
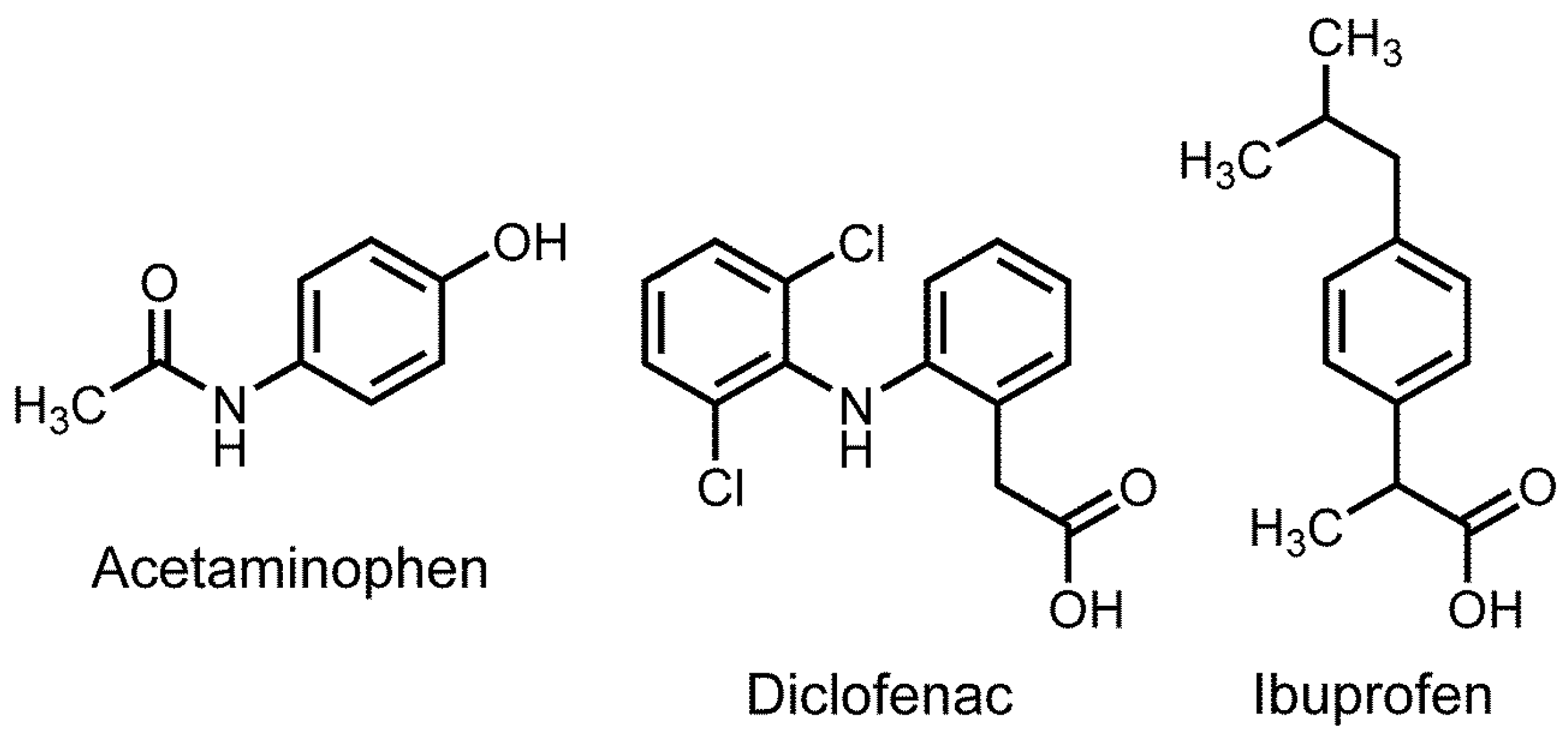
Old and New Analgesic Acetaminophen: Pharmacological Mechanisms Compared with