
Oxide Modified Iron in Electron Beam Powder Bed Fusion—From Processability to Corrosion Properties

 ,
,  , , and
, , and 
Abstract
1. Introduction
- The general chemical composition and any corrosion products must not be toxic for the human body.
- The degradation rate has to be tailorable to maintain an adequate level of structural integrity for the entire bone healing process.
- The material, surface, and geometry have to meet all relevant requirements with respect to stiffness and strength, wherein all characteristics have to be adapted to the prevailing quasistatic and cyclic loading scenario.
- Melting point higher than for the liquid metal.
- Thermodynamic stability in this environment (i.e., no decomposition or reaction with the melt compounds).
- Low wetting angle.
- Low interfacial energy, i.e., lattice parameters similar to the solid material.
2. Materials and Methods
2.1. X-ray Photoelectron Spectroscopy (XPS)
2.2. Modifications
2.3. Processing
2.4. Microstructural Investigations
2.5. Electrochemical Corrosion Properties
2.6. Mechanical Testing
3. Results
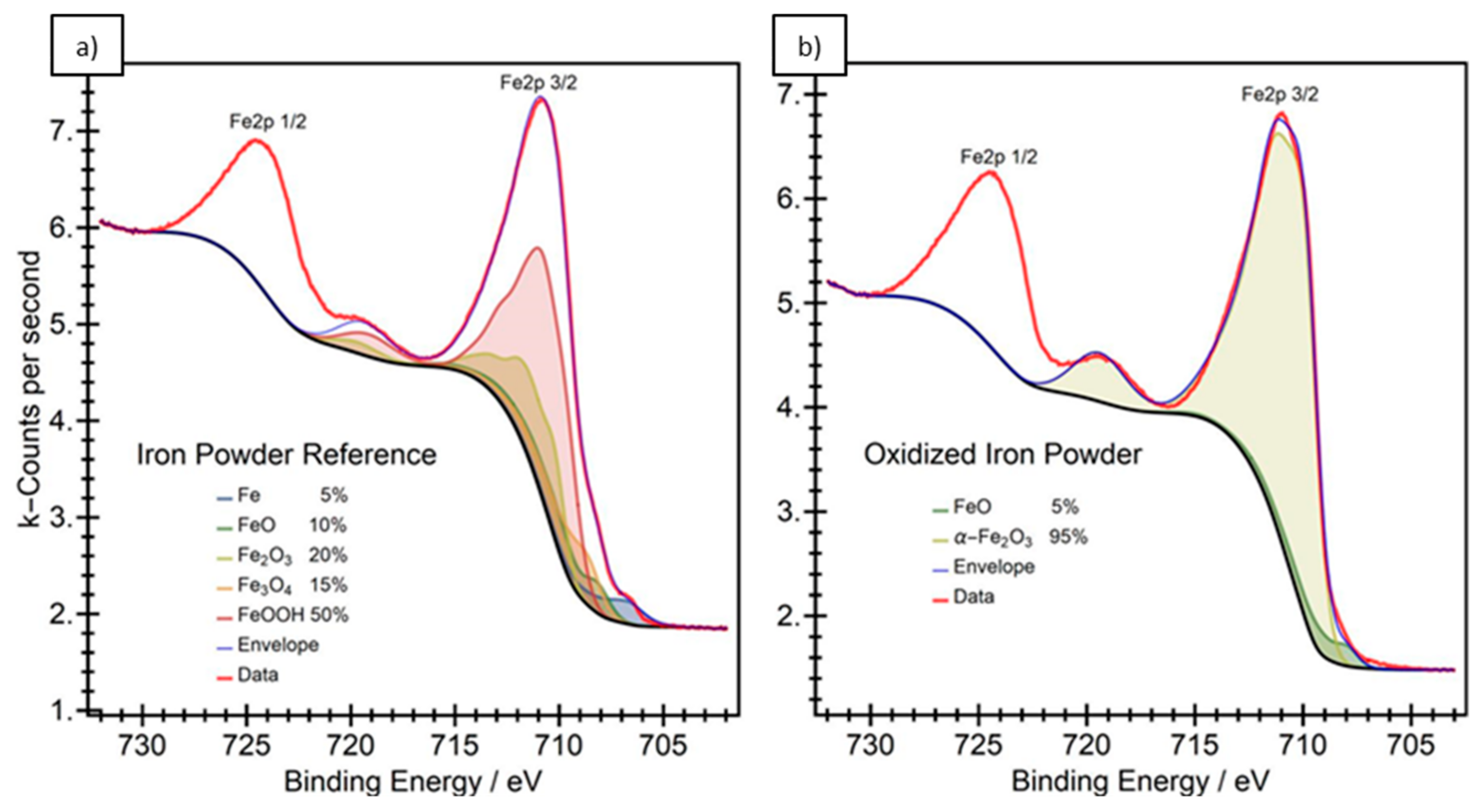
