Abstract
Kawasaki disease (KD) is an acute vasculitis that mainly affects children under 5 years of age, leading to coronary artery alterations (CAAs) in 25% of untreated patients. Macrophage activation syndrome (MAS) is a secondary hemophagocytic lymphohistiocytosis (HLH) that can complicate the acute, subacute, and chronic phases of KD. We retrospectively reviewed three cases of children affected by KD complicated with MAS hospitalized in two pediatric units in Emilia Romagna, a northern region of Italy. Case 1: a previously healthy 23-month-old female with full clinical criteria of KD and a hemorrhagic rash due to MAS during the acute phase of the illness. This patient responded promptly to a high dose of intravenous immune globulin (IVIG) and three pulses of high doses of methylprednisolone (MPD) with improvement in clinical signs and laboratory tests without the development of CAA at any phase of illness. Case 2: a previously healthy 10-month-old female with incomplete KD with persistent fever and maculopapular rash. This patient did not respond to IVIG and developed MAS during the subacute phase, characterized by persistent fever, hypertransaminasemia, hyperferritinemia, and hypofibrinogenemia after two high doses of IVIG and boluses of MPD. The patient responded to the addition of IL-1 blocker and anakinra and did not present CAA alterations during any phase of the illness. Case 3: a previously healthy 26-month-old male with incomplete KD with fever, maculopapular rash, cheilitis, and hyperemic conjunctivitis. This patient developed gallbladder hydrops and CAA in the acute phase and did not respond to two high doses of IVIG and a high dose of MPD. In the subacute phase, this patient was complicated with MAS and responded to intravenous anakinra. During the subacute phase, the patient developed transient aneurysms that regressed during the chronic phase. These cases reiterate that prompt diagnosis and aggressive immunomodulatory treatment can limit the most severe complications of MAS complicating KD. High doses of IVIG and MPD may result in a favorable outcome or more aggressive adjunctive treatment may be needed. Anakinra, cyclosporine, monoclonal antibodies, and plasmapheresis can be used as adjunctive treatment in the case of unresponsive MAS in KD. Notably, MAS, present during the subacute phase in cases 2 and 3, promptly responded to anakinra, an IL-1 blocker, without the use of cyclosporine. Our experience confirms that the IL-1 blocker can be considered an optimal choice after non-response to IVIG and MPD in KD complicating with MAS, avoiding over-treatment with cytotoxic drugs.
1. Introduction
Kawasaki disease (KD) is an acute vasculitis of the medium and small caliber vessels that primarily affects children under 5 years of age [1,2,3]. It remains the leading cause of acquired heart disease in developed countries. However, based on the literature, 10–20% of KD patients do not respond to standard therapy with a high dose of IVIG and have an increased risk of developing CAA [1,2,3]. KD can present as a more severe form, such as KD shock syndrome, or can complicate with macrophage activation syndrome (MAS). According to American and Italian guidelines, diagnostic criteria of complete KD are the presence of >5 days of fever and > four of the following: bilateral non-exudative conjunctivitis, erythema of the lips and oral mucosa, changes in extremities, skin rash, and cervical lymphadenopathy. Incomplete KD occurs in patients presenting fever without a sufficient number of main clinical criteria, while atypical KD presents a fever associated with signs and symptoms that differ from the classical ones (meningeal inflammation, gastrointestinal symptoms, acute abdomen, arthritis, pneumonia) [1,2,3]. The acute phase includes the first ten days of illness, characterized by inflammation of medium-sized vessels; the subacute phase spans from the tenth day of illness to the twentieth day of illness, and the pathognomonic sign is inflammation of the intima of the vessels [1,2,3]. Diagnosis of KD is based on diagnostic clinical criteria and supported by the results of blood and instrumental exams, but the absence of specific clinical findings or tests and the overlap with other diseases can make early diagnosis difficult and increase the risk of complications [1,2,3].
