Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) are a class of drugs commonly used worldwide for their analgesic and antipyretic effects. However, an overdose of NSAIDs can have negative effects on various systems, including the cardiovascular, gastrointestinal, hepatic, renal, and neural systems. The search for new, safer, and more effective anti-inflammatory agents has now become a necessity. The aim of the present study is to identify new natural compounds that act against cyclooxygenase-2 (COX-2), one of the main anti-inflammatory targets, using computational approaches. For this purpose, molecular docking and MM/GBSA binding free energy calculations were utilized to discover new natural inhibitors for COX-2. In addition, several prediction tools, such as SwissADME server, QikProp, and Pro-Tox II, were used in this study to elucidate the pharmacokinetic properties, drug-likeness ability, safety, and the lethal dose (LD50) of the studied compounds. The results of molecular docking have indicated that among all phytochemicals under examination, canniprene, oroxylin A and luteolin show high docking scores and binding affinities toward COX-2 (−10.587, −10.254, and −9.494 Kcal.mol−1, respectively) when compared with the reference inhibitor. Moreover, the top hits demonstrated stability during molecular dynamics simulation and were found to conform to drug-like rules with good bioavailability. Toxicity parameters of the best hits indicate that these compounds could be safe COX-2 inhibitors, but further in vitro and in vivo studies are needed to confirm these findings.
1. Introduction
Cyclooxygenase-2 (COX-2), also known as prostaglandin G/H synthase 2 (PGHS-2), along with cyclooxygenase-1 (COX-1), plays a crucial role in the inflammatory response. These enzymes regulate the synthesis pathway of prostaglandins and thromboxanes, collectively known as prostanoids [1]. Prostanoids are potent lipid mediators generated in response to diverse stimuli. Prostanoids are involved in various physiological processes, including inflammation, fever, and pain [1,2,3], maintain renal function and cardiovascular homeostasis [4]. Beyond their role in inflammation, COX-2 activities have been linked to a range of other pathologies. In fact, numerous studies have demonstrated that enhanced COX-2 expression is associated with various cancers, neurodegenerative disorders, and nephropathies [1,2,5,6].
Due to its pivotal role in both inflammatory and pathological processes, COX-2 has become an interesting target for therapeutic intervention [7]. Existing anti-inflammatory treatments, including non-steroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors, are widely recognized for their effectiveness in attenuating inflammation and are among the most commonly prescribed medications [8]. NSAIDs are frequently utilized for their anti-inflammatory, anti-pyretic, analgesic and anti-rheumatic properties [8]. However, traditional NSAIDs are associated with serious gastrointestinal complications due to their inhibition of COX-1, which led to the development of selective COX-2 inhibitors aimed at reducing these adverse effects [4,8].
Despite their gastrointestinal safety profile compared to non-selective NSAIDs, COX-2 selective inhibitors are associated with an increased risk of myocardial infraction stroke and other cardiovascular complications [4]. These drugs reduce vascular prostacyclin (PGI2) production, which may disrupt its normal homeostasis [9]. This disturbance may lead to an imbalance between COX-2 PGI2, which maintains vasodilation and inhibits platelet aggregation, and thromboxane A2 (TXA2), a COX-1 derived eicosanoid that promotes thrombosis, increasing the risk thrombotic cardiovascular events [9,10]. It has also been suggested that the sulfonamide moiety of coxibs may contribute to the adverse associated with these medications [10].
These side effects highlight the necessity of finding safer alternatives, hence the interest in natural compounds as potential COX-2 inhibitors. Natural compounds, with their diverse chemical structures and generally favorable safety profiles, offer a promising source for the discovery of new COX-2 inhibitors. In this context, the use of computational approaches can significantly accelerate and guide this discovery process.
The present study aimed to identify novel natural inhibitors of COX-2 using advanced computational tools. By performing molecular docking studies, MMGBSA calculations, and molecular dynamics simulation, we provided a comprehensive assessment of the binding affinity and stability of a library of natural compounds toward the active site of COX-2.
2. Materials and Methods
2.1. Library Selection, Pre-Evaluation/Filtration, and Preparation
A library comprising 400 COX inhibitor compounds was selected for structure-based virtual screening against the target protein. These compounds were obtained from the ChemFaces database (https://www.chemfaces.com/, accessed on 10 December 2023), and their 3D structures were then downloaded in SDF file format from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/, accessed on 12 December 2023).
Prior to docking analysis, the library was filtered based on Lipinski’s and Veber rules, with non-conforming compounds automatically excluded. Selected compounds were then prepared using Maestro’s Ligprep module (v11.8), which includes energy minimization of their 3D structures using the OPLS-3e force field, and the generation of tautomeric forms and ionization states at pH 7.0 ± 2.0 using Epik [11].
2.2. Protein Retrieval, Preparation, and Grid Generation
The X-ray crystal structure of cyclooxygenase-2 (COX-2), accessible under the accession code 3LN1, was downloaded as a PDB file from the RCSB Protein Data Bank (https://www.rcsb.org/pdb, accessed on 20 December 2023) [12]. This structure, with a resolution of 2.4 Å, demonstrates the binding interaction between COX-2 and celecoxib at the enzyme’s active site. To ensure a high-quality 3D structure for computational analysis, subsequent protein preparation was conducted using the Protein Preparation Wizard module in Maestro (Schrödinger, LLC, New York, NY, USA, 2018) [11]. This process involved removing unbound water molecules and co-factors, adding hydrogen atoms, completing any missing loops or side chains, and assigning hydrogen bonds. The structure was then optimized using PROPKA at a pH of 7 and subjected to energy minimization with the OPLS-3e force field. The grid box was created around the protein’s active site, defined by the interacting residues and the centroids of the co-crystallized inhibitor within the enzyme’s active site.