3.1. Particle Surface Structure
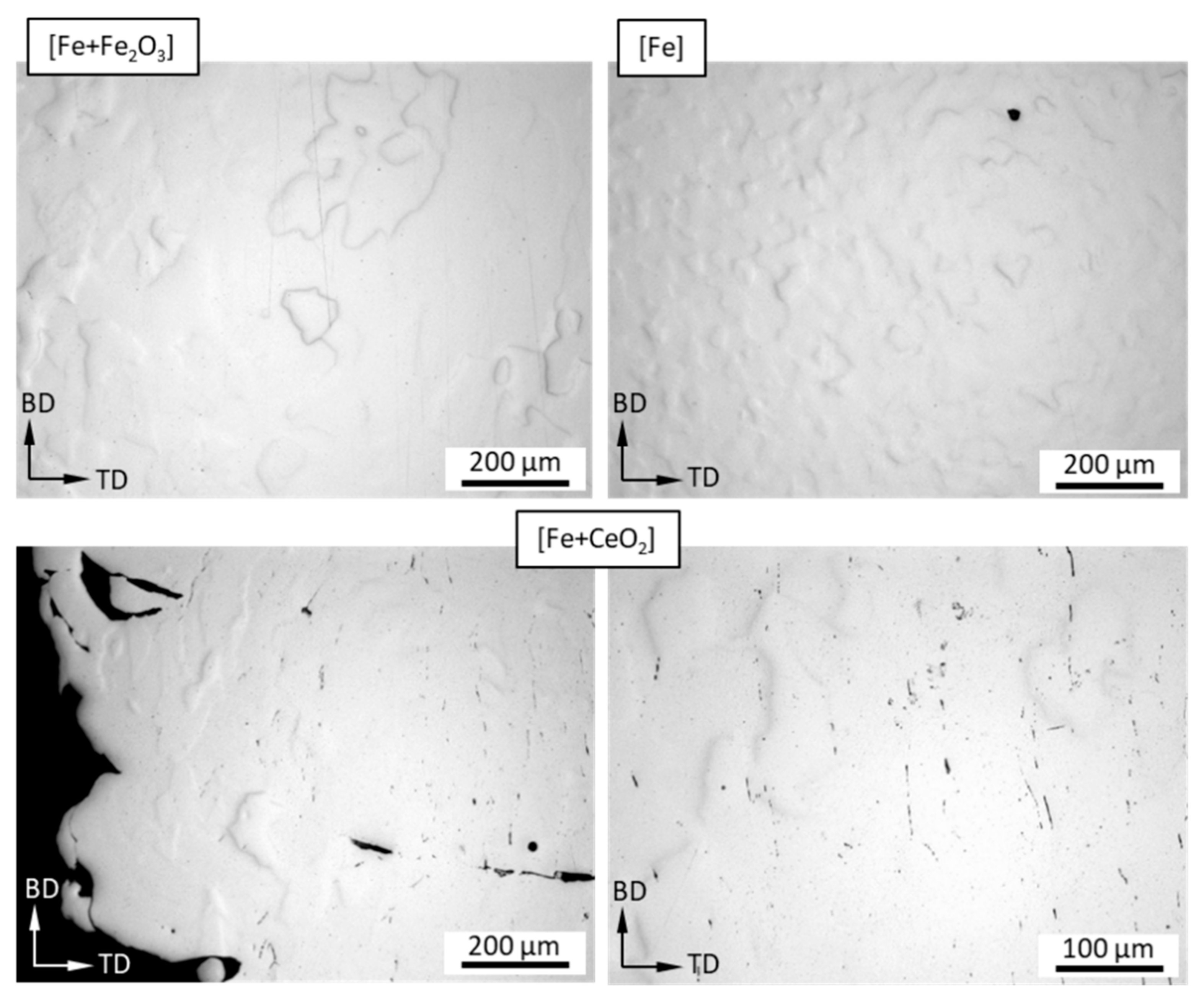
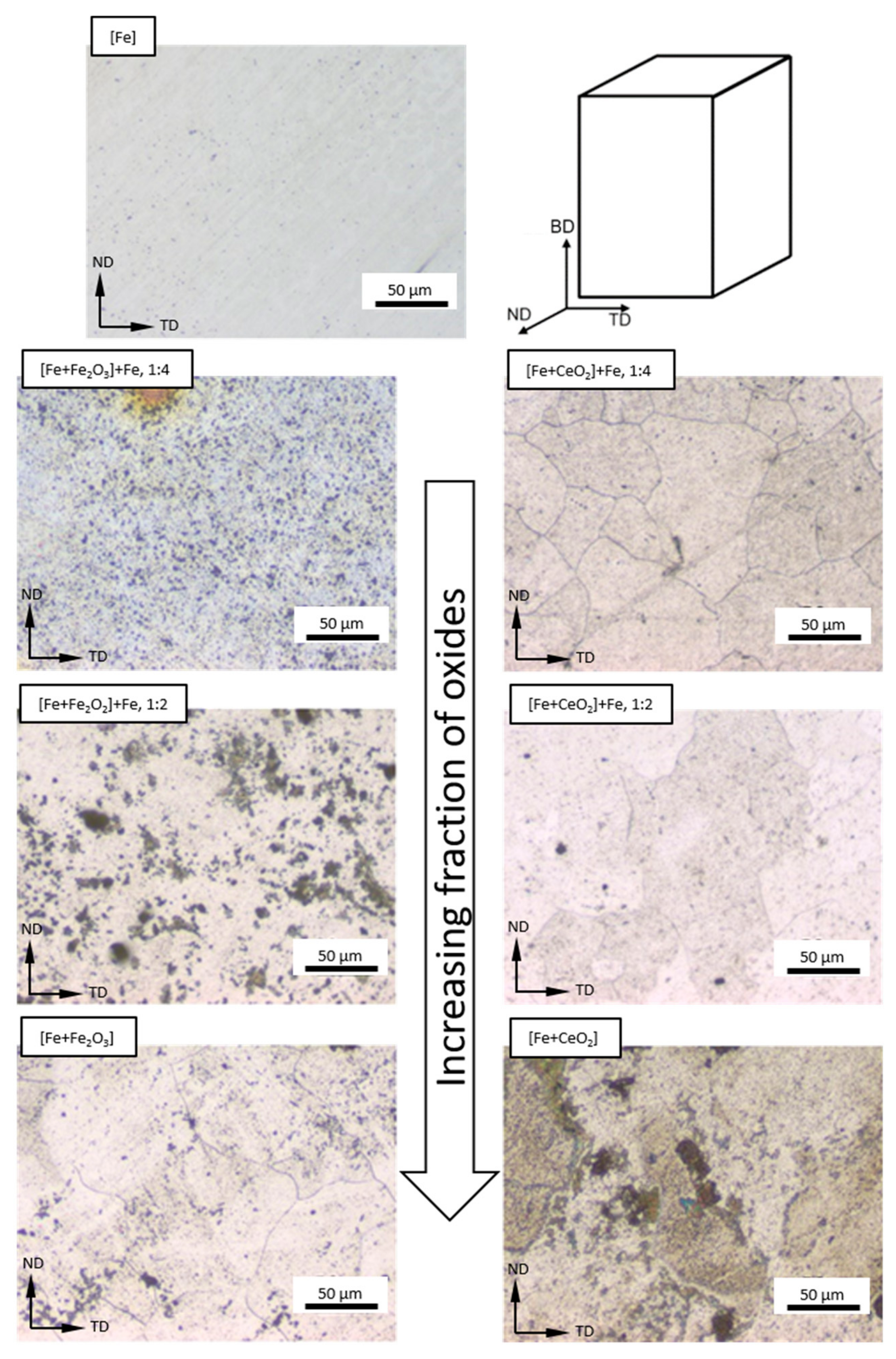
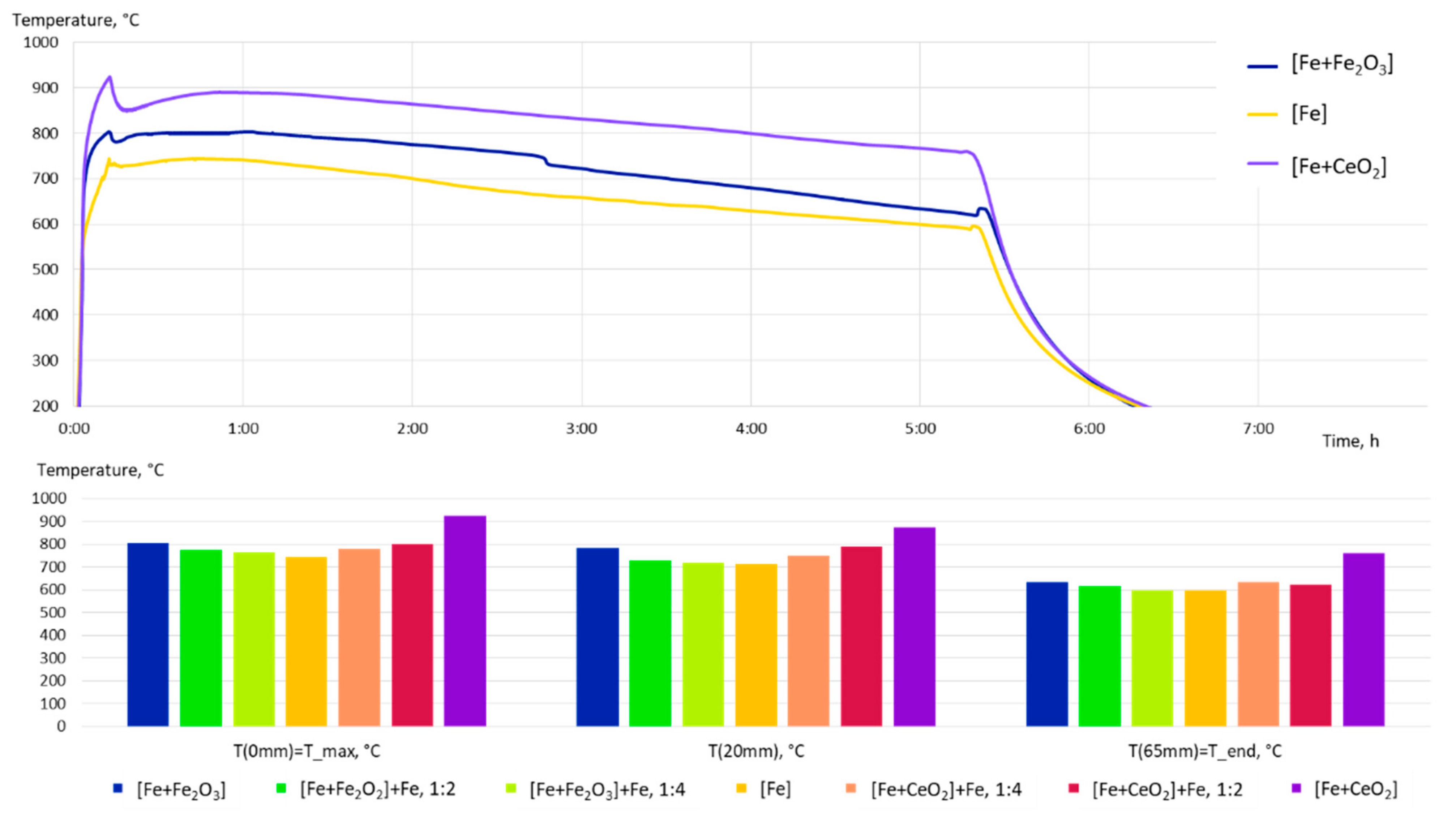
3.2. Microstructure Following E-PBF Processing
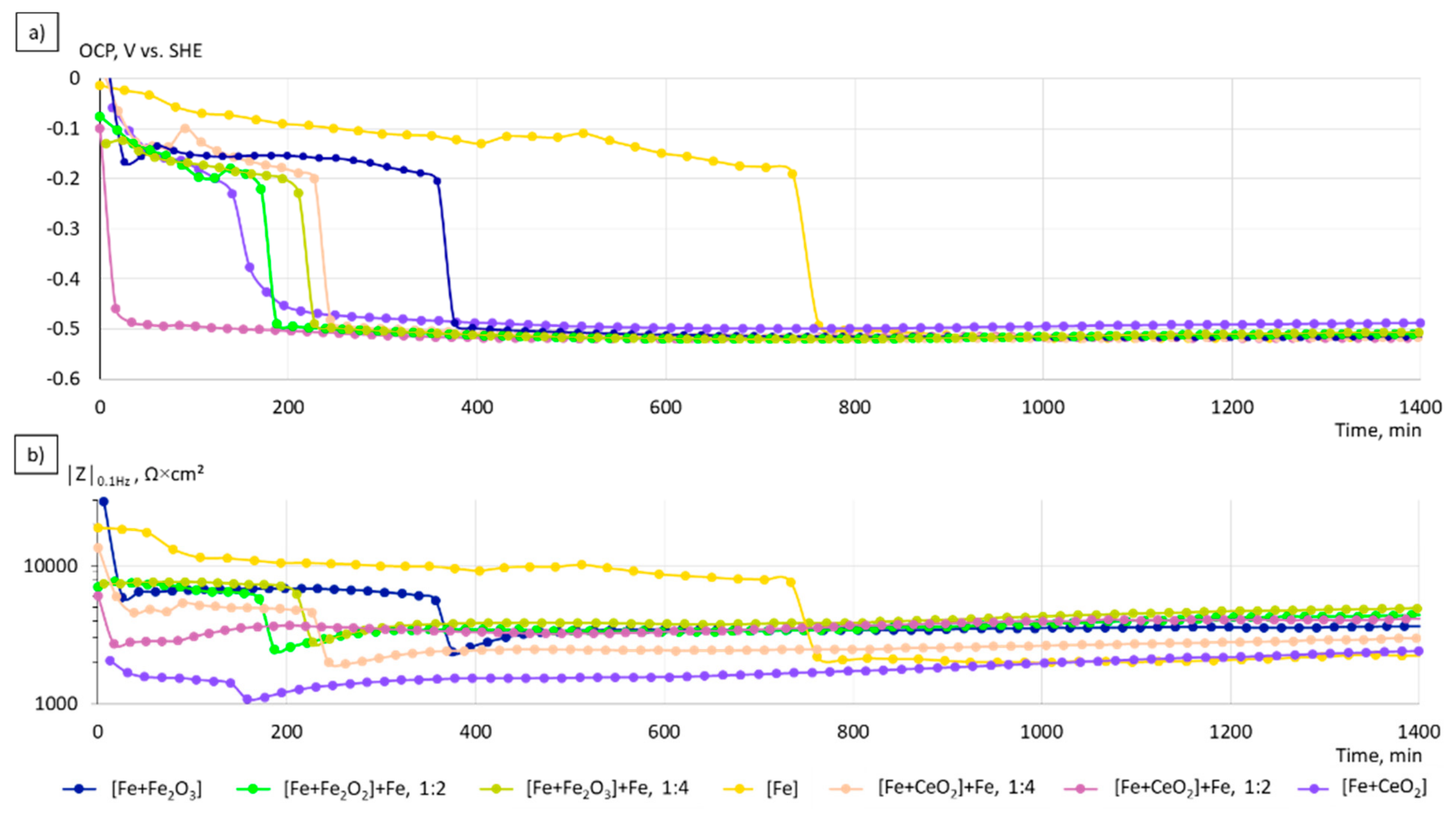
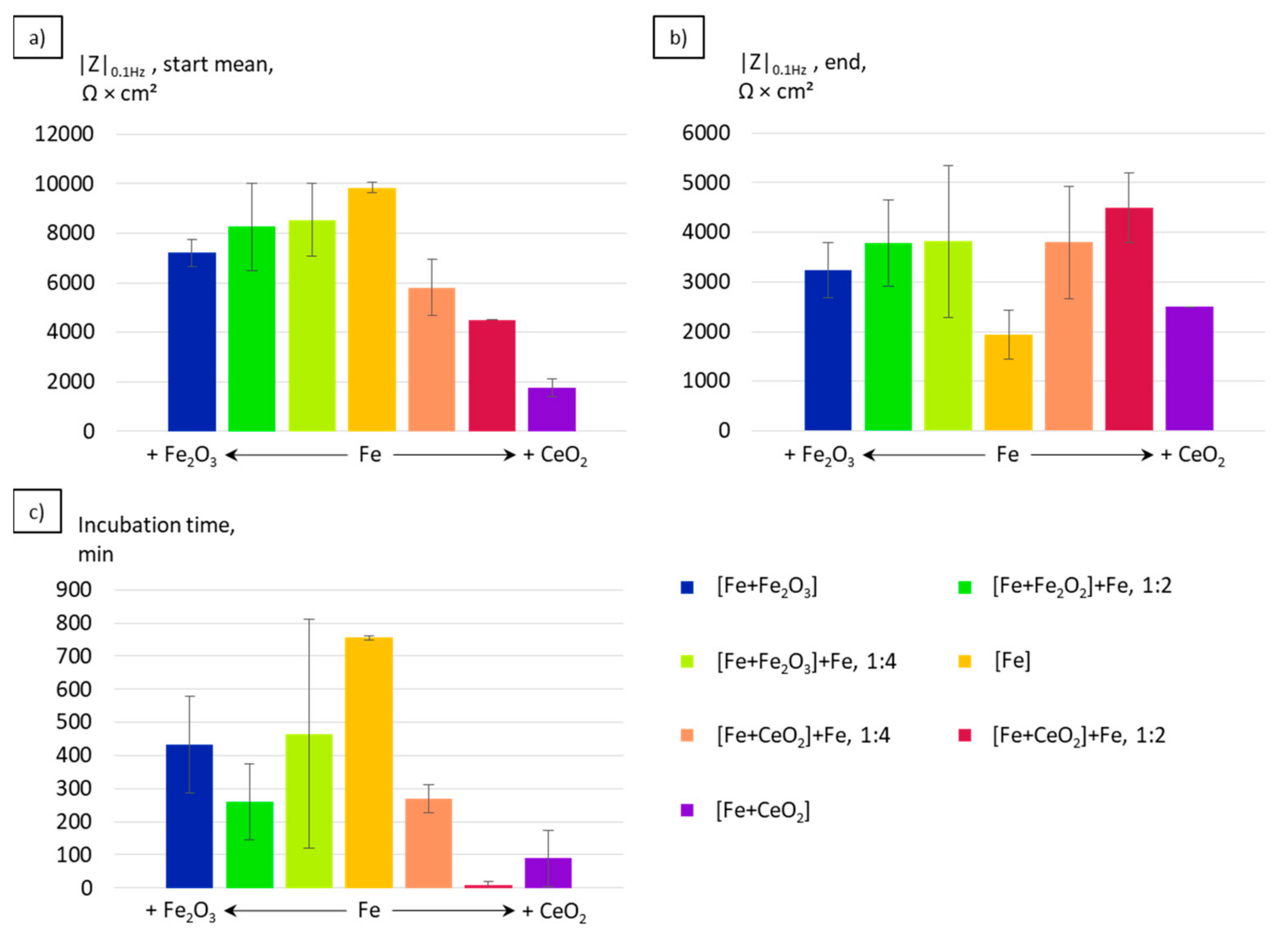
3.3. Electrochemical Corrosion Analysis
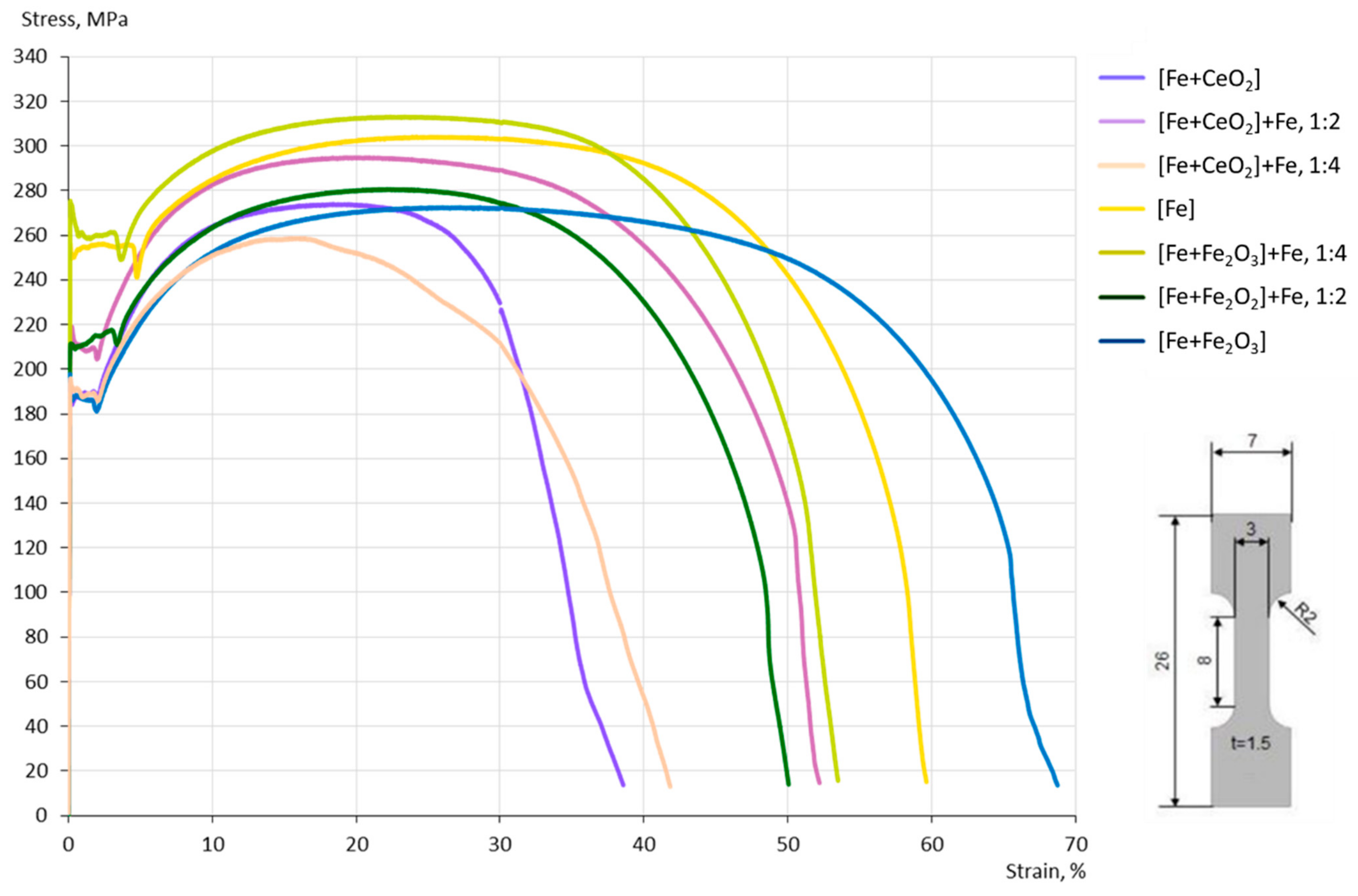
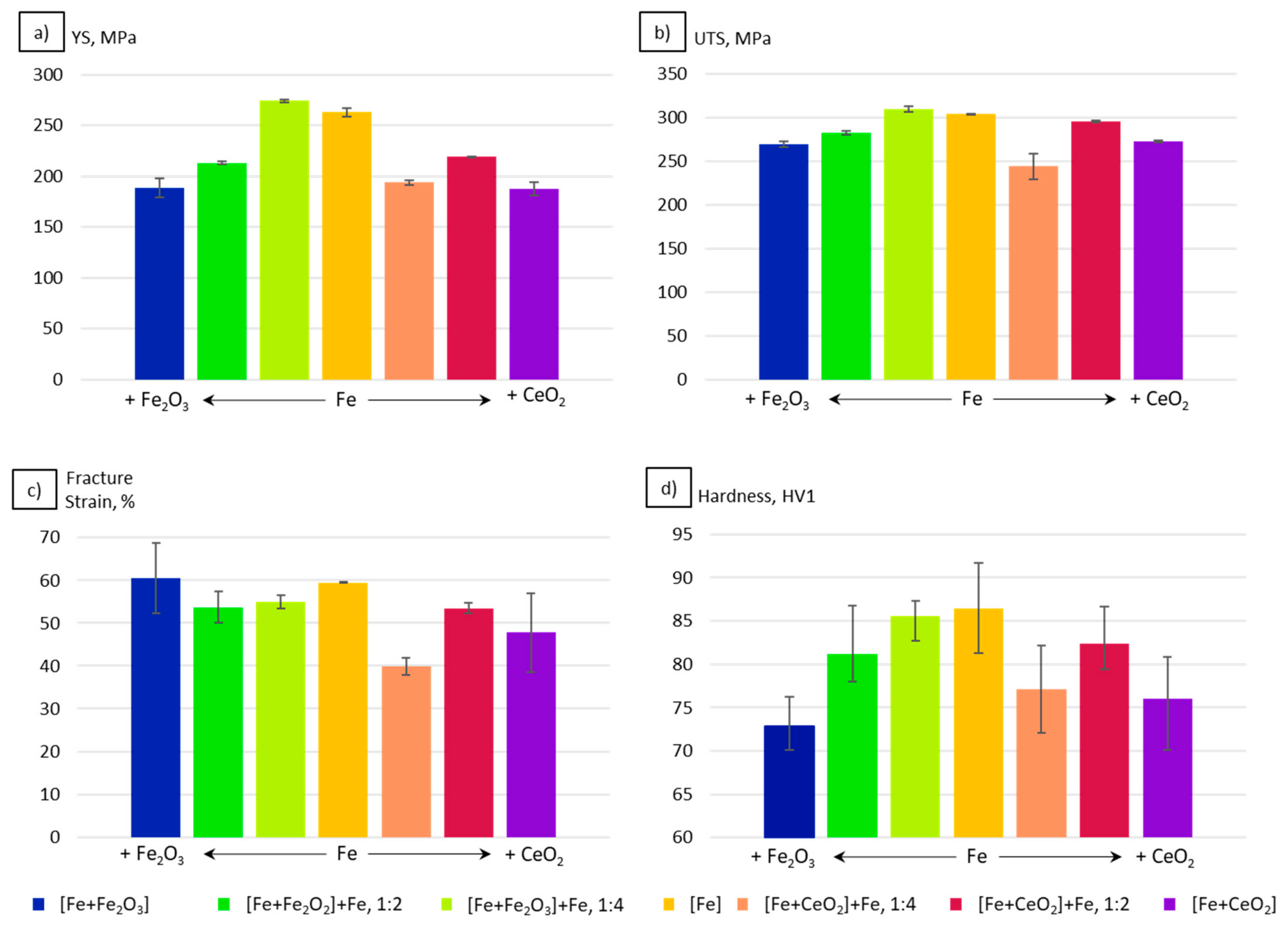
3.4. Mechanical Properties
4. Discussion
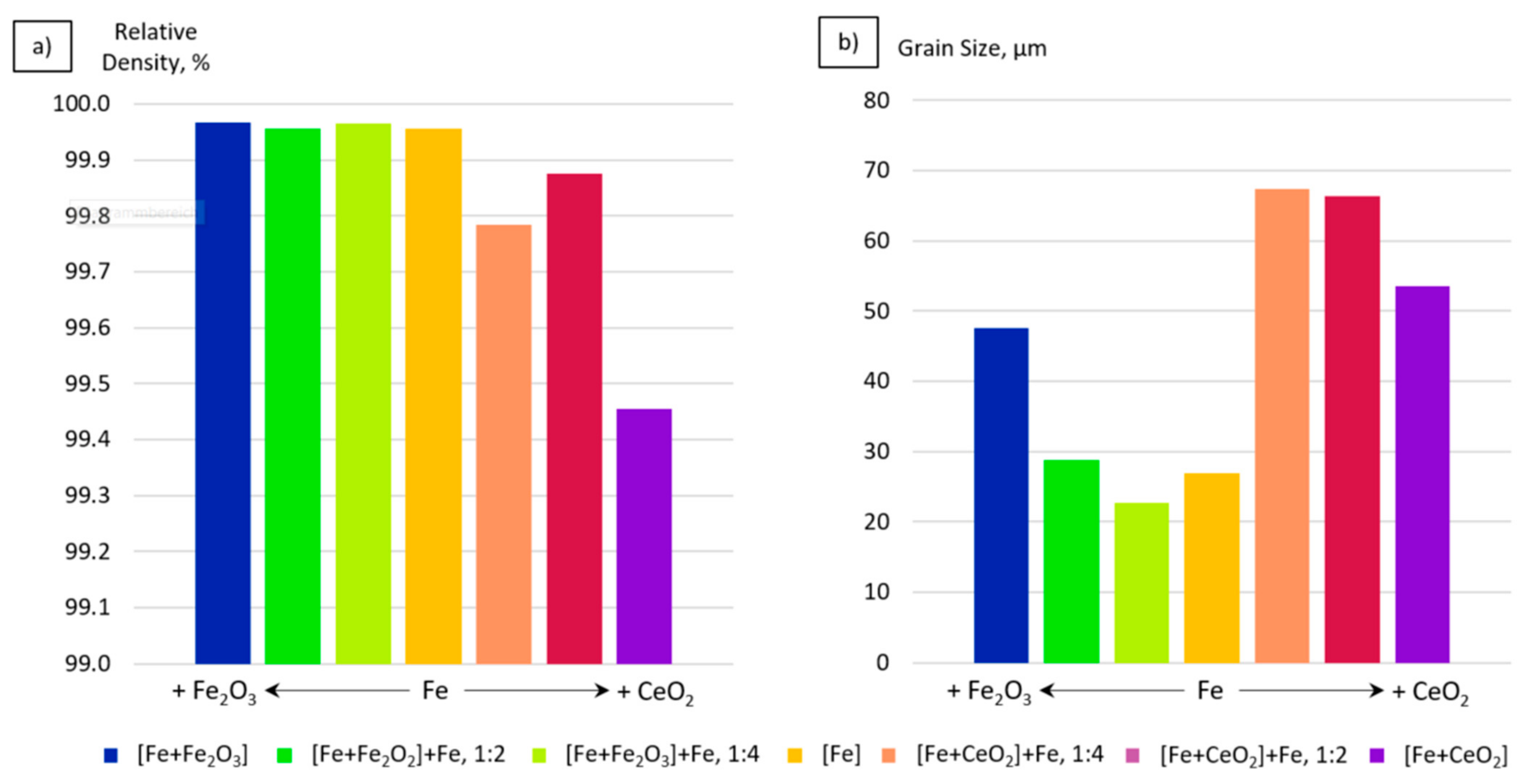
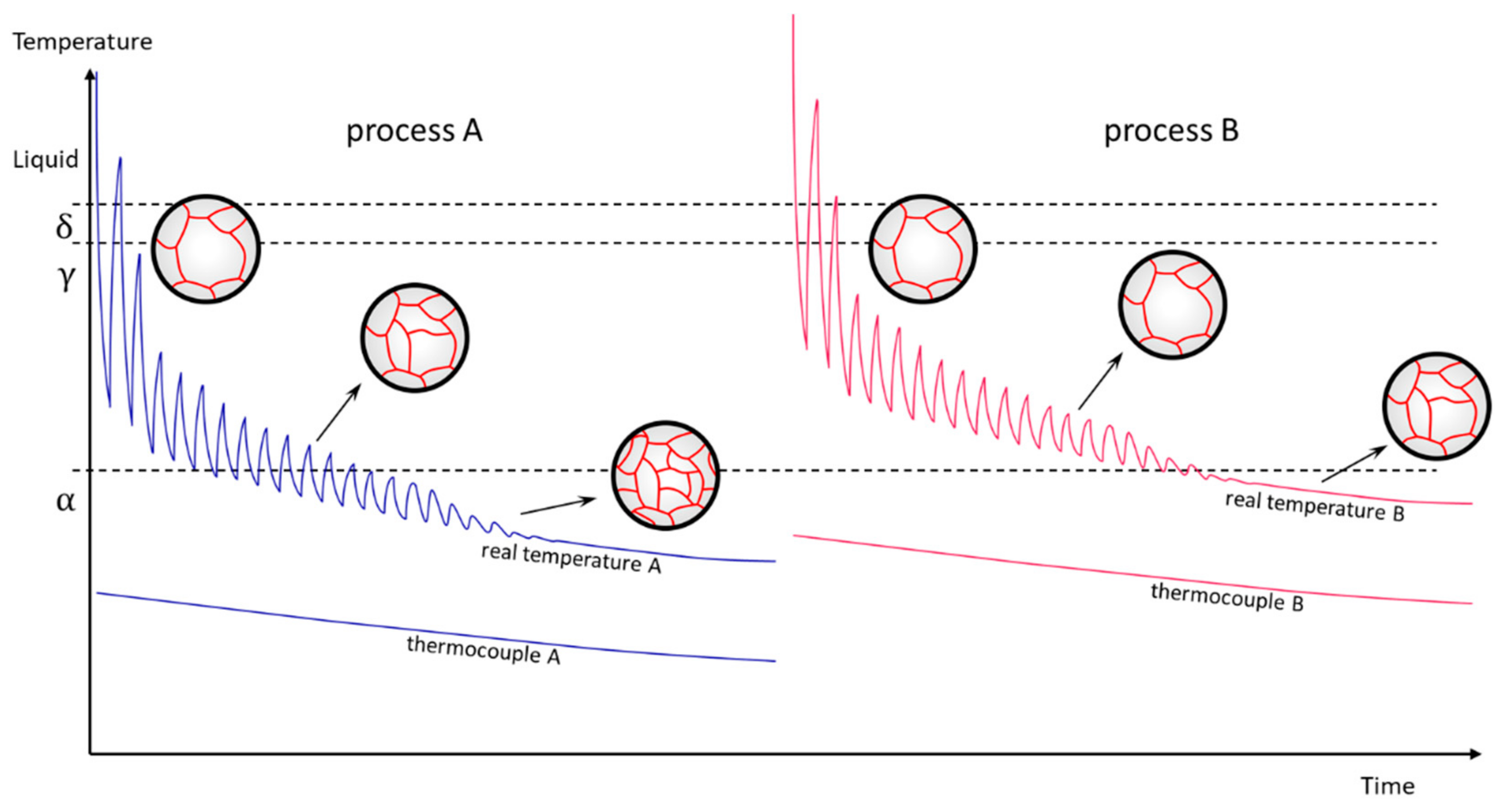
4.1. Porosity and Grain Size Evolution
4.2. Electrochemical Properties
4.3. Mechanical Properties
5. Conclusions
- The grain size evolution mainly followed the temperature evolution during E-PBF processing. The use of CeO2 NP resulted in a severe microstructure coarsening, which could be primarily traced back to higher process temperatures. Grain refinement induced by oxides was hardly seen. The smallest grain sizes after E-PBF processing were found in the pure Fe and in the Fe2O3 modification with the lowest fraction of oxides.
- For the Fe2O3 modifications, the resulting grain sizes were in perfect agreement with the mechanical properties. According to the Hall–Petch relation hardness, UTS and YS increased with decreasing grain sizes.
- In the case of the CeO2-modified conditions, the mechanical properties showed more pronounced scatter, which can be attributed to the higher number of defects. Still, relatively high strength as well as excellent damage tolerance were found in these modifications. This characteristic will allow for manufacturing material modifications featuring a defined porosity and predictable properties.