MAS, also known as a secondary form of HLH, is a serious, potentially fatal complication of rheumatic diseases characterized by excessive activation and expansion of T lymphocytes and macrophages. It exhibits hemophagocytic activity, leading to a hyperinflammatory state associated with cytopenia, liver dysfunction, and coagulopathy resembling disseminated intravascular coagulation [4,5]. Notably, extreme hyperferritinemia is present [4,5]. Mortality rates are reported to be approximately 20–30% [4,5].
MAS can complicate systemic juvenile idiopathic arthritis (sJIA) in 10% of cases [6]. However, it is increasingly reported in various rheumatic diseases in childhood, including pediatric systemic lupus erythematosus, KD, juvenile dermatomyositis, antiphospholipid syndrome, mixed connective tissue disease and, recently, multisystemic inflammatory syndrome in children (MIS-C), a severe systemic inflammation triggered by SARS-CoV-2 [7,8]. MAS is commonly triggered by infections, particularly viral infections, or during periods of high disease activity including disease onset. The incidence of MAS in patients with KD has been estimated to be 1.1–1.9% [9,10].
Diagnosis of MAS respects the HLH-2004 criteria with the presence of five of the following eight clinical and laboratory features: fever, splenomegaly, cytopenia in > two cell lines, hypertriglyceridaemia, hyperferritinaemia, and hemophagocytosis (bone marrow, liver, or lymph nodes), low NK, and high levels of IL-2 receptors.
In 2016, Ravelli et al. have developed new and more sensitive (73%) and specific (99%) criteria (sJIA-MAS 2016) for the classification of MAS in patients with sJIA, including high ferritin (>684 ng/mL) and at least two of the following laboratory abnormalities: thrombocytopenia (PLT < 180.000/mmc), hypofibrinogenemia (<360 mg/dL), increased of aspartate aminotransferase (AST > 48 U/L), and of triglycerides (>160 mg/dL) [6] (Table 1).

Table 1.
Comparison between HLH-2004 diagnostic criteria and 2016 consensus criteria for MAS in sJIA.
Specific criteria for MAS complicating KD are missing, but other authors have suggested that the 2016 consensus criteria for MAS in sJIA can be applied to KD patients due to higher sensitivity and sensibility with respect to HLH 2004 criteria. In addition, the criteria can be used for other inflammatory conditions causing secondary HLH or MAS [6,9]. Splenomegaly is not present in the 2016 Ravelli criteria due to the usual presence of this clinical sign in sJIA.
The under-diagnosis of MAS in KD patients is an important issue. Distinguishing MAS from severe forms of KD as resistant forms can be a challenge, as both share persistent fever, elevated levels of aminotransferases, thrombocytopenia, anemia, and hyperferritinemia. Some authors suggest considering KD-resistant forms as sub-clinical forms of MAS [9,10,11]. Notably, MAS has higher values of ferritin, triglycerides, cytopenia, and the presence of hemorrhagic manifestations with respect to refractory KD, suggesting that resistant forms could be the first step of MAS.
The recognition of MAS and immediate therapeutic intervention is the key to silencing this inflammatory phenomenon. Traditionally, steroids, etoposide, and cyclosporine have been the mainstays of therapy for HLH and MAS. For MAS, most clinicians start with intravenous MPD pulse therapy (e.g., 30 mg/kg for three consecutive days) followed by 2–3 mg/kg/day in two to four divided doses. If a response to steroids is not immediately evident, parenteral administration of cyclosporine A (CsA) (2–7 mg/kg/day) is usually initiated [12,13]. New classes of medications have become available. Cytokines can be directly neutralized with biological agents such as anakinra or emapalumab. Alternatively, the action of cytokines can be inhibited by blocking signaling pathways with JAK inhibitors. These cytokine-directed therapies hold much promise in their ability to control inflammation with less toxicity than chemotherapy [12].