2.3. Structure-Based Virtual Screening
The docking simulations were carried out using the Glide program of Schrödinger software (version 2018) with default parameters [12]. Initially, all 400 compounds were docked against COX-2 using the extra precision (XP) algorithm. Following this, compounds with the highest docking scores from the XP docking were considered as the best hits and subjected to further analysis. The binding modes of these compounds were analyzed using Maestro’s pose viewer (v11.8).
2.4. Binding Free Energy Calculation Using the MMGBSA Method
Utilizing the molecular mechanics energies combined with the generalized Born surface area (MMGBSA) approach, we estimated the binding affinity (ΔGbind) of the top compounds and their potential inhibitory effect toward COX-2. This measure reflects the strength of the binding interaction between a molecule and its target and assesses the stability of the resulting ligand–protein complex. The free binding energy (ΔGbind) of receptor-ligand complexes was calculated using the following formula:
ΔG (binding energy) = ΔG (complex) – (ΔG (protein) + ΔG (ligand) [11].
Here, ΔG (complex) is the free energy of the COX-2-ligand complex. ΔG (protein) represents the free energy of COX-2 without ligand. ΔG (ligand) is the free energy of the ligand alone [11].
2.5. In Silico Prediction of Pharmacokinetics, Toxicological, and Drug-like Properties
To elucidate the ADMET profiles of the lead compounds identified in our study, we employed different computational tools. Pharmacokinetic properties, encompassing parameters such as solubility, human oral absorption, and blood brain barrier permeability, were determined using the online server SwissADME and Qikprop module. Toxicological evaluations were conducted utilizing the Protox-II server to assess potential side effects. Additionally, the drug-like properties of the compounds were evaluated using the SwissADME server.
2.6. Molecular Dynamics (MD) Simulation
Molecular dynamics (MD) simulation was performed to comprehensively analyze the dynamic behavior and stability of the protein–ligand complexes. The simulation was carried out using the Desmond package in Schrödinger software for a duration of 50 nanoseconds (ns) and employing the OPLS-3e force field. The docked complexes were solvated with water molecules utilizing the simple point charge (SPC) scheme as a water model [13]. To mimic physiological conditions, sodium and chloride ions were added to the system. Throughout the simulation, temperature and pressure were maintained at constant values [13,14].
3. Results and Discussion
In this study, we conducted a ligand-based virtual screening to evaluate the potential binding affinities and modes of 400 compounds identified as COX inhibitors against COX-2. To focus our search on potential drug candidates, we performed a pre-filtration step using Lipinski’s and Veber’s rules. As a result, we identified 307 compounds that met the drug criteria. The docking procedure was validated by comparing the lowest energy state of the re-docked co-crystalized inhibitor predicted by Glide with the observed binding mode of the native state of the co-crystalized inhibitor, resulting in an RMSD value less than 2 Å, namely, 1.173 Å, underscoring the accuracy of our study [13] (Supplementary Materials).
3.1. Molecular Docking and Binding Free Energy Calculations
From the XP docking results, three compounds, canniprene, oroxylin A, and luteolin, were identified as potential hits. These compounds were compared to two NSAIDs references, rofecoxib and celecoxib, to assess their interactions with the target protein. Celecoxib, in particular, was used to confirm the binding of the studied candidates to the COX-2 active site.
As summarized in Table 1, canniprene exhibited a notable docking score of −10.587 Kcal/mol with an MMGBSA value of −52.36 Kcal/mol. Similarly, oroxylin A demonstrated a strong docking score of −10.254 Kcal/mol and an MMGBSA value of −32.17 Kcal/mol. Luteolin, with a docking score of −9.494 Kcal/mol and an MMGBSA value of −43.41 Kcal/mol, showed moderate binding affinity. Among the reference drugs, rofecoxib showed a lower docking score of −9.357 Kcal/mol, and it MMGBSA value of −64.67 Kcal/mol was relatively more negative, indicating notable stability in the complex with the protein. In contrast, celecoxib, the second reference drug, exhibited the most favorable results. Its remarkable MMGBSA value of −79.21 Kcal/mol confirms its robust and stable interaction with COX-2, which aligns with its high docking score of −12.882 Kcal/mol.

Table 1.
Molecular docking scores and binding free energy values of the hit compounds and reference drugs.
