparation and Expression of Recombinant HA Proteins
The preparation and expression of recombinant HA protein were described previously [
25]. For the development of the competitive ELISA, we used a recombinantly expressed, full-length hemagglutinin protein of A/Teal/Germany/Wv632/2005 (H5N1) (GenBank accession no. CY061885). This full-length H5 hemagglutinin gene was cloned into a pAB- bee
TM-FH vector (AB Vector, LLC, San Diego, CA, USA) by GenScript (GenScript USA Inc., Piscataway, NJ, USA). The vector containing the desired gene sequences was then purified and co-transfected with linearized baculovirus vector DNA, ProFold
TM-ER1 (AB Vector, LLC, San Diego, CA, USA), into
Spodoptera frugiperda (Sf9) insect cells to generate a recombinant baculovirus containing a full-length HA gene. This recombinant baculovirus was plaque-purified and sequenced to verify the correct nucleotide sequence. The titer of the recombinant H5 baculoviral stock (PFU/mL) was determined by a viral plaque assay.
Trichoplusia ni (
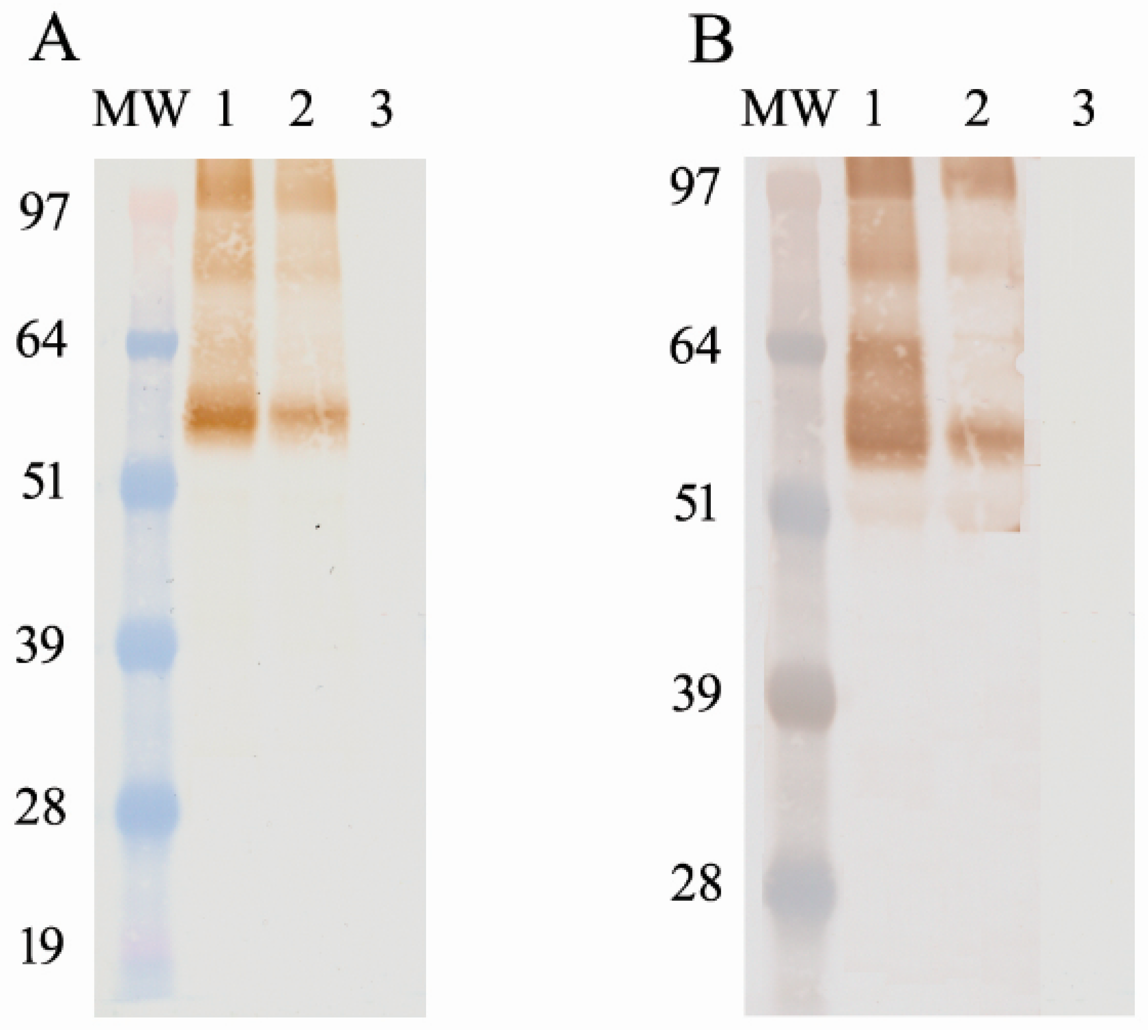
Tni) insect cells were then infected with the recombinant virus at an MOI of 10. The infected insect cells were incubated for 72 h at 27 °C via shaking and were harvested via centrifugation (10,000 rpm for 20 min). The cell pellet was lysed using the I-PER insect cell protein extraction reagent (Pierce Biotechnology, Rockford, IL, USA) on ice for 10 min, and then was centrifuged again at 15,000 rpm for 20 min. The soluble protein in the supernatant was purified using Ni-NTA resin. The recombinant protein was detected and confirmed via Western blotting using a 6× histidine-specific mAb (Novagen, Billerica, MA, USA). After the determination of the correct protein size, the concentration of the antigen was determined using the Qubit 2.0 Flex and bicinchoninic acid assay (BCA). An average between the two methods was used to calculate the antigen-per-well concentration.