Treatment guidelines for KD patients complicating with MAS are lacking. Interestingly, there is considerable clinical and therapeutic overlap between MAS and more severe forms such as resistant KD and MIS-C [14,15,16,17].
The over-treatment of MAS in KD patients can be a problem due to the good response of MAS in KD to immunomodulatory treatment without the use of cytotoxic drugs that lead to several hepatic and renal complications. Successful treatment with the IL-1 blockade, anakinra, for refractory KD complicated with MAS has been reported, and many KD patients have had a positive outcome with immunomodulators instead of intensive chemotherapy [12,13].
Prompt therapy, as well as an earlier diagnosis, are the cornerstone to reducing morbidity and mortality and improving the outcomes of MAS complicating KD [4,5,10]. Reports for KD complicated with MAS are limited [14,16,17]; for this reason, we report three cases, describing clinical presentation, laboratory findings, and response to therapy and highlighting the efficacy of the IL-1 blocker in KD-resistant forms with MAS.
1.1. Case 1
A 23-month-old female was admitted to the Pediatric Unit of Ramazzini Hospital, Carpi, with a history of a 4-day high fever and a rash, initially maculopapular and becoming hemorrhagic on the fourth day and involving the head, trunk, and extremities (Figure 1).
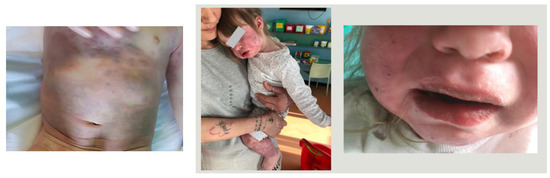
Figure 1.
Clinical manifestations of case 1.
The patient was irritable, presenting bilateral non-purulent conjunctivitis, mucositis, alterations of extremities, and hepatosplenomegaly. Laboratory tests on admission showed white blood cells (WBCs): 4.100/mmc; hemoglobin (Hb): 10.6 g/dl; platelet count (PLT): 143.000/mmc; erythrocyte sedimentation rate (ESR): 7 mm in the first hour; C-reactive-protein (CRP): 6.3 mg/L; P.T. ratio: 1.98; fibrinogen: 329 mg/dL; sodium: 126 mEq/L; albumin: 3 g/dL; and urine was normal. The day after admission, blood tests were reported and revealed a reduction in PLT (115.000/mmc) and fibrinogen (173 mg/dL), and a high level of ferritin (689 ng/mL) and triglycerides (347 mg/dL). A SARS-CoV-2 swab was not performed because the case occurred before the SARS-CoV-2 pandemic. The echocardiography was normal except for a trivial pericardial effusion. The abdominal echography detected splenomegaly without evidence of gallbladder hydrops.
Serological investigations excluded EBV, parvovirus, mycoplasma, and adenovirus infection.
A diagnosis of complete KD with MAS, according to HLA-2004 criteria and sJIA–MAS 2016 criteria, was performed. The patient was promptly treated on the sixth day of illness with a high dose of IVIG at 2 g/kg and boluses of MPD at 30 mg/kg/day for three consecutive days with resolution of fever and improvement of clinical signs.
The laboratory tests improved (WBC: 10.700/mmc, PLT: 192.000/mmc, CRP 0.7 mg/dL, fibrinogen 135 mg/dL, AST: 35 U/L, and ALT: 26 U/L, ferritin: 265 ng/mL). An echocardiography of the subacute phase and chronic phase confirmed the normal size of coronary arteries. The fever resolved on the seventh day of illness. After MPD boluses, steroids were tapered to oral administration with escalation in 4 weeks and acetylsalicylic acid (ASA) at an antiaggregant dose was continued for a total of 8 weeks. On the 10th day of illness, the patient had finger hand desquamation, and all the clinical signs improved except for the persistence of a maculopapular exanthema of the body that persisted until the chronic phase.
