
Programmable Proteins: Target Specificity, Programmability and Future Directions


{kind=link}
Abstract
1. Introduction: What Are Programmable Proteins?
2. Programmable Nucleases, Modifiers, and Nucleic Acid-Binding Proteins
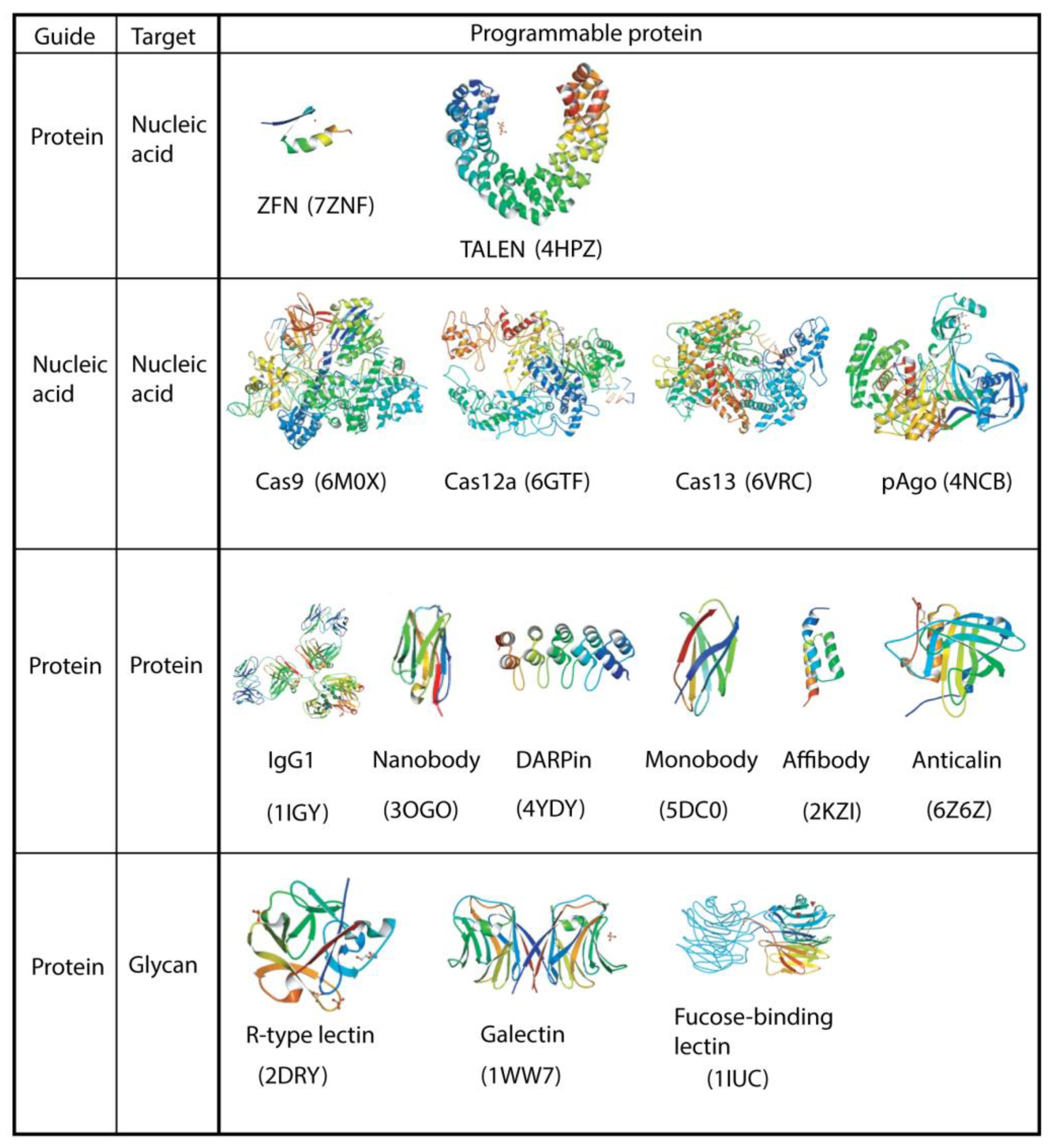
2.1. ZFNs and TALENs
2.2. Cas9 (Type II CRISPR-Cas)
2.3. Cas12 (Type V CRISPR-Cas)
2.4. Cas13 (Type VI CRISPR-Cas)
2.5. OMEGA (TnpB, IscB)
2.6. Argonaute
3. Programmable Protein-Binding Proteins
3.1. Single-Domain Antibody, Nanobody, VHH
3.2. DARPin
3.3. Monobody, Adnectin®, FingR
3.4. Affibody
3.5. Anticalin®
4. Glycan-Binding Proteins (GBPs)
5. Perspectives
Funding
Conflicts of Interest
References
- Micura, R.; Höbartner, C. Fundamental studies of functional nucleic acids: Aptamers, riboswitches, ribozymes and DNAzymes. Chem. Soc. Rev. 2020, 49, 7331–7353. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Du, L.; Li, M. Aptamer-based carbohydrate recognition. Curr. Pharm. Des. 2010, 16, 2269–2278. [Google Scholar] [CrossRef]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, J. Catalytic Nucleic Acids: Biochemistry, Chemical Biology, Biosensors, and Nanotechnology. iScience 2020, 23, 100815. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. The Biochemical Landscape of Riboswitch Ligands. Biochemistry 2022, 61, 137–149. [Google Scholar] [CrossRef]
- Lee, G.; Jang, G.H.; Kang, H.Y.; Song, G. Predicting aptamer sequences that interact with target proteins using an aptamer-protein interaction classifier and a Monte Carlo tree search approach. PLoS ONE 2021, 16, e0253760. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Židek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with Alpha Fold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef]
- Cao, L.; Coventry, B.; Goreshnik, I.; Huang, B.; Sheffler, W.; Park, J.S.; Jude, K.M.; Marković, I.; Kadam, R.U.; Verschueren, K.H.G.; et al. Design of protein-binding proteins from the target structure alone. Nature 2022, 605, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lisanza, S.; Juergens, D.; Tischer, D.; Watson, J.L.; Castro, K.M.; Ragotte, R.; Saragovi, A.; Milles, L.F.; Baek, M.; et al. Scaffolding protein functional sites using deep learning. Science 2022, 377, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Chandrasegaran, S.; Carroll, D. Origins of Programmable Nucleases for Genome Engineering. J. Mol. Biol. 2016, 428, 963–989. [Google Scholar] [CrossRef]
- Kropocheva, E.V.; Lisitskaya, L.A.; Agapov, A.A.; Musabirov, A.A.; Kulbachinskiy, A.V.; Esyunina, D.M. Prokaryotic Argonaute Proteins as a Tool for Biotechnology. Mol. Biol. 2022, 1–20. [Google Scholar] [CrossRef]
- Kim, Y.G.; Chandrasegaran, S. Chimeric restriction endonuclease. Proc. Natl. Acad. Sci. USA 1994, 91, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Hafez, M.; Hausner, G. Homing endonucleases: DNA scissors on a mission. Genome 2012, 55, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Nain, V. TALENs-an indispensable tool in the era of CRISPR: A mini review. J. Genet. Eng. Biotechnol. 2021, 19, 125. [Google Scholar] [CrossRef]