2.3. Hemagglutination Inhibition (HI) Assay
The HI assay was performed according to the OIE manual [
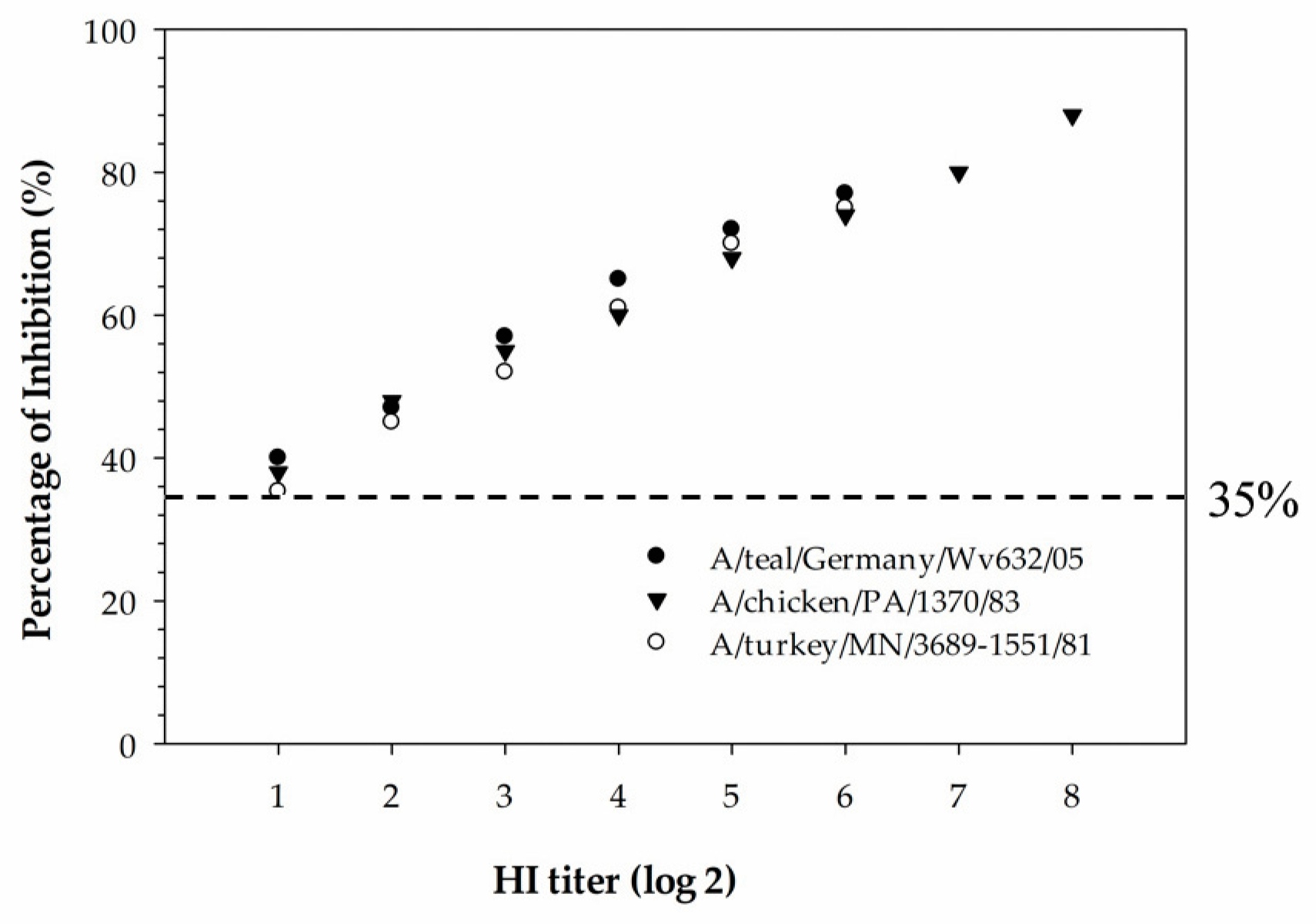
26]. The HI titer was expressed as the highest dilution of serum, resulting in the complete inhibition of 4 hemagglutinin units (HAUs) of the virus. The HI titer was regarded as being positive if there was inhibition at a serum dilution of 1:16 or more against 4 HAUs of the virus. Hemagglutination inhibition titers for the H5-positive chicken antisera were obtained using a homologous virus.