1.2. Case 2
A ten-month-old female patient was admitted to the Pediatric Unit at Ramazzini Hospital, Carpi, presenting with a 4-day high fever, maculo-papular rash, and elevated inflammatory markers (WBC: 19.700/mmc, CRP: 11 mg/dL, fibrinogen: 747 ng/dL, ferritin: 4.704 ng/dL). The acid nucleic of SARS-CoV-2 in the nasopharyngeal swab was negative. Serological investigations excluded EBV, parvovirus, mycoplasma, and adenovirus infection.
An empiric antibiotic treatment was started without success. For the persistence of the fever, an echocardiogram was performed, showing the presence of pericardial effusion. The repeated laboratory tests on the seventh day of fever showed an elevated WBC count (33.600/mmc), platelets (493.000/mmc), AST (115 U/L), ALT (97 U/L), and a low level of albumin (2.8 g/dL) and sodium (132 mEq/L). Upon the suspicion of incomplete KD, the patient received a high dose of IVIG (2 g/kg) and MPD (0.8 mg/kg/day) on the seventh day of illness with the persistence of fever 36 hours after IVIG treatment. Thus, on the tenth day of fever, the patient received a second high dose of IVIG, and for the persistence of fever 48 h later, the patient started boluses of MPD at 30 mg/kg for three days without clinical response. In addition, inflammatory markers (WBC: 38,300/mmc), triglycerideamia (337 mg/dL), levels of aminotransferases (AST 340 U/L), and ferritin (>7500 ng/dL) increased, and anemia (9.4 g/dL) and fibrinogenaemia (156 ng/dL) decreased. For the persistence of fever, upon suspicion of MAS, according to sJIA 2016 criteria, intravenous anakinra (2 mg/kg every 6 h) was started, and the patient was transferred to the Pediatric Unit at Gozzadini Children’s Hospital, University of Bologna, for the continuation of treatment. After anakinra and MPD at 12 mg/kg q 8 h, the fever resolved, and clinical and laboratory markers of inflammation due to MAS reduced (WBC: 9400/mmc, CRP: 0.15 mg/dL, ferritin: 79 ng/dL, triglycerides: 232 mg/dL, AST: 50 U/L).
The patient never developed CAA during any phase of illness.
1.3. Case 3
A previously healthy 26-month-old male was admitted to the Pediatric Unit at Bologna University Hospital on the fourth day of fever, presenting vomiting, maculopapular rash, chelitis, and hyperemic conjunctivitis. The acid nucleic of SARS-CoV-2 in the nasopharyngeal swab was negative. Laboratory tests documented elevated levels of inflammatory markers (WBC: 21.870/mmc, CRP: 32.48 mg/dL, PCT 5 ng/dL), increased AST (201 U/L) and ALT (349 U/L), and cholestasis. The patient was submitted to empiric antibiotic therapy without improvement and with persistent fever. The abdomen US showed gallbladder hydrops. An echocardiography was performed, which showed ectasia of the left anterior descending (LAD) (Z-score 2.3) and left main coronary artery (LMCA) (Z-score 2.2). Upon the suspicion of incomplete KD, the patient was treated on the sixth day of illness with a single dose of IVIG (2 g/kg) and ASA at an anti-inflammatory dose. Because of the recrudescence of fever after 24 h of apyrexia, the patient received a second dose of IVIG (2 g/kg) and boluses of MPD (30 mg/kg/day for three consecutive days) without effectiveness. A bone marrow aspiration was performed, and the cytology documented rare elements of hemophagocytosis. Infective etiologies were excluded, and laboratory tests showed a huge increase in ferritin (1529 ng/dL), hypertrigliceridemia (505 mg/dL), hypofibrinogenemia (156 mg/dL), hypertransaminasemia (AST/ALT: 210/349), thrombocytopenia (PLT: 134.000/mmc), and anemia (7.1 g/dL). Splenomegaly appeared. Suspecting MAS complicating KD, according to HLH- 2004 criteria and 2016 consensus for MAS in sJIA, the patient received anakinra iv (8 mg/kg/die) and other MPD at 30 mg/kg/day for three days. Forty-eight hours later, the patient became afebrile and laboratory tests dramatically improved (WBC: 10.360/mmc, Hb: 11 g/dL, PLT: 641.000/mmc, ferritin: 15 ng/dL, ALT/AST: 22/34, fibrinogen: 241 mg/dL). The echocardiography performed on day 13 showed a multi-coronary involvement with small aneurysms of LMCA: Z-score 2.5; LAD: Z-score 3.4; left circumflex artery (LCA): Z-score 2.85; and right coronary artery (RCA): Z-score 4. During hospitalization, anakinra was slowly decreased and switched to subcutaneous administration. ASA was continued at an antiaggregant dose, and steroids were shifted to oral administration with escalating doses until suspension after 6 weeks. CAA slowly regressed and normalized (Z-score <2 in all sites investigated) at day 29 after fever onset.