- The corrosion kinetics correlated well to the change in the chemical composition and the microstructure of the manufactured specimens. The increase of the grain size, the formation of local galvanic elements due to the incorporation of oxide phases, and the creation of pores in the case of CeO2-modified particles contribute to the overall increased corrosion rates in the initial phase of immersion.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DebRoy, T.; Wei, H.L.; Zuback, J.S.; Mukherjee, T.; Elmer, J.W.; Milewski, J.O.; Beese, A.M.; Wilson-Heid, A.; De, A.; Zhang, W. Additive manufacturing of metallic components—Process, structure and properties. Prog. Mater. Sci. 2018, 92, 112–224. [Google Scholar] [CrossRef]
- Oliveira, J.P.; LaLonde, A.D.; Ma, J. Processing parameters in laser powder bed fusion metal additive manufacturing. Mater. Des. 2020, 193, 108762. [Google Scholar] [CrossRef]
- Ahmadi, S.M.; Hedayati, R.; Li, Y.; Lietaert, K.; Tümer, N.; Fatemi, A.; Rans, C.D.; Pouran, B.; Weinans, H.; Zadpoor, A.A. Fatigue performance of additively manufactured meta-biomaterials: The effects of topology and material type. Acta Biomater. 2018, 65, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jahr, H.; Lietaert, K.; Pavanram, P.; Yilmaz, A.; Fockaert, L.I.; Leeflang, M.A.; Pouran, B.; Gonzalez-Garcia, Y.; Weinans, H.; et al. Additively manufactured biodegradable porous iron. Acta Biomater. 2018, 77, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lietaert, K.; Li, W.; Zhang, X.-Y.; Leeflang, M.A.; Zhou, J.; Zadpoor, A.A. Corrosion fatigue behavior of additively manufactured biodegradable porous iron. Corros. Sci. 2019, 156, 106–116. [Google Scholar] [CrossRef]
- Murr, L.E. Open-cellular metal implant design and fabrication for biomechanical compatibility with bone using electron beam melting. J. Mech. Behav. Biomed. Mater. 2017, 76, 164–177. [Google Scholar] [CrossRef]
- Schouman, T.; Schmitt, M.; Adam, C.; Dubois, G.; Rouch, P. Influence of the overall stiffness of a load-bearing porous titanium implant on bone ingrowth in critical-size mandibular bone defects in sheep. J. Mech. Behav. Biomed. Mater. 2016, 59, 484–496. [Google Scholar] [CrossRef]
- Wieding, J.; Lindner, T.; Bergschmidt, P.; Bader, R. Biomechanical stability of novel mechanically adapted open-porous titanium scaffolds in metatarsal bone defects of sheep. Biomaterials 2015, 46, 35–47. [Google Scholar] [CrossRef]
- Fromme, P.; Blunn, G.W.; Aston, W.J.; Abdoola, T.; Koris, J.; Coathup, M.J. The effect of bone growth onto massive prostheses collars in protecting the implant from fracture. Med. Eng. Phys. 2017, 41, 19–25. [Google Scholar] [CrossRef]
- Özbek, I.; Konduk, B.A.; Bindal, C.; Ucisik, A.H. Characterization of borided AISI 316L stainless steel implant. Vacuum 2002, 65, 521–525. [Google Scholar] [CrossRef]
- Hryniewicz, T.; Rokosz, K.; Filippi, M. Biomaterial Studies on AISI 316L Stainless Steel after Magnetoelectropolishing. Materials 2009, 2, 129–145. [Google Scholar] [CrossRef]
- Shah, F.A.; Omar, O.; Suska, F.; Snis, A.; Matic, A.; Emanuelsson, L.; Norlindh, B.; Lausmaa, J.; Thomsen, P.; Palmquist, A. Long-term osseointegration of 3D printed CoCr constructs with an interconnected open-pore architecture prepared by electron beam melting. Acta Biomater. 2016, 36, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Harun, W.; Kamariah, M.; Muhamad, N.; Ghani, S.; Ahmad, F.; Mohamed, Z. A review of powder additive manufacturing processes for metallic biomaterials. Powder Technol. 2018, 327, 128–151. [Google Scholar] [CrossRef]
- Okazaki, Y.; Gotoh, E. Comparison of metal release from various metallic biomaterials in vitro. Biomaterials 2005, 26, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Witte, F. The history of biodegradable magnesium implants: A review. Acta Biomater. 2010, 6, 1680–1692. [Google Scholar] [CrossRef]
- Yusop, A.H.; Bakir, A.A.; Shaharom, N.A.; Abdul Kadir, M.R.; Hermawan, H. Porous biodegradable metals for hard tissue scaffolds: A review. Int. J. Biomater. 2012, 2012, 641430. [Google Scholar] [CrossRef] [PubMed]
- Geetha, M.; Singh, A.K.; Asokamani, R.; Gogia, A.K. Ti based biomaterials, the ultimate choice for orthopaedic implants—A review. Prog. Mater. Sci. 2009, 54, 397–425. [Google Scholar] [CrossRef]
- Schinhammer, M.; Hänzi, A.C.; Löffler, J.F.; Uggowitzer, P.J. Design strategy for biodegradable Fe-based alloys for medical applications. Acta Biomater. 2010, 6, 1705–1713. [Google Scholar] [CrossRef]
- Hofstetter, J.; Martinelli, E.; Weinberg, A.M.; Becker, M.; Mingler, B.; Uggowitzer, P.J.; Löffler, J.F. Assessing the degradation performance of ultrahigh-purity magnesium in vitro and in vivo. Corros. Sci. 2015, 91, 29–36. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, Y.; Chen, X.-H.; Yang, J.-A.; Pan, H.; Chen, D.; Wang, L.; Zhang, J.; Zhu, D.; Wu, S.; et al. Fundamental Theory of Biodegradable Metals—Definition, Criteria, and Design. Adv. Funct. Mater. 2019, 29, 1805402. [Google Scholar] [CrossRef]
- Witte, F.; Kaese, V.; Haferkamp, H.; Switzer, E.; Meyer-Lindenberg, A.; Wirth, C.J.; Windhagen, H. In vivo corrosion of four magnesium alloys and the associated bone response. Biomaterials 2005, 26, 3557–3563. [Google Scholar] [CrossRef] [PubMed]
- Kujur, M.S.; Manakari, V.; Parande, G.; Prasadh, S.; Wong, R.; Mallick, A.; Gupta, M. Development of rare-earth oxide reinforced magnesium nanocomposites for orthopaedic applications: A mechanical/immersion/biocompatibility perspective. J. Mech. Behav. Biomed. Mater. 2021, 114, 104162. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.H.; Yung, M.M.N.; Ng, A.M.C.; Ma, A.P.Y.; Wong, S.W.Y.; Chan, C.M.N.; Ng, Y.H.; Djurišić, A.B.; Guo, M.; Wong, M.T.; et al. Toxicity of CeO2 nanoparticles—The effect of nanoparticle properties. J. Photochem. Photobiol. B 2015, 145, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Hufenbach, J.; Kochta, F.; Wendrock, H.; Voß, A.; Giebeler, L.; Oswald, S.; Pilz, S.; Kühn, U.; Lode, A.; Gelinsky, M.; et al. S and B microalloying of biodegradable Fe-30Mn-1C—Effects on microstructure, tensile properties, in vitro degradation and cytotoxicity. Mater. Des. 2018, 142, 22–35. [Google Scholar] [CrossRef]
- Hufenbach, J.; Sander, J.; Kochta, F.; Pilz, S.; Voss, A.; Kühn, U.; Gebert, A. Effect of Selective Laser Melting on Microstructure, Mechanical, and Corrosion Properties of Biodegradable FeMnCS for Implant Applications. Adv. Eng. Mater. 2020, 22, 2000182. [Google Scholar] [CrossRef]