2.4. Sera Used in This Study
Chickens and turkeys of specific-pathogen-free flocks were used in the production of Influenza A virus antisera. All procedures involving experimental animal inoculations and care complied with the Canadian Council of Animal Care guidelines. These animals were allowed to acclimate for 5–7 days prior to inoculations. For LPIA viruses, fresh allantoic fluid was collected that contained an HA of greater than 1:16, which was used and diluted 1:10 in PBS. For HPAI viruses, fresh allantoic fluid with an HA of greater than 1:16 was collected and inactivated via binary ethylenimine inactivation (BEI) or β-propiolactone (BPL) virus inactivation procedures, as stated above. The inactivated HPAI viruses were then formulated with 50 µg of QuilA/mL of allantoic fluid prior to inoculation. Before inoculation of the birds, the undiluted virus was plated on blood agar plates and incubated at 37 °C ± 2 °C overnight. If plates presented positive for bacterial presence, the inoculum was then filtered through a 0.45 µm syringe filter. If not already diluted, the inoculum was diluted 1:10 in PBS prior to inoculation (LPIA viruses were already previously diluted).
LPIA viruses were inoculated intravenously via the wing vein in conjunction with three drops of inoculum that were administrated via the oral, nasal, and ocular routes of the birds. HPAI viruses were injected intramuscularly along with Emulsigen adjuvant into different regions of the pectoral muscle. Approximately 0.25 mL of HPAI inoculum was administrated into each injection site. A predetermined schedule was made in consultation with veterinarians for bleeding. These serum samples were then collected from each infected/immunized bird and tested for the presence/absence of antibodies against the inoculated virus by HI assay. Booster doses were determined using the HI reactivity results. Once the desired antibody response and titer had been achieved, the animals were anesthetized and exsanguinated via predesignated protocols. Serum pools were then created and tested again for HI reactivity to obtain a greater than 1:16 positive result. The serum was then aliquoted and stored at −20 °C for later usage.
Sera used to assess the diagnostic sensitivity and specificity include experimental anti-H5 sera and field sera submitted through the Canadian Notifiable Avian Influenza Surveillance System (CanNAISS). The IAV-positive status was confirmed by IAV-NP cELISA [
23] and the subtype status was confirmed by HI assay.
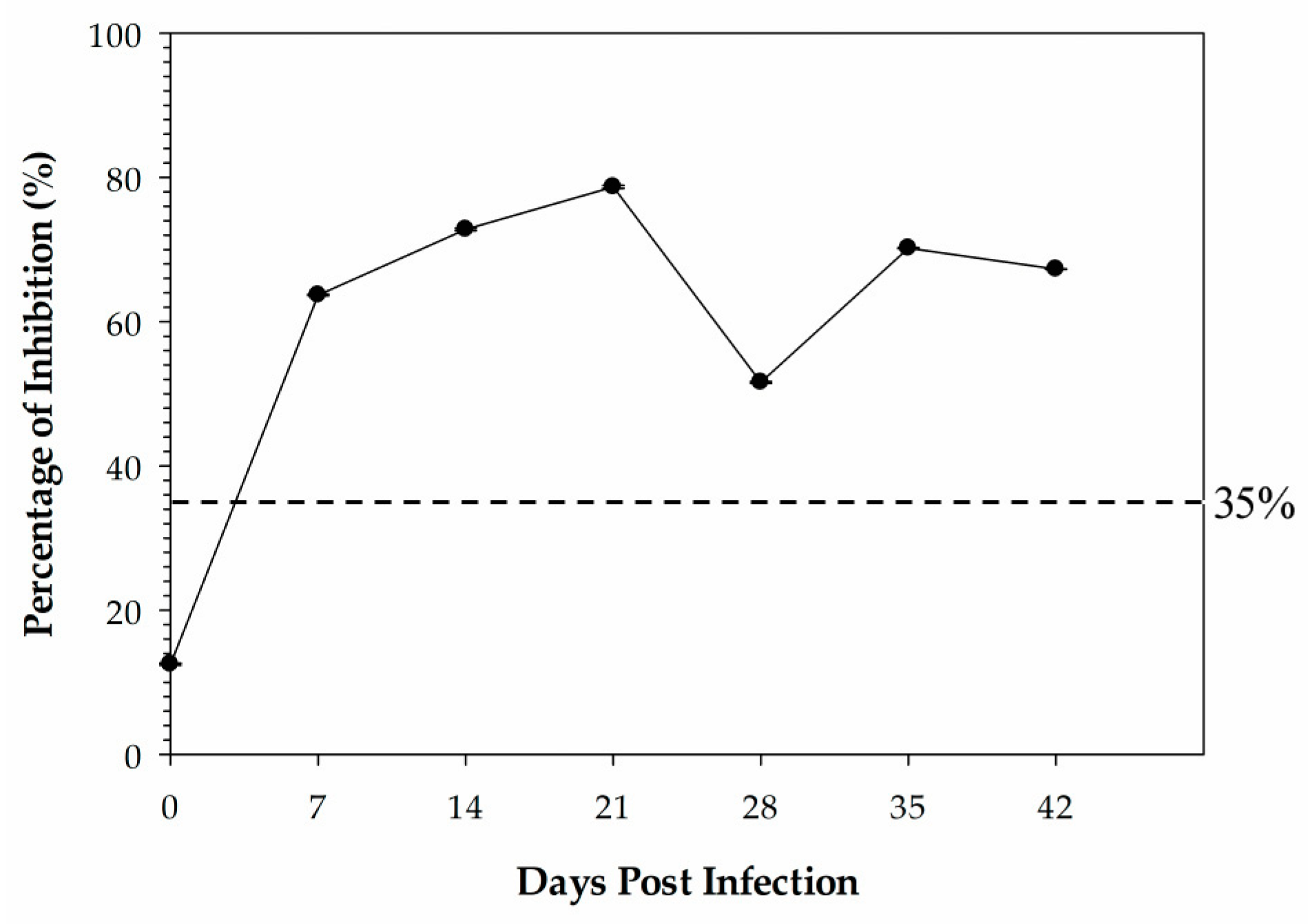
Sera used for the kinetic time course antibody response study were derived from 4 specific-pathogen-free (SPF) leghorn chickens that were inoculated with 0.5 mL of the β-propiolactone-inactivated antigen (A/Teal/Germany/Wv632/2005 (H5N1)) by intramuscular injection and housed in the same room. Chicken sera were collected at 0 days post-vaccination (DPI), 14 DPI, 21 DPI, 28 DPI, 35 DPI, and 42 DPI.