In Table 2 and Table 3, the principal demographic, clinical, and laboratory findings of the three cases are shown.
Table 2.
Demographic, clinical, and cardiac findings of the described cases.
Table 3.
Laboratory tests confirming diagnosis of MAS complicating KD in the three described cases.
2. Discussion
MAS is a severe and rare complication of KD involving 1.1–1.9% of cases that can cause organ failure, disseminated coagulopathy, cytopenia, and intensive care unit admissions [15]. The mortality rate of MAS in KD was reported to be 8–22%. Despite 85% of KD occurring in children under the age of 2 years old, published reports show that MAS complicates KD in patients older than 5 years old: on the opposite, our three patients were younger than 5 years old: two of the three were less than 2-years-old and the third was slightly older [14,15,16]. Usually, male predominance has been reported, but two of the three described cases were female [14,15,16]. MAS may occur during any stage of KD and may also precede KD diagnosis, but in most cases, it appears simultaneously with KD [14]. MAS occurred during the acute phase in case 1 and during the subacute phase in the other two cases. The incidence of MAS is higher in resistant KD (7%), in Kawasaki shock syndrome (4.8%), and in incomplete KD presentation (3.2%) with respect to typical KD [18]. Our first reported case had a complete KD presentation, while cases 2 and 3 were incomplete resistant KD forms. Continuous fever and splenomegaly are the main clinical findings of MAS in patients with KD, which can help to diagnose this condition [10,11]. Indeed, splenomegaly is rarely present in patients with KD but occurs in 69% of patients with KD complicated with MAS and can be present in resistant KD: it was present in cases 1 and 3. In addition, about 20% of patients can present hemorrhagic manifestations and multiorgan dysfunctions (cardiovascular, respiratory, hepatic, renal, or neurological dysfunctions) mimicking a sepsis condition [17]. In case 1, the presence of a hemorrhagic rash and splenomegaly in the acute phase suggested expanding laboratory tests with screening for MAS (ferritin, fibrinogen, triglycerides, and aminotransferases) as recommended by the 2016 consensus for MAS in sJIA. The diagnosis of MAS complicating KD would be suspected in unresponsive KD patients with hemorrhagic rash, splenomegaly, and alteration of specific laboratory tests for MAS, after the exclusion of other infectious diseases. Notably, persistent fever, skin rash, thrombocytopenia, abnormal liver enzymes, anemia, and hypertriglyceridaemia can be found in both resistant KD and MAS, but usually, the levels of ferritin and triglycerides are more increased in MAS than in KD. In any case, echocardiograms are crucial: when altered, they support the primitive diagnosis of KD and help make treatment decisions to add ASA and anticoagulation, in the case of large aneurysms, and in follow-up. Specific criteria for the diagnosis of MAS in KD are lacking. The 2016 criteria for MAS in sJIA include easier laboratory tests that can confirm the suspicion of MAS and allow an earlier diagnosis, avoiding the possibility of delayed therapy and a more severe clinical outcome and higher risk of mortality due to MAS. All three cases met the criteria proposed by Ravelli in the 2016 consensus for MAS in sJIA. In addition, cases 1 and 3 met the HLH-2004 criteria. Bone marrow aspiration with signs of hemophagocytosis is included in HLH-2004 criteria for MAS, and it is a sign of over-activation of macrophages but does not always occur in the early stage of illness and could not be detected at any phase of MAS (25–100%) using bone examination [6,19,20]. For this reason, the presence of hemophagocytosis is not present in the 2016 consensus criteria for MAS in sJIA. Only the third case made a bone marrow aspiration with the presence of rare elements of hemophagocytosis.