- Hermawan, H.; Purnama, A.; Dube, D.; Couet, J.; Mantovani, D. Fe-Mn alloys for metallic biodegradable stents: Degradation and cell viability studies. Acta Biomater. 2010, 6, 1852–1860. [Google Scholar] [CrossRef]
- Kraus, T.; Moszner, F.; Fischerauer, S.; Fiedler, M.; Martinelli, E.; Eichler, J.; Witte, F.; Willbold, E.; Schinhammer, M.; Meischel, M.; et al. Biodegradable Fe-based alloys for use in osteosynthesis: Outcome of an in vivo study after 52 weeks. Acta Biomater. 2014, 10, 3346–3353. [Google Scholar] [CrossRef]
- Otto, M.; Pilz, S.; Gebert, A.; Kühn, U.; Hufenbach, J. Effect of Build Orientation on the Microstructure, Mechanical and Corrosion Properties of a Biodegradable High Manganese Steel Processed by Laser Powder Bed Fusion. Metals 2021, 11, 944. [Google Scholar] [CrossRef]
- Niendorf, T.; Brenne, F.; Hoyer, P.; Schwarze, D.; Schaper, M.; Grothe, R.; Wiesener, M.; Grundmeier, G.; Maier, H.J. Processing of New Materials by Additive Manufacturing: Iron-Based Alloys Containing Silver for Biomedical Applications. Met. Mat Trans. A 2015, 46, 2829–2833. [Google Scholar] [CrossRef]
- Berger, P.; Ricco, J.B.; Liqui Lung, P.; Moll, F.L. Localized argyria caused by metallic silver aortic grafts: A unique adverse effect. Eur. J. Vasc. Endovasc. Surg. 2013, 46, 565–568. [Google Scholar] [CrossRef][Green Version]
- Glehr, M.; Leithner, A.; Friesenbichler, J.; Goessler, W.; Avian, A.; Andreou, D.; Maurer-Ertl, W.; Windhager, R.; Tunn, P.-U. Argyria following the use of silver-coated megaprostheses: No association between the development of local argyria and elevated silver levels. Bone Jt. J. 2013, 95, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Drynda, A.; Hassel, T.; Bach, F.W.; Peuster, M. In vitro and in vivo corrosion properties of new iron-manganese alloys designed for cardiovascular applications. J. Biomed. Mater. Res. 2015, 103, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Schinhammer, M.; Gerber, I.; Hänzi, A.C.; Uggowitzer, P.J. On the cytocompatibility of biodegradable Fe-based alloys. Mater. Sci. Eng. C Mater. Biol. Appl. 2013, 33, 782–789. [Google Scholar] [CrossRef]
- Bowen, P.K.; Drelich, J.; Goldman, J. Zinc exhibits ideal physiological corrosion behavior for bioabsorbable stents. Adv. Mater. 2013, 25, 2577–2582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Xia, D.; Wu, S.; Zheng, Y.; Guan, Z.; Rau, J.V. A review on current research status of the surface modification of Zn-based biodegradable metals. Bioact. Mater. 2022, 7, 192–216. [Google Scholar] [CrossRef] [PubMed]
- Waksman, R.; Pakala, R.; Baffour, R.; Seabron, R.; Hellinga, D.; Tio, F.O. Short-term effects of biocorrodible iron stents in porcine coronary arteries. J. Interv. Cardiol. 2008, 21, 15–20. [Google Scholar] [CrossRef]
- Peuster, M.; Hesse, C.; Schloo, T.; Fink, C.; Beerbaum, P.; von Schnakenburg, C. Long-term biocompatibility of a corrodible peripheral iron stent in the porcine descending aorta. Biomaterials 2006, 27, 4955–4962. [Google Scholar] [CrossRef]
- Pan, F.; Zhang, J.; Chen, H.-L.; Su, Y.-H.; Kuo, C.-L.; Su, Y.-H.; Chen, S.-H.; Lin, K.-J.; Hsieh, P.-H.; Hwang, W.-S. Effects of Rare Earth Metals on Steel Microstructures. Materials 2016, 9, 417. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, M.-X.; Ren, H. Roles of Lanthanum and Cerium in Grain Refinement of Steels during Solidification. Metals 2018, 8, 884. [Google Scholar] [CrossRef]
- Li, M.; Li, J.; Qiu, D.; Zheng, Q.; Wang, G.; Zhang, M.-X. Crystallographic study of grain refinement in low and medium carbon steels. Philos. Mag. 2016, 96, 1556–1578. [Google Scholar] [CrossRef]
- Tuttle, R. Understanding the Mechanism of Grain Refinement in Plain Carbon Steels. Inter Met. 2013, 7, 7–16. [Google Scholar] [CrossRef]
- Thewlis, G. Effect of cerium sulphide particle dispersions on acicular ferrite microstructure development in steels. Mater. Sci. Technol. 2006, 22, 153–166. [Google Scholar] [CrossRef]
- Koseki, T.; Thewlis, G. Overview Inclusion assisted microstructure control in C–Mn and low alloy steel welds. Mater. Sci. Technol. 2005, 21, 867–879. [Google Scholar] [CrossRef]
- Grong, O.; Matlock, D.K. Microstructural development in mild and low-alloy steel weld metals. Int. Met. Rev. 1986, 31, 27–48. [Google Scholar] [CrossRef]
- Ali, Y.; Qiu, D.; Jiang, B.; Pan, F.; Zhang, M.-X. Current research progress in grain refinement of cast magnesium alloys: A review article. J. Alloys Compd. 2015, 619, 639–651. [Google Scholar] [CrossRef]
- Kelly, P.M.; Zhang, M.-X. Edge-to-edge matching—The fundamentals. Met. Mat Trans. A 2006, 37, 833–839. [Google Scholar] [CrossRef]
- Zhang, M.-X.; Kelly, P.M. Edge-to-edge matching model for predicting orientation relationships and habit planes—The improvements. Scr. Mater. 2005, 52, 963–968. [Google Scholar] [CrossRef]
- AlMangour, B.; Grzesiak, D.; Yang, J.-M. Rapid fabrication of bulk-form TiB2/316L stainless steel nanocomposites with novel reinforcement architecture and improved performance by selective laser melting. J. Alloys Compd. 2016, 680, 480–493. [Google Scholar] [CrossRef]
- AlMangour, B.; Grzesiak, D.; Jenn-MingYang. Selective laser melting of TiC reinforced 316L stainless steel matrix nanocomposites: Influence of starting TiC particle size and volume content. Mater. Des. 2016, 104, 141–151. [Google Scholar] [CrossRef]
- Boegelein, T.; Dryepondt, S.N.; Pandey, A.; Dawson, K.; Tatlock, G.J. Mechanical response and deformation mechanisms of ferritic oxide dispersion strengthened steel structures produced by selective laser melting. Acta Mater. 2015, 87, 201–215. [Google Scholar] [CrossRef]
- Gao, R.; Zeng, L.; Ding, H.; Zhang, T.; Wang, X.; Fang, Q. Characterization of oxide dispersion strengthened ferritic steel fabricated by electron beam selective melting. Mater. Des. 2016, 89, 1171–1180. [Google Scholar] [CrossRef]
- Zhai, W.; Zhu, Z.; Zhou, W.; Nai, S.M.L.; Wei, J. Selective laser melting of dispersed TiC particles strengthened 316L stainless steel. Compos. Part B Eng. 2020, 199, 108291. [Google Scholar] [CrossRef]
- Martin, J.H.; Yahata, B.D.; Hundley, J.M.; Mayer, J.A.; Schaedler, T.A.; Pollock, T.M. 3D printing of high-strength aluminium alloys. Nature 2017, 549, 365–369. [Google Scholar] [CrossRef]
- Torrent, C.; Wackenrohr, S.; Richter, R.; Sobrero, C.E.; Degener, S.; Krooß, P.; Maier, H.J.; Niendorf, T. On the microstructural and cyclic mechanical properties of pure iron processed by electron beam melting. Adv. Eng. Mater. 2021, 23, 2100018. [Google Scholar] [CrossRef]