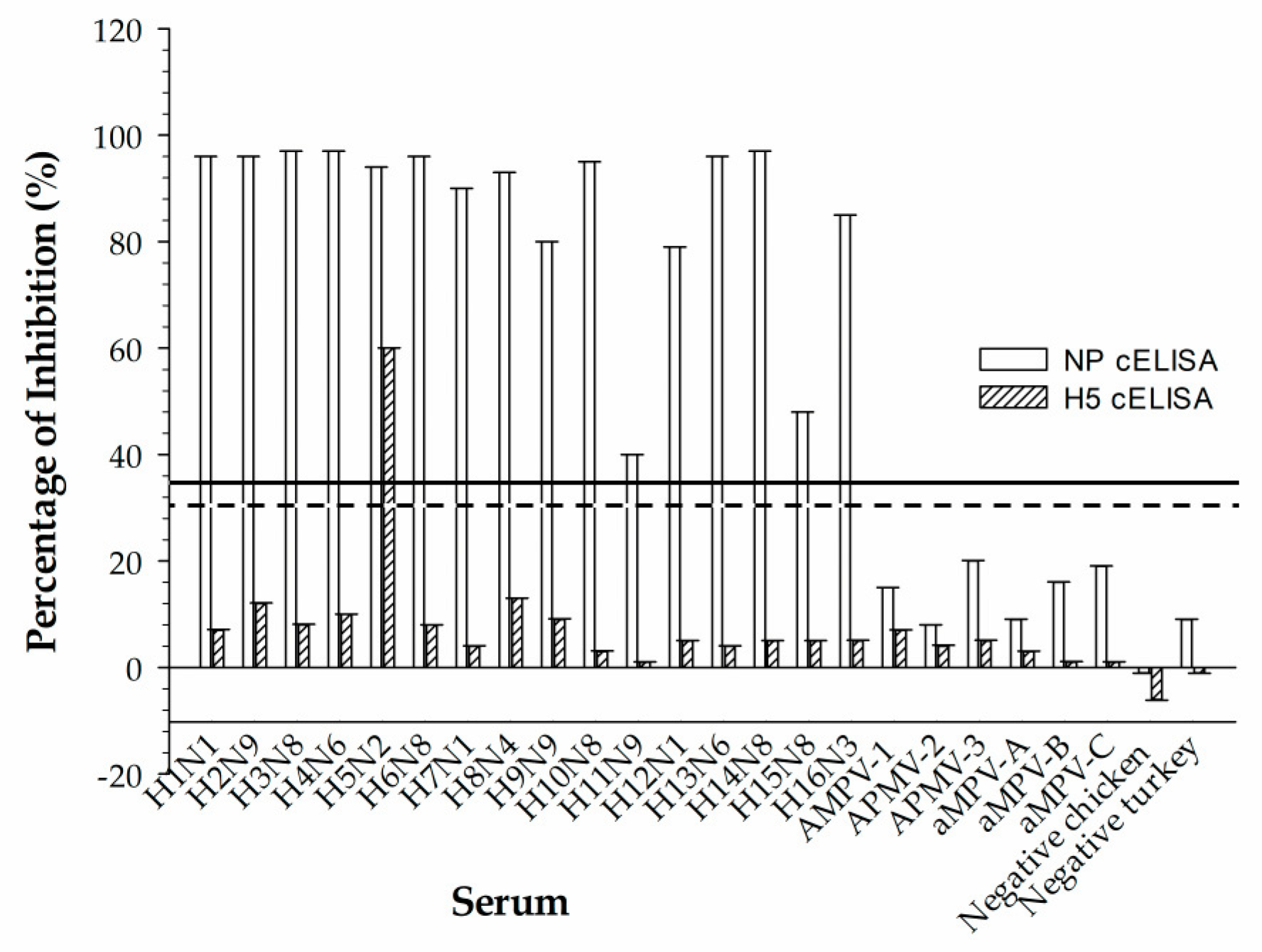
Reference chicken sera that represent 16 serotypes of avian influenza virus (IAV); 3 paramyxoviruses (APMV), types 1, 2, and 3; and avian metapneumovirus subtypes A, B, and C were used to assess cross-reactivity in the rec-H5 cELISA. The virus strains used to produce 16 IAV reference sera were A/Swine/Iowa/31 (H1N1), A/Pintail/AB/293/77 (H2N9), A/Duck/UK/1/63 (H3N8), A/Duck/Czech/56 (H4N6), A/Turkey/BC/FAV5-52/09 (H5N2), A/Turkey/Mass/65 (H6N2), A/Chicken/BC/514/04 (H7N3), A/Turkey/ON/6118/162 (H8N4), A/Turkey/MN/12877/1285/81 (H9N2), A/Chicken/Germany/N/49 (H10N7), A/Duck/Memphis/546/74 (H11N9), A/Duck/AB/60/76 (H12N5), A/Gull/MD/704/77/BEL-42 (H13N1), A/Mallard/Gurjev/263/82 (H14N5), A/Duck/Australia/341/83 (H15N8) and A/BHG/Sweden/5/99 (H16N3). The virus strains used to produce three APMV reference sera were Newcastle disease virus (NDV), LaSota (AMPV-1), Yucaipa (AMPV-2), and Ty6661/67 (AMPV-3). The virus strains used to produce three aMPV reference sera were 14/1 UK (aMPV-A), Hungary/657/4 (aMPV-B), and Colorado (aMPV-C).