- Nussenzweig, P.M.; Marraffini, L.A. Molecular Mechanisms of CRISPR-Cas Immunity in Bacteria. Annu. Rev. Genet. 2020, 54, 93–120. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S. Evolutionary plasticity and functional versatility of CRISPR systems. PLoS Biol. 2022, 20, e3001481. [Google Scholar] [CrossRef]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, a012757. [Google Scholar] [CrossRef]
- Yamagata, M.; Sanes, J.R. CRISPR-mediated Labeling of Cells in Chick Embryos Based on Selectively Expressed Genes. Bio Protoc. 2021, 11, e4105. [Google Scholar] [CrossRef]
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Safari, F.; Zare, K.; Negahdaripour, M.; Barekati-Mowahed, M.; Ghasemi, Y. CRISPR Cpf1 proteins: Structure, function and implications for genome editing. Cell Biosci. 2019, 9, 36. [Google Scholar] [CrossRef]
- Teng, F.; Cui, T.; Feng, G.; Guo, L.; Xu, K.; Gao, Q.; Li, T.; Li, J.; Zhou, Q.; Li, W. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 2018, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- O’Connell, M.R. Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR-Cas Systems. J. Mol. Biol. 2019, 431, 66–87. [Google Scholar] [CrossRef]
- Tang, T.; Han, Y.; Wang, Y.; Huang, H.; Qian, P. Programmable System of Cas13-Mediated RNA Modification and Its Biological and Biomedical Applications. Front. Cell Dev. Biol. 2021, 9, 677587. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Abudayyeh, O.O.; Gootenberg, J.S.; Zhang, F.; Collins, J.J. CRISPR-based diagnostics. Nat. Biomed. Eng. 2021, 5, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Karvelis, T.; Druteika, G.; Bigelyte, G.; Budre, K.; Zedaveinyte, R.; Silanskas, A.; Kazlauskas, D.; Venclovas, Č.; Siksnys, V. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 2021, 599, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Altae-Tran, H.; Kannan, S.; Demircioglu, F.E.; Oshiro, R.; Nety, S.P.; McKay, L.J.; Dlakic, M.; Inskeep, W.P.; Makarova, K.S.; Macrae, R.K.; et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 2021, 374, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute proteins: Structural features, functions and emerging roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, W.; Wang, J.; Sheng, G.; Xiang, G.; Zhang, T.; Shi, W.; Li, C.; Wang, Y.; Zhao, F.; et al. Argonaute proteins from human gastrointestinal bacteria catalyze DNA-guided cleavage of single- and double-stranded DNA at 37 °C. Cell Discov. 2019, 5, 38. [Google Scholar] [CrossRef]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef]