- Torrent, C.J.J.; Krooß, P.; Niendorf, T. On the Impact of Build Envelope Sizes on E-PBF Processed Pure Iron. Met. Mater Trans. B 2021. [Google Scholar] [CrossRef]
- Bachmann, F.; Hielscher, R.; Schaeben, H. Texture analysis with MTEX- Free and open source software toolbox. Solid State Phenom. 2010, 160, 63–68. [Google Scholar] [CrossRef]
- Oyane, A.; Kim, H.-M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. A 2003, 65, 188–195. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Laschuk, N.O.; Easton, E.B.; Zenkina, O.V. Reducing the resistance for the use of electrochemical impedance spectroscopy analysis in materials chemistry. RSC Adv. 2021, 11, 27925–27936. [Google Scholar] [CrossRef]
- Ulum, M.F.; Caesarendra, W.; Alavi, R.; Hermawan, H. In-Vivo Corrosion Characterization and Assessment of Absorbable Metal Implants. Coatings 2019, 9, 282. [Google Scholar] [CrossRef]
- Cordero, Z.C.; Meyer, H.M.; Nandwana, P.; Dehoff, R.R. Powder bed charging during electron-beam additive manufacturing. Acta Mater. 2017, 124, 437–445. [Google Scholar] [CrossRef]
- King, W.E.; Barth, H.D.; Castillo, V.M.; Gallegos, G.F.; Gibbs, J.W.; Hahn, D.E.; Kamath, C.; Rubenchik, A.M. Observation of keyhole-mode laser melting in laser powder-bed fusion additive manufacturing. J. Mater. Processing Technol. 2014, 214, 2915–2925. [Google Scholar] [CrossRef]
- Tammas-Williams, S.; Zhao, H.; Léonard, F.; Derguti, F.; Todd, I.; Prangnell, P.B. XCT analysis of the influence of melt strategies on defect population in Ti–6Al–4V components manufactured by Selective Electron Beam Melting. Mater. Charact. 2015, 102, 47–61. [Google Scholar] [CrossRef]
- Marchese, G.; Basile, G.; Bassini, E.; Aversa, A.; Lombardi, M.; Ugues, D.; Fino, P.; Biamino, S. Study of the Microstructure and Cracking Mechanisms of Hastelloy X Produced by Laser Powder Bed Fusion. Materials 2018, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; McLean, A.; Rutter, J.W.; Sommerville, I.D. Influence of rare earth metals on the nucleation and solidification behavior of iron and 1045 steel. Metall. Trans. B 1988, 19, 383–395. [Google Scholar] [CrossRef]
- HAO, F.; LIAO, B.; LI, D.; DAN, T.; REN, X.; YANG, Q.; LIU, L. Effects of rare earth oxide on hardfacing metal microstructure of medium carbon steel and its refinement mechanism. J. Rare Earths 2011, 29, 609–613. [Google Scholar] [CrossRef]
- Ding, X.; Koizumi, Y.; Aoyagi, K.; Kii, T.; Sasaki, N.; Hayasaka, Y.; Yamanaka, K.; Chiba, A. Microstructural control of alloy 718 fabricated by electron beam melting with expanded processing window by adaptive offset method. Mater. Sci. Eng. A 2019, 764, 138058. [Google Scholar] [CrossRef]
- Günther, J.; Brenne, F.; Droste, M.; Wendler, M.; Volkova, O.; Biermann, H.; Niendorf, T. Design of novel materials for additive manufacturing—Isotropic microstructure and high defect tolerance. Sci. Rep. 2018, 8, 1298. [Google Scholar] [CrossRef]
- Körner, C. Additive manufacturing of metallic components by selective electron beam melting—A review. Int. Mater. Rev. 2016, 61, 361–377. [Google Scholar] [CrossRef]
- Lejček, P.; Roudnická, M.; Čapek, J.; Dvorský, D.; Drahokoupil, J.; Šimek, D.; Čížek, J.; Svora, P.; Molnárová, O.; Vojtěch, D. Selective laser melting of pure iron: Multiscale characterization of hierarchical microstructure. Mater. Charact. 2019, 154, 222–232. [Google Scholar] [CrossRef]
- Hansen, N. Hall–Petch relation and boundary strengthening. Scr. Mater. 2004, 51, 801–806. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Fe + Fe2O3] | [Fe + Fe2O3] + Fe, 1:2 | [Fe + Fe2O3] + Fe, 1:4 | Fe | [Fe + CeO2] + Fe, 1:4 | [Fe + CeO2] + Fe, 1:2 | [Fe + CeO2] |
|---|---|---|---|---|---|---|
| 0.045% | 0.022% | 0.011% | - | 0.008% | 0.016% | 0.031% |
| Strategy | Preheat | Melt |
|---|---|---|
| Meander, 90° | I = 15 mA | I = 12.25 mA |
| Hatch only | v = 12,000 mm/s | v = 4000 mm/s |
| Layer = 0.05 mm | 12 repetitions | hatch = 0.08 mm |
| Reagents | Provider | Quality | Proportion |
|---|---|---|---|
| NaCl | VWR | p.a. | 5.403 g/L |
| NaHCO3 | VWR | p.a. | 0.504 g/L |
| Na2CO3 | VWR | p.a. | 0.426 g/L |
| KCl | Merck | p.a. | 0.225 g/L |
| K2HPO4*3 H2O | Merck | p.a. | 0.230 g/L |
| MgCl*6 H2O | Merck | p.a. | 0.311 g/L |
| C8H18N2O4S | Roth | p.a. | 17.892 g/L |
| CaCl2 | Merck | p.a. | 0.293 g/L |
| Na2SO4 | Merck | p.a. | 0.072 g/L |
| Analyzed Powder | C 1s | O 1s | Fe2p3/2 | Ce 4d |
|---|---|---|---|---|
| Iron powder | 44.4 | 45.8 | 9.8 | - |
| Oxidized powder | 39.6 | 49.0 | 11.4 | - |
| CeO2 modified | 43.5 | 46.1 | 8.3 | 2.1 |
| Material Condition | 0 min | 1435 min | ||
|---|---|---|---|---|
| [Fe + Fe2O3] | 2.3 × 10−7 | 2.7 × 10−3 | 6.2 × 10−6 | 7.2 × 10−2 |
| [Fe + Fe2O3] + Fe, 1:1 | 2.4 × 10−6 | 2.8 × 10−2 | 5.1 × 10−6 | 5.9 × 10−2 |
| [Fe + Fe2O3] + Fe, 1:4 | 1.9 × 10−6 | 2.2 × 10−2 | 4.6 × 10−6 | 5.3 × 10−2 |
| [Fe] | 5.2 × 10−7 | 6.0 × 10−3 | 1.0 × 10−5 | 1.2 × 10−1 |
| [Fe + CeO2] + Fe, 1:4 | 9.6 × 10−7 | 1.1 × 10−2 | 7.7 × 10−6 | 8.9 × 10−2 |
| [Fe + CeO2] + Fe, 1:1 | 3.4 × 10−6 | 3.9 × 10−2 | 5.4 × 10−6 | 6.3 × 10−2 |
| [Fe + CeO2] | 1.0 × 10−5 | 1.2 × 10−1 | 1.0 × 10−5 | 1.2 × 10−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrent, C.J.J.; Krooß, P.; Huang, J.; Voigt, M.; Ebbert, C.; Knust, S.; Grundmeier, G.; Niendorf, T. Oxide Modified Iron in Electron Beam Powder Bed Fusion—From Processability to Corrosion Properties. Alloys 2022, 1, 31-53. https://doi.org/10.3390/alloys1010004
Torrent CJJ, Krooß P, Huang J, Voigt M, Ebbert C, Knust S, Grundmeier G, Niendorf T. Oxide Modified Iron in Electron Beam Powder Bed Fusion—From Processability to Corrosion Properties. Alloys. 2022; 1(1):31-53. https://doi.org/10.3390/alloys1010004
Chicago/Turabian StyleTorrent, Christof J. J., Philipp Krooß, Jingyuan Huang, Markus Voigt, Christoph Ebbert, Steffen Knust, Guido Grundmeier, and Thomas Niendorf. 2022. "Oxide Modified Iron in Electron Beam Powder Bed Fusion—From Processability to Corrosion Properties" Alloys 1, no. 1: 31-53. https://doi.org/10.3390/alloys1010004
APA StyleTorrent, C. J. J., Krooß, P., Huang, J., Voigt, M., Ebbert, C., Knust, S., Grundmeier, G., & Niendorf, T. (2022). Oxide Modified Iron in Electron Beam Powder Bed Fusion—From Processability to Corrosion Properties. Alloys, 1(1), 31-53. https://doi.org/10.3390/alloys1010004