Another crucial difficulty in the diagnosis of MAS is the overlap of this condition with severe forms of KD that are resistant to therapy.
Risk factors for IVIG resistance in KD include prolonged fever, young age, leukocytosis, thrombocytopenia, and high CRP levels, which allow more aggressive therapies to resolve inflammation and prevent the appearance of CAA [18,21,22]. MAS and refractory KD share a similar severe phenotype, and many reports suggest considering MAS in KD patients with high risk for IVIG resistance, proposing MAS screening in routine laboratory tests used for refractory KD, particularly in children with younger age, the male gender, and those with increased markers of inflammation [22]. Case 2 and case 3 developed MAS after unresponsiveness to second-line therapy for KD.
The goal in treating MAS is to promptly reduce the aberrant systemic inflammation as quickly as possible and prevent organ damage. This includes KD patients, who have a higher risk of cardiac complications as the appearance of CAA. Thrombocytopenia, usually present in MAS and in severe forms of KD may be a risk factor for coronary artery aneurysms (CAAs).
The standard treatment for MAS is high doses of intravenous steroids such as MDP for three to five days. Depending on the child’s response, additional therapy such as cyclosporine, a drug that suppresses the immune system, may be added. In some cases, cyclosporine has been reported to reduce symptoms within 12 to 24 hours [4,5]. The IL-1 blocker, anakinra, has also been demonstrated to be effective in the treatment of MAS and sJIA and has been proposed for refractory cases to IVIG and MPD in MIS-C [23]. Unfortunately, standard recommendations for MAS complicating KD are lacking, although most clinicians consider first-line treatment to be IVIG infusion with a high doses pulse of steroids. In our experience, the first case responded to IVIG and boluses of MDP at 30 mg/kg/day, and the other two cases responded to addition of anakinra iv, a IL-1 blocker, after non-response to the first-line treatment. Despite this being only a small case series, cases 2 and 3 showed an excellent response to anakinra iv in both cases of refractory KD complicating with MAS after unresponsiveness to IVIG and boluses of MPD. Indeed, the clinical signs and laboratory tests improved soon after the start of anakinra iv with the resolution of hyperinflammatory condition and regressions of aneurysms in the chronic phase in case 3. Importantly, no side effects occurred. Unresponsive KD or KD patients with risk factors for CAA usually early start steroids in addition to IVIG and ASA, and this practice can mask the occurrence of MAS. We suggest considering MAS in the case of IVIG unresponsiveness and performing specific laboratory tests, especially in the presence of continuous fever, peculiar hemorrhagic skin rash, and splenomegaly. The severity of manifestations can guide the treatment in the case of the occurrence of MAS in KD, In the case of severe cases, anakinra could be safely added to high-dose MPD boluses [24], avoiding the possibility of over-treatment of KD cases complicating with MAS with strong immunosuppressing drugs.
Other additional treatments, such as tumor necrosis factor (TNF) inhibitors, immunosuppressant agents, or plasma exchange can be considered [24] in cases of unresponsiveness. Echocardiograms to focus on the size of coronary arteries are mandatory to eventually add anticoagulation therapy in the case of large aneurysms.