2.5. Selection of Anti-H5 HA Monoclonal Antibody
A panel of nine H5-subtype monoclonal antibodies generated from mice immunized with the inactivated virus A/Turkey/ON/6213/66 (H5N1) was described previously [
24]. The characteristics of the mAbs are summarized as follows: Eight out of nine had conformational epitopes, including mAb #10 (F37H5N1-56). The #10 mAb was one of the two mAbs that reacted with all thirteen H5 strains tested in the indirect ELISA, double-antibody sandwich (DAS) ELISA, and dot blot assay. Eight out of nine mAbs were non-neutralizing mAbs by HI assay, including mAb #10. mAb #10 was one of the three mAbs that worked in immunohistochemistry and immunofluorescence assays for H5 antigen detection. The isotype of mAb #10 is IgG2b/κ. Given the characteristics of mAb #10 described above, it was chosen to explore its suitability within the cELISA immunoassay format in this study.
2.6. Competitive ELISA Condition Parameters
The cELISA condition parameters were described previously [
25]. The optimum conditions of the cELISA were assessed by various factors, such as microtiter plate type, blocking/diluent buffer, pre-assay blocking, incubation temperatures and/or agitation, coating antigen/mAb, and testing serum concentration.
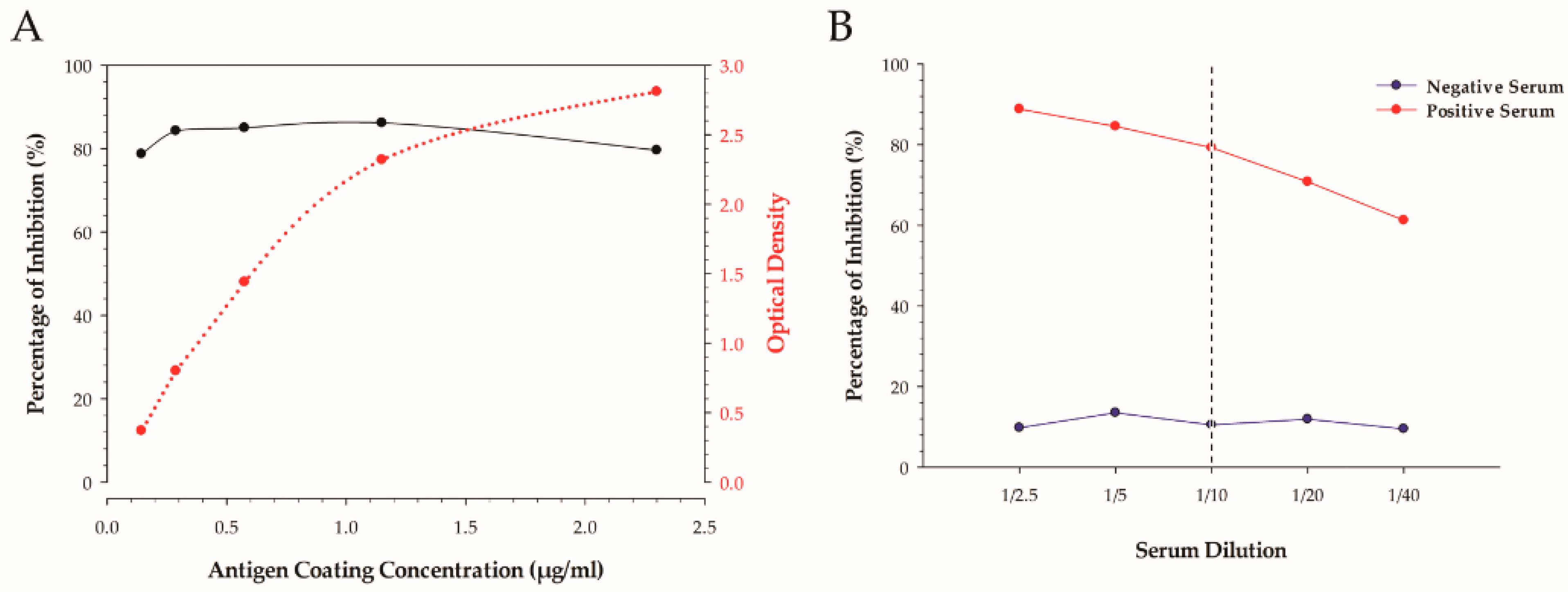
The determination of the optimal amount of coating antigen per well was achieved by utilizing a checkerboard-style titration by indirect ELISA. The amount of coating antigen per well is reported in µg/mL, and was calculated from the stock protein concentration that was determined by finding the average between the Qubit 2.0 and BCA methods. The concentration of monoclonal and secondary antibodies was determined by a checkerboard titration and expressed as µg/mL per well.