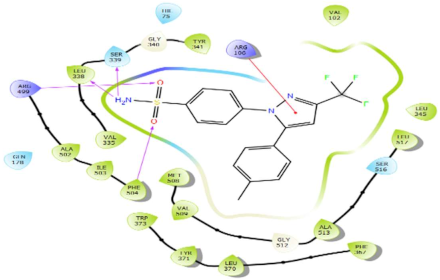
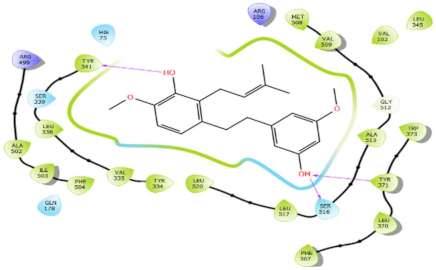
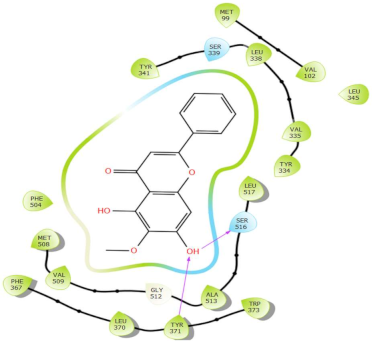
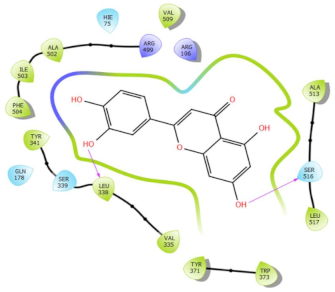
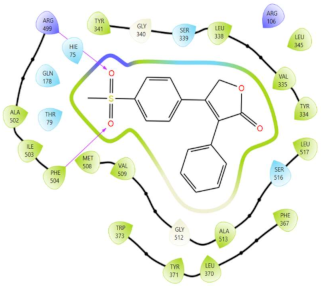
In Table 2, we present the 2D interactions and types of molecular interactions between the investigated compounds and COX-2 protein, as well as those of the reference inhibitors. These results highlight the various mechanisms by which these compounds interact with COX-2, providing valuable insights into their inhibitory potential compared with known reference compounds. All three compounds established notable hydrogen bonding and hydrophobic interactions, suggesting good binding affinity with the enzyme’s active site. In particular, these compounds showed hydrophobic interactions almost similar to those of the reference drugs (Table 2).
Table 2.
Molecular interactions of ligands–COX-2 complexes with the interacting residues.
Structurally, COX-2 shares important similarities with COX-1, but they differ in the specific amino acid compositions of their active sites [15,16]. The COX-2 enzyme has a larger and more flexible drug-binding cavity due to the amino acid residue Val509 instead of Ile523 and Arg499 in place of His513 found in COX-1 [15,17]. This structural variation results in the formation of a distinct side-pocket in COX-2, influencing the enzyme’s selectivity for certain drugs [17,18]. For instance, selective COX-2 NSAIDs, such as celecoxib and rofecoxib, use their sulfonamide group to bind deeply into the polar side of this pocket, which is inaccessible in COX-1 and remains unoccupied in COX-2 complexes with non-selective inhibitors [15,17]. The residues Gln178, Leu338, Ser339, His75, and Arg499 are common amino acids that mediate the interaction of Coxibs with COX-2 [15,17,19]. Our analysis showed that the interaction profiles of both celecoxib and rofecoxib align with these findings, improving our docking results. In addition, the hit compounds were found to occupy the active site of COX-2 and interact with critical residues. As detailed in Table 2, they show potential polar interactions with Arg499, Ser339, Leu338 (only luteolin via hydrogen bond), and Gln178, which are key residues in the polar side pocket of COX-2 active site. Interactions with these small amino acids are essential for the selective inhibition of the enzyme [20,21].
3.2. ADME/T Profiles of the Best Hits
The ADME results analysis revealed that the hit compounds possess promising drug-like properties, conforming to both Lipinski’s and Veber’s rules (Table 3). The three compounds demonstrated acceptable TPSA values, lipophilicity, and good water solubility, with the exceptions of canniprene and oroxylin A, which displayed moderate solubility (Table 3). Notably, all candidates exhibited high gastrointestinal absorption, indicating potential for effective oral bioavailability. In terms of blood–brain barrier (BBB) permeability, only canniprene showed the capacity to cross the barrier, suggesting its suitability for targeting the nervous system (Table 4). Toxicity assessment results are presented in Table 5. In general, all three compounds exhibit no hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, or cytotoxicity, with the exceptions of canniprene which showed immunotoxicity and luteolin that showed carcinogenicity and mutagenicity. The LD50 values of canniprene, oroxylin A, and luteolin were in the order of 1930, 4000, and 3919 mg/kg, respectively.
Table 3.
Physicochemical characteristics and drug-like properties of the best hits.
Table 4.
Predicted pharmacokinetics parameters.
Table 5.
Toxicity profiles and LD50 of canniprene, oroxylin A, and luteolin.