3. Conclusions
An early diagnosis of MAS in KD is crucial to start a prompt and aggressive therapy to halt severe MAS-related inflammation and limit severe complications, morbidity, and mortality. Diagnosis of the occurrence of MAS can be challenging due to overlapping features of the severe forms of KD, but the typical clinical manifestations along with laboratory tests can help.
Despite the small number of cases reported here, in our experience, an IL-1 blocker is a valid therapy after the ineffectiveness of first-line treatment of resistant KD complicating with MAS, which can avoid the use of cytotoxic drugs. Further evidence is needed to support our findings and to define more specific criteria for the diagnosis of MAS in KD and guidelines to treat this severe complication.
Author Contributions
Conceptualization: E.C.; methodology: E.C. and M.F.; software: I.S.; validation: F.T. and M.L.; resources: E.C.; data curation: I.S. and E.R.P.; writing—original draft preparation: E.C.; writing—review and editing: E.C. and M.F.; visualization: L.A. and E.R.P.; supervision: M.F.; project administration: E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
All subjects provided their informed consent for inclusion before they participated in this study. This study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Area Vasta Emilia Nord. Approval Code: 850/2019/OSS/AUSLMO—ERKAWA, Date: 5 November 2019.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mc Crindle, B.W.; Rowley, A.H.; Newburger, J.W. Diagnosis treatment, and long term management of Kawasaki Disease: A scientific statement for health professionals from the American Heart Association. Circulation 2017, 135, 927–999. [Google Scholar]
- Marchesi, A.; Rigante, D.; Cimaz, R.; Ravelli, A.; Tarissi de Jacobis, I.; Rimini, A.; Cardinale, F.; Cattalini, M.; De Zorzi, A.; Dellepiane, R.M.; et al. Revised recommendations of the Italian Society of Pediatrics about the general management of Kawasaki disease. Ital. J. Pediatr. 2021, 47, 16. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, A.; Tarissi de Jacobis, I.; Rigante, D. Kawasaki disease: Guidelines of the Italian Society of Pediatrics, part I—Definition, epidemiology, etiopathogenesis, clinical expression and management of the acute phase. Ital. J. Pediatr. 2018, 44, 102. [Google Scholar] [CrossRef] [PubMed]
- Ziaee, V.; Moardinejad, M.H. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Pediatr. Rheumatol. 2008, 6, P62. [Google Scholar] [CrossRef]
- Mashuku, S.; Hibi, S.; Todo, S. Hemophagocytic lymphohistiocytosis in infancy and childhood. J. Pediatr. 1997, 130, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Minoia, F.; Davı, S.; Horne, C.; Bovis, F.; Pistorio, A.; Arico, M.; Avcin, M.; Behrens, E.; De Benedetti, F.; et al. Classification criteria for Macrophage Activation Syndrome complicating systemic Juvenile idiopathic arthritis: A European meague against rheumatism. Ann. Rheum. Dis. 2016, 75, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Smith, J.J.; Verweyen, E.L.; Clay, G.M.; Esteban, Y.M.; de Loizaga, S.R.; Baker, E.J.; Do, T.; Dhakal, S.; Lang, S.M.; Grom, A.A.; et al. Inflammatory biomarkers in COVD-19 associated multisystem inflammatory syndrome in children, Kawasaki disease, and macrophage activation syndrome: A cohort study. Lancet Rheumatol. 2021, 3, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.S.; Villarreal, E.G.; Flores, S. COVID-19 and Hyperinflammatory Syndrome in Children: Kawasaki disease with Macrophage Activation Syndrome in Disguise? Cureus 2020, 12, e9515. [Google Scholar] [CrossRef] [PubMed]