A two-fold dilution series of neat positive H5 IAV serum and negative normal chicken serum at a ratio ranging from a 1:2.5 to 1:40 was undertaken and compared.
Two different variants of a 96-well microtiter plate were used to determine the best platform for the cELISA test. A non-treated, flat-bottom, clear polystyrene plate (Thermo Fisher Scientific, Rochester, NY, USA) that has a naturally hydrophobic surface property was tested in parallel with a specially treated, flat-bottom polystyrene plate that was designed to have a high-protein-binding surface property for maximum capture (Maxisorp, Thermo Fisher). Assays with the plate types were run in parallel using 14 reference sera in duplicate to determine differences in the resulting outcomes using the identical protocol and lot-numbered reagents.
Utilizing a blocking buffer as a pre-step treatment was studied to determine its effects on the assay. This procedure was carried out on non-treated 96-well microtiter plates with 3% heat-inactivated FBS in PBS-T as a blocking buffer.
Blocking/diluent buffers tested included 3% heat-inactivated fetal bovine serum (FBS) in PBS-T, 5% skim milk in PBS-T, and 2% bovine + 2% rabbit serum in PBS-T. These buffers were tested on both antigens against the same seven H5 chicken antisera in duplicate to observe any changes the buffers may cause in the assay.
The effect of agitation during the incubation periods of the cELISA was examined both at 37 °C and at an ambient temperature.
The effect of the antigen coating time was assessed to determine if the overnight coating was necessary due to its time restraint in regard to testing delays and planning, or if coating the antigen at a higher temperature for a shorter time would yield similar results. In this regard, antigens coated overnight at 4 °C and antigens coated for 2 h at 37 °C were tested and compared.
2.7. Development of H5 cELISA
The working concentration of the recombinant H5 proteins was diluted in a 0.06 M carbonate buffer with pH 9.6 (45 mM sodium bicarbonate and 18 mM sodium carbonate). Each well of non-treated 96-well microtiter plates was coated overnight at 4 °C with 100 µL of the above-diluted antigen and then was washed 5× with PBS plus 0.05% Tween20 (PBS-T). Each well was blocked with 100 µL of 3% heat-inactivated FBS in PBS-T and incubated for 1 h at 37 °C. After washing 5× with PBS-T, equal volumes (50 µL) of test serum samples and competitive mAb diluted to the working concentration with the blocking buffer were mixed and incubated simultaneously at 37 °C for 1 h. Wells containing competitive mAb mixed with standard anti-H5 sera, negative sera, or no serum were utilized as a positive, negative, or dilution control (DC), respectively. Each serum sample was tested in duplicate. After 5× washing, 100 µL of horseradish peroxidase-labeled goat anti-mouse IgG conjugate (Jackson Immunoresearch Laboratories, West Grove, PA, USA) was added at a dilution of 1/2000, incubated for 1 h at 37 °C, and then washed and developed with 3,3′,5,5′-tetramethylbenzidine as a peroxidase substrate (TMB) (Sigma-Aldrich, St. Louis, MO, USA). The reaction was carried out for 15 min at room temperature, which was followed by adding 50 μL of 2 M H2SO4 to each well to stop the reaction. Colorimetric development was quantified spectrophotometrically at 450 nm with a Molecular Devices EMax precision microplate reader (Molecular Devices, LLC, San Jose, CA, USA). Results were interpreted as the percentage of inhibition (PI) calculated according to the following formula: % inhibition = ((1 − (OD450 of serum sample/OD450 of DC)) × 100.