- Yu, X.; Yang, Y.P.; Dikici, E.; Deo, S.K.; Daunert, S. Beyond Antibodies as Binding Partners: The Role of Antibody Mimetics in Bioanalysis. Annu. Rev. Anal. Chem. 2017, 10, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A. Exploring alternative antibody scaffolds: Antibody fragments and antibody mimics for targeted drug delivery. Drug Discov. Today Technol. 2018, 30, 35–46. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Engineered Protein Scaffolds as Next-Generation Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 391–415. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Lipovsek, D.; Plückthun, A. In-vitro protein evolution by ribosome display and mRNA display. J. Immunol. Methods 2004, 290, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Cherf, G.M.; Cochran, J.R. Applications of Yeast Surface Display for Protein Engineering. Methods Mol. Biol. 2015, 1319, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef]
- Chauhan, V.M.; Pantazes, R.J. MutDock: A computational docking approach for fixed-backbone protein scaffold design. Front. Mol. Biosci. 2022, 9, 933400. [Google Scholar] [CrossRef]
- Urvoas, A.; Guellouz, A.; Valerio-Lepiniec, M.; Graille, M.; Durand, D.; Desravines, D.C.; van Tilbeurgh, H.; Desmadril, M.; Minard, P. Design, production and molecular structure of a new family of artificial alpha-helicoidal repeat proteins (αRep) based on thermostable HEAT-like repeats. J. Mol. Biol. 2010, 404, 307–327. [Google Scholar] [CrossRef]
- Lee, S.C.; Park, K.; Han, J.; Lee, J.J.; Kim, H.J.; Hong, S.; Heu, W.; Kim, Y.J.; Ha, J.S.; Lee, S.G.; et al. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc. Natl. Acad. Sci. USA 2012, 109, 3299–3304. [Google Scholar] [CrossRef]
- Grabulovski, D.; Kaspar, M.; Neri, D. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J. Biol. Chem. 2007, 282, 3196–3204. [Google Scholar] [CrossRef]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Lorey, S.; Fiedler, E.; Kunert, A.; Nerkamp, J.; Lange, C.; Fiedler, M.; Bosse-Doenecke, E.; Meysing, M.; Gloser, M.; Rundfeldt, C.; et al. Novel ubiquitin-derived high affinity binding proteins with tumor targeting properties. J. Biol. Chem. 2014, 289, 8493–8507. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.; Liu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Hughes, A.L.; Guo, J.; Avila, D.; McKinney, E.C.; Flajnik, M.F. A novel “chimeric” antibody class in cartilaginous fish: IgM may not be the primordial immunoglobulin. Eur. J. Immunol. 1996, 26, 1123–1129. [Google Scholar] [CrossRef]
- Matz, H.; Munir, D.; Logue, J.; Dooley, H. The immunoglobulins of cartilaginous fishes. Dev. Comp. Immunol. 2021, 115, 103873. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Applications of Nanobodies. Annu. Rev. Anim. Biosci. 2021, 9, 401–421. [Google Scholar] [CrossRef]
- Muyldermans, S. A guide to: Generation and design of nanobodies. FEBS J. 2021, 288, 2084–2102. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef]