3.3. Molecular Dynamics Simulation Results
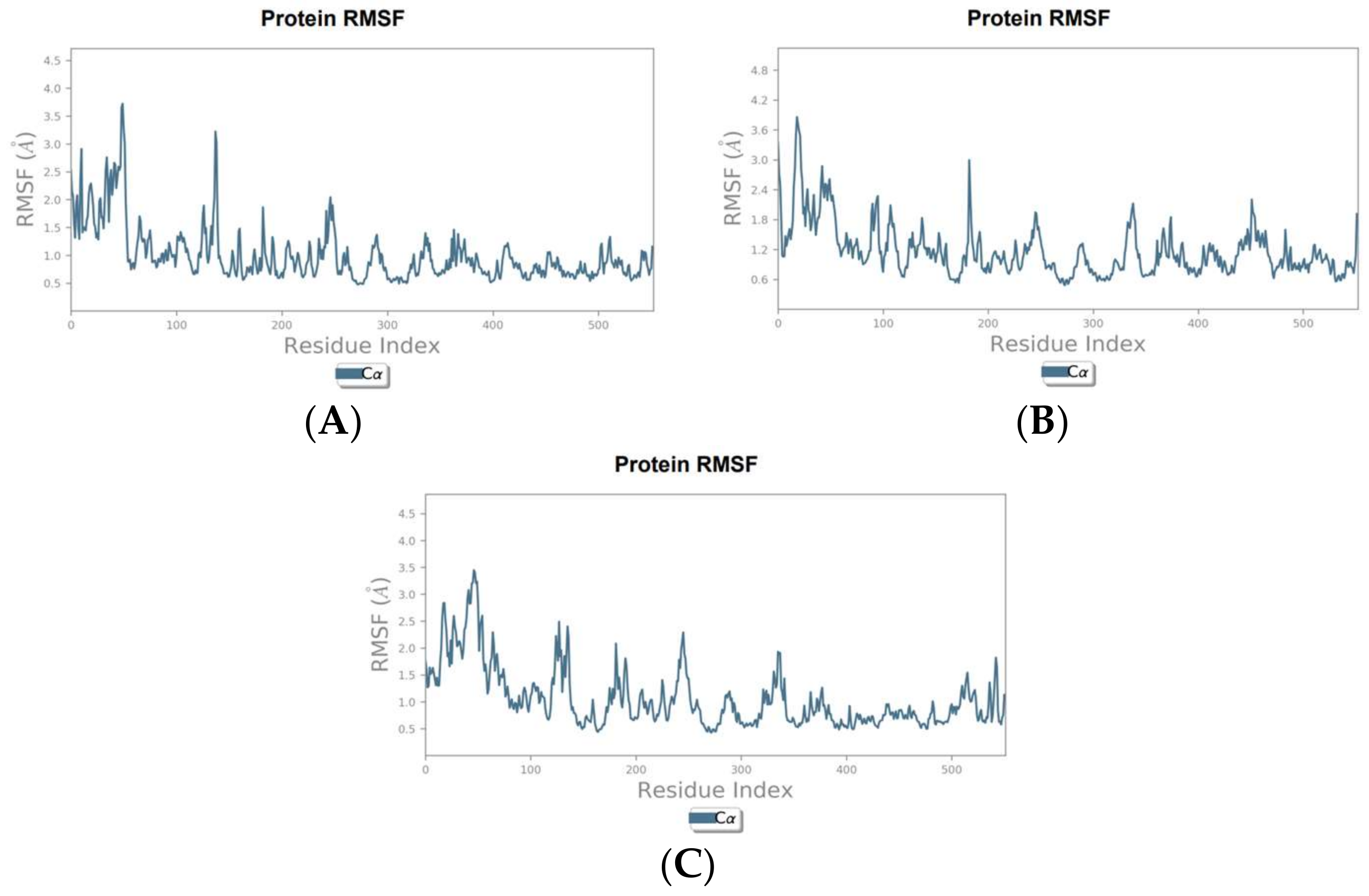
To validate and further investigate the interactions and stability of the identified hit compounds within the COX-2 binding site, molecular dynamics (MD) simulations were performed. These simulations provide a deeper understanding of the compounds’ efficacy as COX-2 inhibitors by analyzing the dynamic behavior and stability of the protein-ligand complexes over time and under more realistic conditions. The root means square deviation (RMSD) plots were generated to assess the stability of the complexes over the simulation period, while the root square fluctuation (RMSF) plots were generated to analyze the flexibility of COX-2 amino acid residues in the presence of the hit compounds.
The RMSD analysis of the canniprene–COX-2 complex revealed that the system remained approximately stable for the first 25 ns of the simulation. During this period, canniprene exhibited minor fluctuations for the first 10 ns, followed by a phase of stabilization. After 25 ns, canniprene displayed a diffusion behavior, with RMSD values ranging from 2 to ~3 Å, indicating mobility from the binding site. The COX-2 backbone RMSD plots showed minimal deviation during the initial 5 ns, corresponding to the equilibration phase. Following this, the protein stabilized for the remainder of the simulation, with RMSD values ranging from 1.5 to 2 Å, reflecting a stable protein structure in presence of this compound with only minor fluctuations (Figure 1A). For the oroxylin A–COX-2 complex, moderate stability was observed during the first 20 ns, with the ligand showing diffusion behavior with a maximum deviation of 2.9 Å. During this period, low RMSD fluctuations were noted for both the protein and the ligand, with RMSD values ranging from 0.8 to 2.9 Å for the ligand and from 1.4 to 3.2 Å for the protein. After 20 ns, both the ligand and the protein showed slight fluctuations before stabilizing for the rest of the simulation. However, oroxylin A displayed slight diffusion behavior again after 40 ns (Figure 1B). The luteolin–COX-2 complex exhibited weak stability throughout the simulation period. Luteolin displayed significant fluctuations during the first 5 ns, followed by stabilization. Similarly, the COX-2 protein reached equilibrium after approximately 11 ns (Figure 1C). The RMSF analysis of the COX-2 protein in complex with canniprene, oroxylin A, and luteolin revealed low fluctuations in the key amino acid residues within the binding site throughout the simulation period. This indicates that the interactions between the ligands and the protein remained relatively stable. The most significant fluctuations were observed in the N-terminal region of the protein, which is typically more flexible than the binding site (Figure 2).