2.8. Validation Methods for the Determination of Diagnostic Sensitivity, Specificity, Repeatability, and Threshold Cut-Off Level
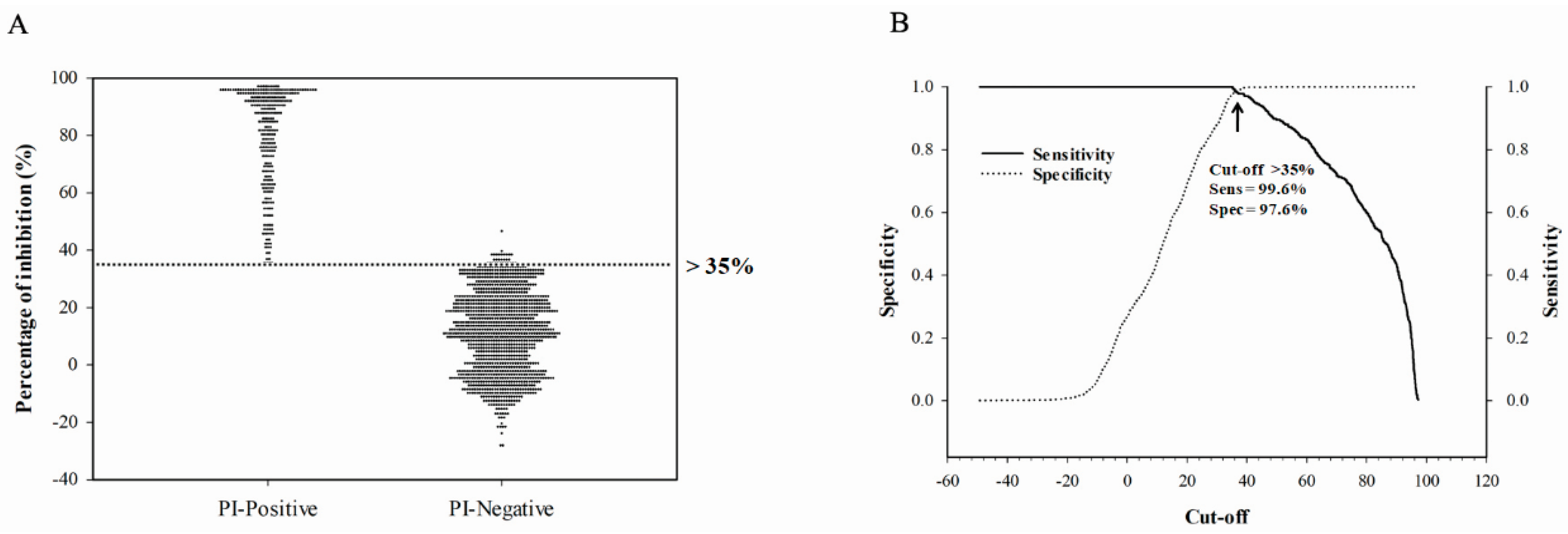
To assess the diagnostic sensitivity and specificity of the assay, all sera with known serostatus (confirmed by HI assay) were used. This included experimental chicken and turkey serum samples produced at the NCFAD and field serum samples submitted to NCFAD through CanNAISS. For validating the assay, confirmed negative serum samples and positive serum samples were used in a receiver operating characteristic (ROC) analysis and calculated to assess diagnostic performance, which included the determination of sensitivity, specificity, and the threshold cut-off using SigmaPlot version 14.0 (Systat Software, San Jose, CA, USA).
The repeatability of the assay was assessed by running the reference sera in multiple replicates within the same run or between runs. The intra-assay repeatability was calculated for 45 replicates on 2 separate plates and then repeated over 2 days for inter-assay repeatability assessment. The values were expressed as a mean, standard deviation, and percent coefficient of variation (CV%) for the repeated measure.
2.9. Statistical Analysis
The rec-H5 antigen-based cELISA was validated using ROC analysis and was used to calculate diagnostic performance, which included the determination of sensitivity, specificity, and the threshold cut-off using SigmaPlot version 14.0 (Systat Software, San Jose, CA, USA) for statistical analysis. The measurement of agreement between the gold-standard HI test and the cELISA tests was assessed using Cohen’s kappa (κ) test. To interpret the given κ value, the criteria laid out by Landis and Koch was utilized [
27]. κ ≤ 0.00 was designated as poor agreement, 0.00 < κ ≤ 0.20 as slight agreement, 0.21 < κ ≤ 0.40 as fair agreement, 0.41 < κ ≤ 0.60 as moderate agreement, 0.61 < κ ≤ 0.80 as substantial agreement, and 0.81 < κ < 1.00 as almost perfect agreement.
4. Discussion
Influenza A viruses of the H5 subtype are widely distributed and have caused numerous outbreaks in poultry and wild birds worldwide [
28]. Highly pathogenic avian influenza viruses (HPIAVs) of the H5 subtype are a constant threat to public health due to their potential to cause zoonotic disease. Avian influenza outbreaks associated with Gs/Gd-lineage H5Nx viruses belonging to clade 2.3.4.4b have been reported in Europe, Africa, Asia and America since 2020 and have continued to threaten the poultry industry and wildlife, accounting for an unprecedentedly large amount of mortality in birds [
17]. Effective prevention and control of H5 outbreaks require active serological surveillance in wild birds and poultry. Since 2008, Canada has designed and implemented the Canadian Notifiable Avian Influenza Surveillance System (CanNAISS) with six surveillance activities (i.e., wild bird surveillance; passive surveillance in domestic poultry when clinical signs suggestive of notifiable avian influenza are reported; targeted surveillance when notifiable avian influenza is detected; pre-slaughter surveillance in commercial poultry; hatchery supply flock surveillance; and voluntary enhanced surveillance in the poultry genetic exporters sector) aiming to detect, prevent and eliminate the presence of H5 and H7 subtypes (notifiable avian influenza) in Canada’s domestic poultry flocks [
28]. Due to the increase in the sampling for serosurveillance, we would like to replace the labor-intensive HI assay with a simple ELISA assay for determining serotype specificity that is more cost-effective and more robust, and that does not require the use of live IAVs as in the HI assay.