- Yamagata, M.; Sanes, J.R. Reporter-nanobody fusions (RANbodies) as versatile, small, sensitive immunohistochemical reagents. Proc. Natl. Acad. Sci. USA 2018, 115, 2126–2131. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Harmand, T.J.; Wijne, C.; Schwartz, T.U.; Ploegh, H.L. Exploring cellular biochemistry with nanobodies. J. Biol. Chem. 2020, 295, 15307–15327. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Promises and Challenges as Lifesaving Treatments. Int. J. Mol. Sci. 2022, 23, 5009. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.; Jost, C.; Ilie, I.M.; Schnabl, J.; Buechi, O.; Eapen, R.S.; Truffer, R.; Caflisch, A.; Forrer, P. Thermostable designed ankyrin repeat proteins (DARPins) as building blocks for innovative drugs. J. Biol. Chem. 2022, 298, 101403. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Veesler, D.; Dreier, B.; Blangy, S.; Lichière, J.; Tremblay, D.; Moineau, S.; Spinelli, S.; Tegoni, M.; Plückthun, A.; Campanacci, V.; et al. Crystal structure and function of a DARPin neutralizing inhibitor of lactococcal phage TP901-1: Comparison of DARP in and camelid VHH binding mode. J. Biol. Chem. 2009, 284, 30718–30726. [Google Scholar] [CrossRef] [PubMed]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef]
- Akkapeddi, P.; Teng, K.W.; Koide, S. Monobodies as tool biologics for accelerating target validation and druggable site discovery. RSC Med. Chem. 2021, 12, 1839–1853. [Google Scholar] [CrossRef]
- Lipovsek, D. Adnectins: Engineered target-binding protein therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef]
- Gross, G.G.; Junge, J.A.; Mora, R.J.; Kwon, H.B.; Olson, C.A.; Takahashi, T.T.; Liman, E.R.; Ellis-Davies, G.C.; McGee, A.W.; Sabatini, B.L.; et al. Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 2013, 78, 971–985. [Google Scholar] [CrossRef]
- Nygren, P.A. Alternative binding proteins: Affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008, 275, 2668–2676. [Google Scholar] [CrossRef]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012, 503, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Rothe, C.; Skerra, A. Anticalin® Proteins as Therapeutic Agents in Human Diseases. BioDrugs 2018, 32, 233–243. [Google Scholar] [CrossRef]
- Griffin, M.E.; Hsieh-Wilson, L.C. Tools for mammalian glycoscience research. Cell 2022, 185, 2657–2677. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, J.; Audfray, A.; Imberty, A. Binding sugars: From natural lectins to synthetic receptors and engineered neolectins. Chem. Soc. Rev. 2013, 42, 4798–4813. [Google Scholar] [CrossRef] [PubMed]
- Tommasone, S.; Allabush, F.; Tagger, Y.K.; Norman, J.; Köpf, M.; Tucker, J.H.R.; Mendes, P.M. The challenges of glycan recognition with natural and artificial receptors. Chem. Soc. Rev. 2019, 48, 5488–5505. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.M.; Kizer, M.E.; Imperiali, B. Strategies and Tactics for the Development of Selective Glycan-Binding Proteins. ACS Chem. Biol. 2021, 16, 1795–1813. [Google Scholar] [CrossRef]
- Cummings, R.D.; Etzler, M.E. Antibodies and Lectins in Glycan Analysis. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 45. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1919/ (accessed on 27 October 2022).