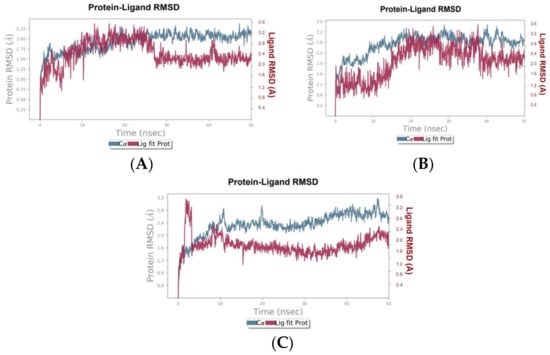
Figure 1.
RMSD plots of the hit compounds in complexes with COX-2 during 50 ns. (A) Canniprene–COX-2 complex, (B) oroxylin A–COX-2 complex, (C) luteolin–COX-2 complex.
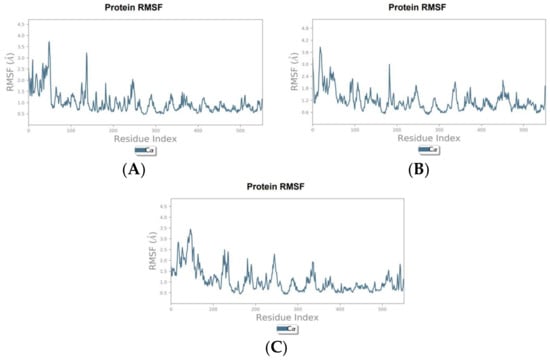
Figure 2.
RMSF graphs of the hit compounds during MD simulation. (A) Canniprene–COX-2 complex, (B) oroxylin A–COX-2 complex, (C) luteolin–COX-2 complex.
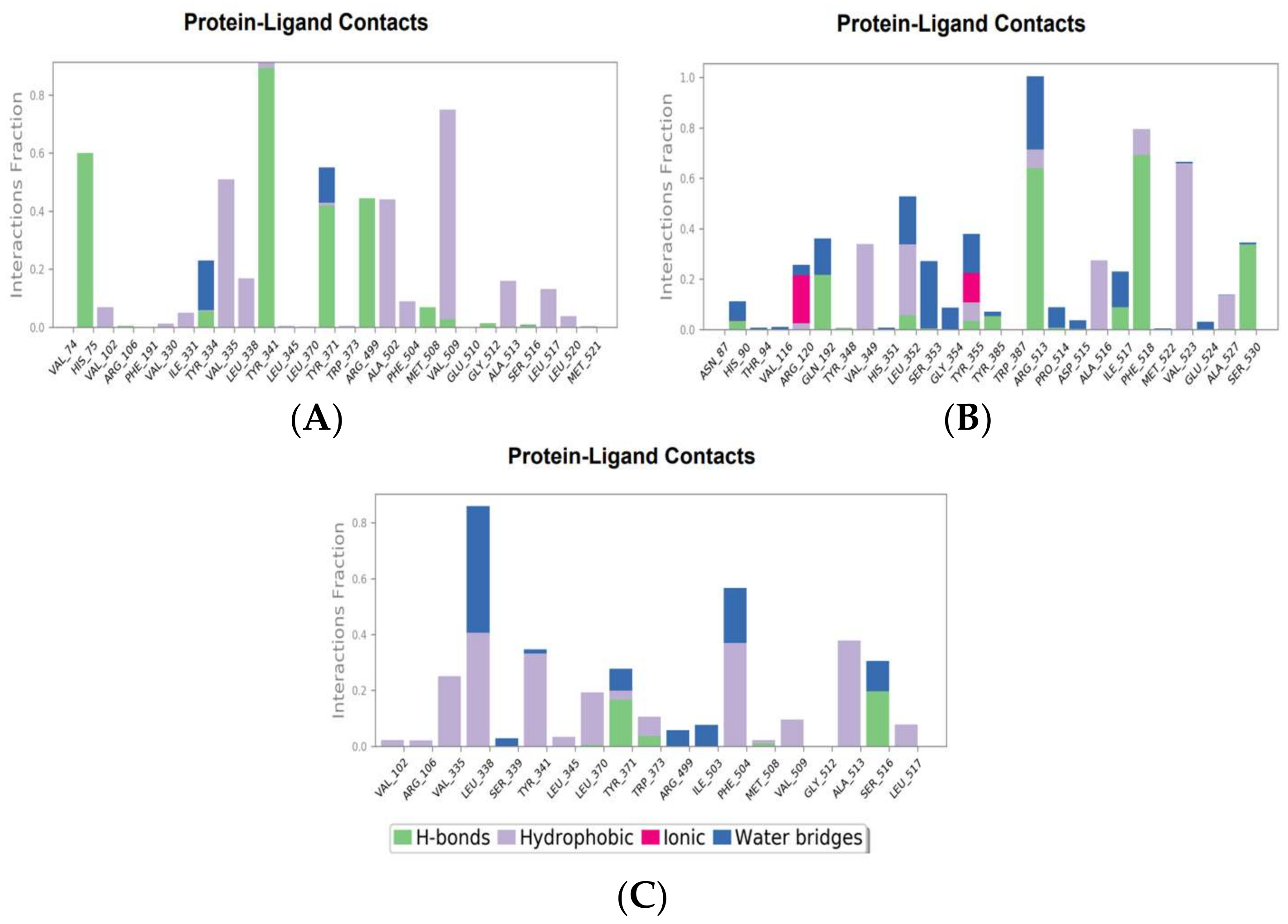
The molecular interactions between COX-2 and the hit compounds, canniprene, oroxylin A, and luteolin, were analyzed using protein–ligand contact histograms over the simulation period. Canniprene demonstrated hydrogen bonds with the residues Tyr341, Tyr371, and Ser516, consistent with its interaction profile from docking results. Additionally, other hydrogen bonds were observed, including mainly interaction with the amino acid Arg499, resembling the binding of celecoxib (Figure 3A). Oroxylin A exhibited hydrogen contacts with Tyr371 and Ser516, along with nearly conserved hydrophobic interactions, as demonstrated by docking analysis.