In the current study, we describe the development and validation of the H5 cELISA which was developed using a recombinant Eurasian-origin, full-length H5 protein as a coating antigen and a competitor H5 mAb developed from mice immunized with the North American-lineage H5N1 strain. Using the H5 cELISA, we were able to detect H5 antibodies developed against different H5 strains belonging to both North American- and Eurasian-lineage viruses including Gs/GD-lineage H5-subtype viruses belonging to 2.3.4.4c and 2.3.4.4b clades. We have, by far, extensively expanded our detection spectrum compared to previously described assays [
24,
29,
30,
31]. The H5 mAb used in this study was previously characterized as an isotype of the IgG2b/κ that binds with a conformational epitope of the H5 HA protein [
24]. In this study, we further confirmed that mAb #10 does not demonstrate anti-hemagglutinin activity and is not suitable for use in the HI assay. We confirmed that the epitope location of this mAb is within the HA1 subunit of the HA protein, as it did not react with the recombinant HA2 protein in a dot blot or indirect ELISA test. The absence of anti-hemagglutinin activity or features in this mAb might have contributed to its broader reactivity spectrum, which is possibly due to a smaller burden of immune selection pressure on its binding epitope. Studies have shown that frequent residue alterations at the HA antigenic sites were observed with vaccinations implemented in poultry [
32,
33]. Thus, if the mAb epitope is within the antigenic sites, it has more chance to lose reactivity due to antigenic evolution.
The Gaussian distribution approach is commonly used to determine the cut-off value for diagnostic tests, in which a cut-off value is defined as the mean plus two standard deviations of the negative reference samples. This approach ensures a diagnostic specificity without considering the diagnostic sensitivity [
34]. In this study, we determined the cut-off value by use of ROC analysis to allow for a combined measure of diagnostic sensitivity and specificity. The HI is regarded as the gold-standard subtyping test for IAV in routine diagnostics recommended by the WOAH. The H5 cELISA developed in this study showed higher accordance with the HI assay for H5 antibody detection with the determined cut-off value.
The newly developed cELISA has proven to be more sensitive compared to the HI assay and also more suitable for the mass screening of serum samples collected from different species of animals. In the hemagglutinin inhibition assay, a serum sample with an HI titer ≥16 is considered positive. The detection limit of the H5 cELISA was determined using three serially diluted H5-positive chicken sera that were tested with the HI assay against homologous H5-subtype viruses. The detection limit of the assay was determined to be below the diagnostic detection limit of the HI assay for all three serum samples tested, indicating that the cELISA was superior in analytical sensitivity compared to the HI assay. Despite being the “gold standard” assay for subtyping IAVs, the HI assay has some drawbacks compared to the ELISA. These include: (1) the HI assay is more laborious and time-consuming in selecting the virus antigen, red blood cell preparations, serum dilution and the manual reading of results; (2) the selection of a homologous viral antigen is difficult when the infection history of the animals from which the serum samples were collected is unknown; and (3) due to the zoonotic potential of IAVs, all procedures involving live virus antigens require the use of a biological safety cabinet. In contrast to the HI assay, the developed H5 cELISA in our study is easy to automate for the large-scale monitoring of H5-subtype-specific antibodies in sera samples collected from different species of animals. In addition, the established rec-H5 cELISA has a biosafety advantage over the HI assay as it can be performed in a regular lab without the use of a biological safety cabinet.
An attractive feature of the developed H5 cELISA is its ability to detect H5 antibodies of the currently circulating H5Nx viruses belonging to clade 2.3.4.4b. To assess the agreement between the developed H5 cELISA and HI assay, we tested 48 field serum samples collected from mallard ducks in 2022 using both assays. A perfect agreement was observed in Cohen’s κ analysis. Experimental studies have shown that mallard ducks can excrete abundant viruses without clinical or pathologic evidence of debilitating disease, supporting the potential role of these ducks as long-distance vectors of HPIAV [
35]. When these migratory birds are in close proximity to domestic poultry facilities, poultry birds can become infected through direct contact with secretions or feces of infected birds, contaminated surfaces, or infected food and water supplies. The H5 subtype responsible for the outbreak that began in 2021 is highly pathogenic and thus is of heightened concern. The ability of our H5 cELISA to detect anti-H5 antibodies in the sera derived from the current H5N1 HPIAVs makes it a rapid and effective serological screening tool for the early detection and control of subclinical H5 infection. The H5 cELISA could be used in the serosurveillance of H5 antibodies in different species of birds and also in mammalian species. The assay could also be used for monitoring the vaccination status of domestic poultry where the disease is endemic and vaccination is used to control the spread of the virus.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}