- Tsaneva, M.; Van Damme, E.J.M. 130 years of Plant Lectin Research. Glycoconj. J. 2020, 37, 533–551. [Google Scholar] [CrossRef]
- Lundstrøm, J.; Korhonen, E.; Lisacek, F.; Bojar, D. LectinOracle: A Generalizable Deep Learning Model for Lectin-Glycan Binding Prediction. Adv. Sci. 2022, 9, e2103807. [Google Scholar] [CrossRef]
- Armenta, S.; Moreno-Mendieta, S.; Sánchez-Cuapio, Z.; Sánchez, S.; Rodríguez-Sanoja, R. Advances in molecular engineering of carbohydrate-binding modules. Proteins 2017, 85, 1602–1617. [Google Scholar] [CrossRef]
- Warkentin, R.; Kwan, D.H. Resources and Methods for Engineering “Designer” Glycan-Binding Proteins. Molecules 2021, 26, 380. [Google Scholar] [CrossRef] [PubMed]
- Yabe, R.; Itakura, Y.; Nakamura-Tsuruta, S.; Iwaki, J.; Kuno, A.; Hirabayashi, J. Engineering a versatile tandem repeat-type alpha2-6sialic acid-binding lectin. Biochem. Biophys. Res. Commun. 2009, 384, 204–209. [Google Scholar] [CrossRef]
- Hu, D.; Tateno, H.; Sato, T.; Narimatsu, H.; Hirabayashi, J. Tailoring GalNAcα1-3Galβ-specific lectins from a multi-specific fungal galectin: Dramatic change of carbohydrate specificity by a single amino-acid substitution. Biochem. J. 2013, 453, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Norton, P.; Comunale, M.A.; Herrera, H.; Wang, M.; Houser, J.; Wimmerova, M.; Romano, P.R.; Mehta, A. Development and application of a novel recombinant Aleuria aurantia lectin with enhanced core fucose binding for identification of glycoprotein biomarkers of hepatocellular carcinoma. Proteomics 2016, 16, 3126–3136. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, J.; Arai, R. Lectin engineering: The possible and the actual. Interface Focus 2019, 9, 20180068. [Google Scholar] [CrossRef] [PubMed]
- Notova, S.; Bonnardel, F.; Lisacek, F.; Varrot, A.; Imberty, A. Structure and engineering of tandem repeat lectins. Curr. Opin. Struct. Biol. 2020, 62, 39–47. [Google Scholar] [CrossRef]
- Katoch, R.; Tripathi, A. Research advances and prospects of legume lectins. J. Biosci. 2021, 46, 104. [Google Scholar] [CrossRef]
- Mattox, D.E.; Bailey-Kellogg, C. Comprehensive analysis of lectin-glycan interactions reveals determinants of lectin specificity. PLoS Comput. Biol. 2021, 17, e1009470. [Google Scholar] [CrossRef]
- Montalbán-López, M.; Scott, T.A.; Ramesh, S.; Rahman, I.R.; van Heel, A.J.; Viel, J.H.; Bandarian, V.; Dittmann, E.; Genilloud, O.; Goto, Y.; et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, N.; Cho, S.; Palsson, B.; Cho, B.K. Repurposing Modular Polyketide Synthases and Non-ribosomal Peptide Synthetases for Novel Chemical Biosynthesis. Front. Mol. Biosci. 2020, 7, 87. [Google Scholar] [CrossRef]
- Wu, C.; van der Donk, W.A. Engineering of new-to-nature ribosomally synthesized and post-translationally modified peptide natural products. Curr. Opin. Biotechnol. 2021, 69, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Bayer, T.; Badenhorst, C.P.S.; Wu, S.; Doerr, M.; Höhne, M.; Bornscheuer, U.T. Recent trends in biocatalysis. Chem. Soc. Rev. 2021, 50, 8003–8049. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamagata, M. Programmable Proteins: Target Specificity, Programmability and Future Directions. SynBio 2023, 1, 65-76. https://doi.org/10.3390/synbio1010005
Yamagata M. Programmable Proteins: Target Specificity, Programmability and Future Directions. SynBio. 2023; 1(1):65-76. https://doi.org/10.3390/synbio1010005
Chicago/Turabian StyleYamagata, Masahito. 2023. "Programmable Proteins: Target Specificity, Programmability and Future Directions" SynBio 1, no. 1: 65-76. https://doi.org/10.3390/synbio1010005
APA StyleYamagata, M. (2023). Programmable Proteins: Target Specificity, Programmability and Future Directions. SynBio, 1(1), 65-76. https://doi.org/10.3390/synbio1010005