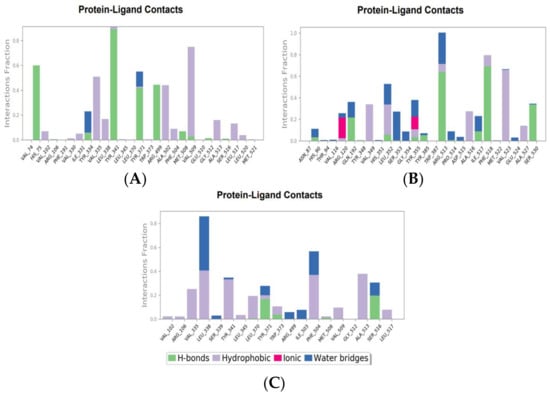
Figure 3.
Molecular interaction of canniprene (A), oroxylin A (B), and luteolin (C) with COX-2 during MD simulation.
These compounds have demonstrated promising interactions with COX-2, suggesting that they may possess therapeutic efficacy in attenuating inflammation. For instance, canniprene, an iso-prenylated bibenzyl found in Cannabis sativa, has been highlighted by [22] for its anti-inflammatory properties through effective inhibition of the activities of 5-lipoxygenase (5-LO) and the microsomal prostaglandin E2 synthase (mPGES). Although other molecular mechanisms underlying canniprene’s anti-inflammatory effects are not extensively documented in the literature. Extracts from Cannabis sativa have shown anti-inflammatory potential by reducing transcriptional levels of pro-inflammatory cytokines, such as IL-1β, IL-6, TNF-α [23], and IL-8 [24]; suppressing inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) expression [23,25]; and modulating NF-κB expression [25]. On the other hand, oroxylin A exhibits well-documented anti-inflammatory effects. Studies have demonstrated that oroxylin A can modulate key signaling pathways involved in regulating inflammatory responses, such as NF-κB [26,27,28] and MAPK pathways [26,27]. Additionally, oroxylin A has shown the ability to suppress the production of cytokines, including IL-1β, IL-6, and TNF-α [26,27]. Furthermore, it effectively inhibits the expression of iNOS and COX-2 [26,27]. Similarly, luteolin has been reported to possess promising anti-inflammatory properties. For example, it suppresses the production of various pro-inflammatory mediators such as IL1β, IL-2, IL-6, IL-8, IL-12, IL-17, TNF-α, COX-2, and iNOS [29,30]. Moreover, luteolin can modulate several signaling pathways, including the NF-κB, MAPK, and JAK-STAT pathways [29,30]. In a study by [31], luteolin exhibited notable inhibition of COX-2 in vitro with an IC50 of 36.6 µmol/l. Our findings are consistent with these results, as other computational studies have similarly indicated luteolin’s good binding affinity for COX-2 and its interaction with crucial residues [32,33].
While our study provides valuable insights into the anti-inflammatory effects of canniprene, oroxylin A, and luteolin via computational approaches targeting the COX-2 enzyme, several limitations need to be considered. First, the computational models employed may oversimplify the biological process, as real cellular responses are more complex and involve additional factors. Moreover, the results of in silico studies require further validation using in vitro and in vivo experiments to confirm their biological potential, safety profile, and selectivity. Expanding the findings and incorporating supplementary validation would provide a more comprehensive understanding and improve the robustness of our conclusions.
4. Conclusions
In this study, a comprehensive analysis of the molecular interactions, stability, and dynamic behavior of the hit compounds canniprene, oroxylin A, and luteolin in complex with COX-2 has provided valuable insights into their potential as COX-2 inhibitors. Docking results identified celecoxib as the most effective reference drug in terms of both binding affinity and stability. However, canniprene and oroxylin A have emerged as promising natural alternatives with significant binding potential. Both canniprene and oroxylin A exhibited good binding affinity and stable interactions with COX-2 during MD simulation, showing molecular interactions consistent with known drugs and maintaining relatively stable RMSD profiles. These findings suggest that canniprene and oroxylin A hold promise as potential COX-2 inhibitors. Nevertheless, further investigations using in vitro and in vivo studies are required to validate their efficacy and safety profiles.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/blsf2024035006/s1, Figure S1: Superimposition of the docked co-crystal ligand and the co-crystal ligand (celecoxib) with an RMSD of 1.173 Å. The co-crystalized inhibitor appears in green, and the re-docked co-crystalized inhibitor appears in red.