- Han, S.B.; Lee, S.Y.; Jeong, D.C.; Kang, J.H. Should 2016 criteria for Macrophage Activation Syndrome be applied in children with Kawasaki disease, as well as with systemic-onset juvenil idiopathic arthritis? Ann. Rheum. Dis. 2016, 75, 7. [Google Scholar] [CrossRef]
- Muise, A.; Tallett, S.E.; Silverman, E.D. Are Children With Kawasaki Disease and Prolonged Fever at Risk for Macrophage Activation Syndrome? Pediatrics 2003, 112, e495–e497. [Google Scholar] [CrossRef]
- Sawhney, S.; Woo, P.; Murray, K.J. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch. Dis. Child. 2001, 85, 421–426. [Google Scholar] [CrossRef] [PubMed]
- McClain, K.L. Treatment and Prognosis of Hemophagocytic Lymphohistiocytosis; UpToDate: Waltham, MA, USA, 2014. [Google Scholar]
- Henderson, L.A.; Cron, R.Q. Macrophage activation syndrome and secondary Hemophagocytic Lymphohistiocytosis in childhood inflammatory disorders: Diagnosis and treatment. Pediatr. Drugs 2020, 22, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Latino, G.A.; Manlhiot, C.; Yeung, R.S.; Chahal, N.; McCrindle, B.W. Macrophage activation syndrome in the acute phase of Kawasaki disease. J. Pediatr. Hematol. Oncol. 2010, 32, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gong, F.; Zhu, W.; Fu, S.; Zhang, Q. Macrophage activation syndrome in Kawasaki Disease: More common that we thought? Semin. Arthritis Rheum. 2015, 44, 405–410. [Google Scholar] [CrossRef] [PubMed]
- García-Pavón, S.; Yamazaki-Nakashimada, M.A.; Báez, M.; Borjas-Aguilar, K.L.; Murata, C. Kawasaki Disease Complicated with Macrophage Activation Syndrome: A systemic Review. J. Pediatr. Hematol. Oncol. 2017, 39, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Luo, Y.; Liu, X.; Xu, J.; Liu, C. Kawasaki disease complicated with macrophage activation syndrome: Case reports and literature review. Front. Pediatr. 2019, 4, 423. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Han, S.B.; Lee, S.Y.; Jeong, D.C. Should refractory Kawasaki disease be considered occult macrophage activation syndrome? Semin. Arthritis Rheum. 2017, 46, e17. [Google Scholar] [CrossRef] [PubMed]
- Crayne, C.; Cron, R.Q. Pediatric macrophage activation syndrome, recognizing the tip of the Iceberg. Eur. J. Rheumatol. 2020, 7 (Suppl. S1), S13–S20. [Google Scholar] [CrossRef]
- Han, S.B.; Lee, S.-Y. Macrophage activation syndrome in children with Kawasaki disease: Diagnostic and therapeutic approches. World J. Pediatr. 2020, 16, 566–574. [Google Scholar] [CrossRef]
- Rhee, S.; Kim, D.; Cho, K.; Rhim, J.W.; Lee, S.Y.; Jeong, D.C. Under-recognized Macrophage Activation Syndrome in Refractory Kawasaki Disease: A Wolf in Sheep’s Clothing. Children 2022, 9, 1588. [Google Scholar] [CrossRef]
- Panaro, S.; Cattalini, M. The spectrum of manifestations of severe acute respiratory syndrome-Coronavirus 2 (SARS-CoV2) Infection in children: What we can learn from multisystem inflammatory syndrome in children (MIS-C). Front. Med. 2021, 8, 747190. [Google Scholar] [CrossRef]
- Mehta, P.; Cron, R.Q.; Hartwell, J.; Manson, J.J.; Tattersall, R.S. Silencing the cytokine storm: The use of intravenous anakinra in haemaphagovytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020, 2, 358–367. [Google Scholar] [CrossRef]
- Schulert, G.S.; Grom, A.A. Macrophage activation syndrome and cytokine directed therapies. Best Pract. Res. Clin. Rheumatol. 2014, 28, 277–292. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).