Author Contributions
I.D.: Writing—Original Draft, Software, Formal Analysis, Methodology, Investigation, Visualization. R.R.: Writing—Original Draft, Writing—Review & Editing, Software, Formal Analysis, Supervision, Validation, Visualization. M.E.T.: Writing—Review & Editing, Supervision, Methodology, Validation. F.F.K.: Writing—Review & Editing, Supervision, Methodology, Validation. A.B.: Conceptualization, Writing—Review & Editing, Supervision, Validation. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Acknowledgments
The software and computational resources used in this study were provided by Redouane Rebai.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Martín-Sanz, P.; Hortelano, S.; Bosca, L.; Casado, M. Cyclooxygenase 2: Understanding the pathophysiological role through genetically altered mouse models. Front. Biosci. 2006, 11, 2876–2888. [Google Scholar] [CrossRef]
- Kharwar, A.; Mishra, A.; Singh, V.K.; Tiwari, A.K. In silico approach to design new cyclooxygenase-2 (COX-2) inhibitors based on MM/QM and ADMET analysis. Chem. Phys. Impact 2024, 8, 100509. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. 2011, 10, 655. [Google Scholar] [PubMed]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghulikah, H.A.; El-Sebaey, S.A.; Bass, A.K.; El-Zoghbi, M.S. New pyrimidine-5-carbonitriles as COX-2 inhibitors: Design, synthesis, anticancer screening, molecular docking, and in silico ADME profile studies. Molecules 2022, 21, 7485. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.Y.; Abou-Amra, E.S.; El-Sebaey, S.A. Design and synthesis of new series of chiral pyrimidine and purine analogs as COX-2 inhibitors: Anticancer screening, molecular modeling, and in silico studies. J. Mol. Struct. 2023, 1278, 134930. [Google Scholar] [CrossRef]
- Ju, Z.; Li, M.; Xu, J.; Howell, D.C.; Li, Z.; Chen, F.E. Recent development on COX-2 inhibitors as promising anti-inflammatory agents: The past 10 years. Acta Pharm. Sin. B 2022, 6, 2790–2807. [Google Scholar] [CrossRef]
- Al-Turki, D.A.; Al-Omar, M.A.; Abou-Zeid, L.A.; Shehata, I.A.; Al-Awady, M.S. Design, synthesis, molecular modeling and biological evaluation of novel diaryl heterocyclic analogs as potential selective cyclooxygenase-2 (COX-2) inhibitors. Saudi Pharm. J. 2017, 25, 59–69. [Google Scholar] [CrossRef]
- Sharma, J.N.; Jawad, N.M. Adverse effects of COX-2 inhibitors. Sci. World J. 2005, 5, 629–645. [Google Scholar] [CrossRef]
- Rayar, A.M.; Lagarde, N.; Ferroud, C.; Zagury, J.F.; Montes, M.; Veitia, M.S.I. Update on COX-2 Selective Inhibitors: Chemical Classification, Side Effects and their Use in Cancers and Neuronal Diseases. Curr. Top. Med. Chem. 2017, 17, 2935–2956. [Google Scholar] [CrossRef]
- Murugesan, D.K.; Rajagopal, K.; Vijayakumar, A.R.; Sundararajan, G.; Raman, K.; Byran, G.; Emran, T.B. Design and Synthesis of Pyrazole-Substituted 9-Anilinoacridine Derivatives and Evaluation against Breast Cancer. J. Biol. Regul. Homeost. Agents 2024, 4, 2845–2859. [Google Scholar]
- Pan, T.; He, M.; Deng, L.; Li, J.; Fan, Y.; Hao, X.; Mu, S. Design, synthesis, and evaluation of the COX-2 inhibitory activities of new 1, 3-dihydro-2H-indolin-2-one derivatives. Molecules 2023, 12, 4668. [Google Scholar] [CrossRef] [PubMed]
- Rebai, R.; Carmena-Bargueño, M.; Toumi, M.E.; Derardja, I.; Jasmin, L.; Pérez-Sánchez, H.; Boudah, A. Identification of potent inhibitors of kynurenine-3-monooxygenase from natural products: In silico and in vitro approaches. Heliyon 2024, 10, e30287. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Kalirajan, R.; Islam, F.; Zehravi, M.; Pratap Singh, L.; Rana, R.; Barua, R. Potential Inhibitors from Natural Compounds against SARS-CoV-2 Main Protease: A Systematic Molecular Modelling Approach. ChemistrySelect 2024, 7, e202303729. [Google Scholar] [CrossRef]
- Kassab, S.E.; Khedr M., A.; Ali H., I.; Abdalla M., M. Discovery of new indomethacin-based analogs with potentially selective cyclooxygenase-2 inhibition and observed diminishing to PGE2 activities. Eur. J. Med. Chem. 2017, 141, 306–321. [Google Scholar] [CrossRef]
- Faki, Y.; Er, A. Different chemical structures and physiological/pathological roles of cyclooxygenases. Rambam Maimonides Med. J. 2021, 12, e0003. [Google Scholar] [CrossRef]
- Dwivedi, A.K.; Gurjar, V.; Kumar, S.; Singh, N. Molecular basis for nonspecificity of nonsteroidal anti-inflammatory drugs (NSAIDs). Drug Discov. Today 2015, 20, 863–873. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef]
- Dershaby, N.H.; El-Hawash, S.A.; Kassab, S.E.; Daabees, H.G.; Abdel Moneim, A.E.; El-Miligy, M.M. Rational design and synthesis of new selective COX-2 inhibitors with In Vivo PGE2-lowering activity by tethering benzenesulfonamide and 1, 2, 3-triazole pharmacophores to some NSAIDs. Pharmaceuticals 2022, 15, 1165. [Google Scholar] [CrossRef]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Stallings, W.C. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef]
- El-Miligy, M.M.; Al-Kubeisi, A.K.; Bekhit, M.G.; El-Zemity, S.R.; Nassra, R.A.; Hazzaa, A.A. Towards safer anti-inflammatory therapy: Synthesis of new thymol–pyrazole hybrids as dual COX-2/5-LOX inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Allegrone, G.; Pollastro, F.; Magagnini, G.; Taglialatela-Scafati, O.; Seegers, J.; Koeberle, A.; Appendino, G. The bibenzyl canniprene inhibits the production of pro-inflammatory eicosanoids and selectively accumulates in some Cannabis sativa strains. J. Nat. Prod. 2017, 80, 731–734. [Google Scholar] [CrossRef]
- Shin, J.; Choi, S.; Park, A.Y.; Ju, S.; Kweon, B.; Kim, D.U.; Kim, S. In Vitro and In Vivo Anti-Inflammatory and Antidepressant-like Effects of Cannabis sativa L. Extracts. Plants 2024, 13, 1619. [Google Scholar] [CrossRef]
- Zaiachuk, M.; Suryavanshi, S.V.; Pryimak, N.; Kovalchuk, I.; Kovalchuk, O. The anti-inflammatory effects of Cannabis sativa extracts on LPS-induced cytokines release in human macrophages. Molecules 2023, 28, 4991. [Google Scholar] [CrossRef] [PubMed]
- Frusciante, L.; Geminiani, M.; Olmastroni, T.; Mastroeni, P.; Trezza, A.; Salvini, L.; Santucci, A. Repurposing Castanea sativa Spiny Burr By-Products Extract as a Potentially Effective Anti-Inflammatory Agent for Novel Future Biotechnological Applications. Life 2024, 14, 763. [Google Scholar] [CrossRef] [PubMed]
- Gera, A.; Yadav, L.; Patil, C.R.; Posa, M.K.; Chandrakanth, B.; Kumar, S. Oroxylin A: Nature’s Arsenal Against Liver Fibrosis, Cancer, and Inflammatory Diseases. Health Sci. Rev. 2023, 10, 100143. [Google Scholar] [CrossRef]
- Ji, Y.; Han, J.; Lee, N.; Yoon, J.H.; Youn, K.; Ha, H.J.; Jun, M. Neuroprotective effects of baicalein, wogonin, and oroxylin A on amyloid beta-induced toxicity via NF-κB/MAPK pathway modulation. Molecules 2020, 25, 5087. [Google Scholar] [CrossRef]
- Yao, J.; Hu, R.; Sun, J.; Lin, B.; Zhao, L.; Sha, Y.; Guo, Q.L. Oroxylin a prevents inflammation-related tumor through down-regulation of inflammatory gene expression by inhibiting NF-κB signaling. Mol. Carcinog. 2014, 53, 145–158. [Google Scholar] [CrossRef]
- Aziz, N.; Kim, M.Y.; Cho, J.Y. Anti-inflammatory effects of luteolin: A review of in vitro, in vivo, and in silico studies. J. Ethnopharmacol. 2018, 225, 342–358. [Google Scholar] [CrossRef]
- Gendrisch, F.; Esser, P.R.; Schempp, C.M.; Wölfle, U. Luteolin as a modulator of skin aging and inflammation. Biofactors 2021, 47, 170–180. [Google Scholar] [CrossRef]
- Li, Y.; Frenz, C.M.; Chen, M.; Wang, Y.; Li, F.; Luo, C.; Liang, N.; Yang, H.; Bohlin, L.; Wang, C. Primary virtual and in vitro bioassay screening of natural inhibitors from flavonoids against COX-2. Chin. J. Nat. Med. 2011, 9, 156–160. [Google Scholar]
- Dong, R.; Huang, R.; Shi, X.; Xu, Z.; Mang, J. Exploration of the mechanism of luteolin against ischemic stroke based on network pharmacology, molecular docking and experimental verification. Bioengineered 2021, 12, 12274–12293. [Google Scholar] [CrossRef]
- Shahzadi, A.; Tariq, N.; Sonmez, H.; Waquar, S.; Zahid, A.; Javed, M.A.; Ozturk, M. Potential effect of luteolin, epiafzelechin, and albigenin on rats under cadmium-induced inflammatory insult: In silico and in vivo approach. Front. Chem. 2023, 11, 1036478. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).