Non-Steroidal Anti-Inflammatory Drugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
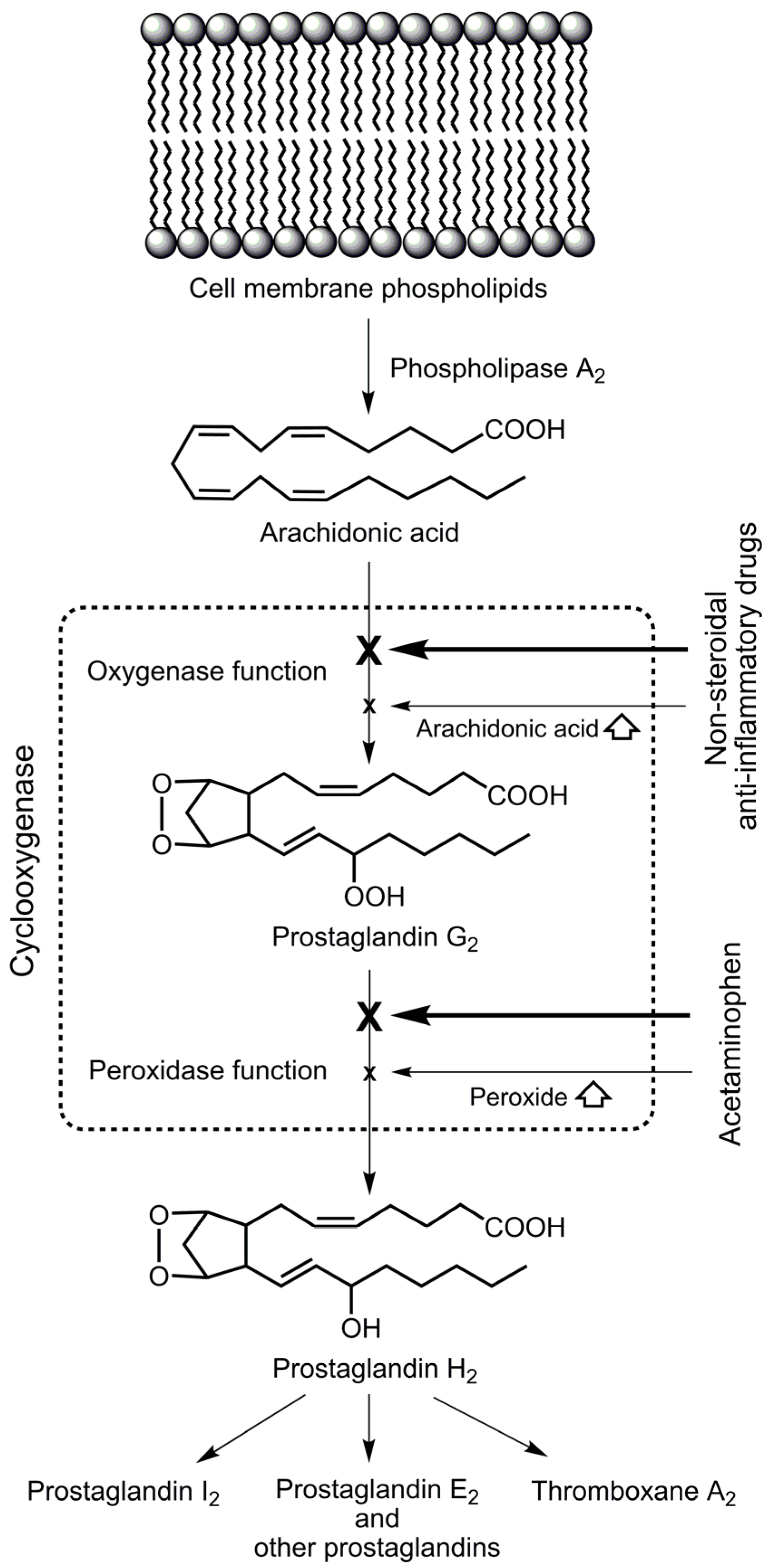
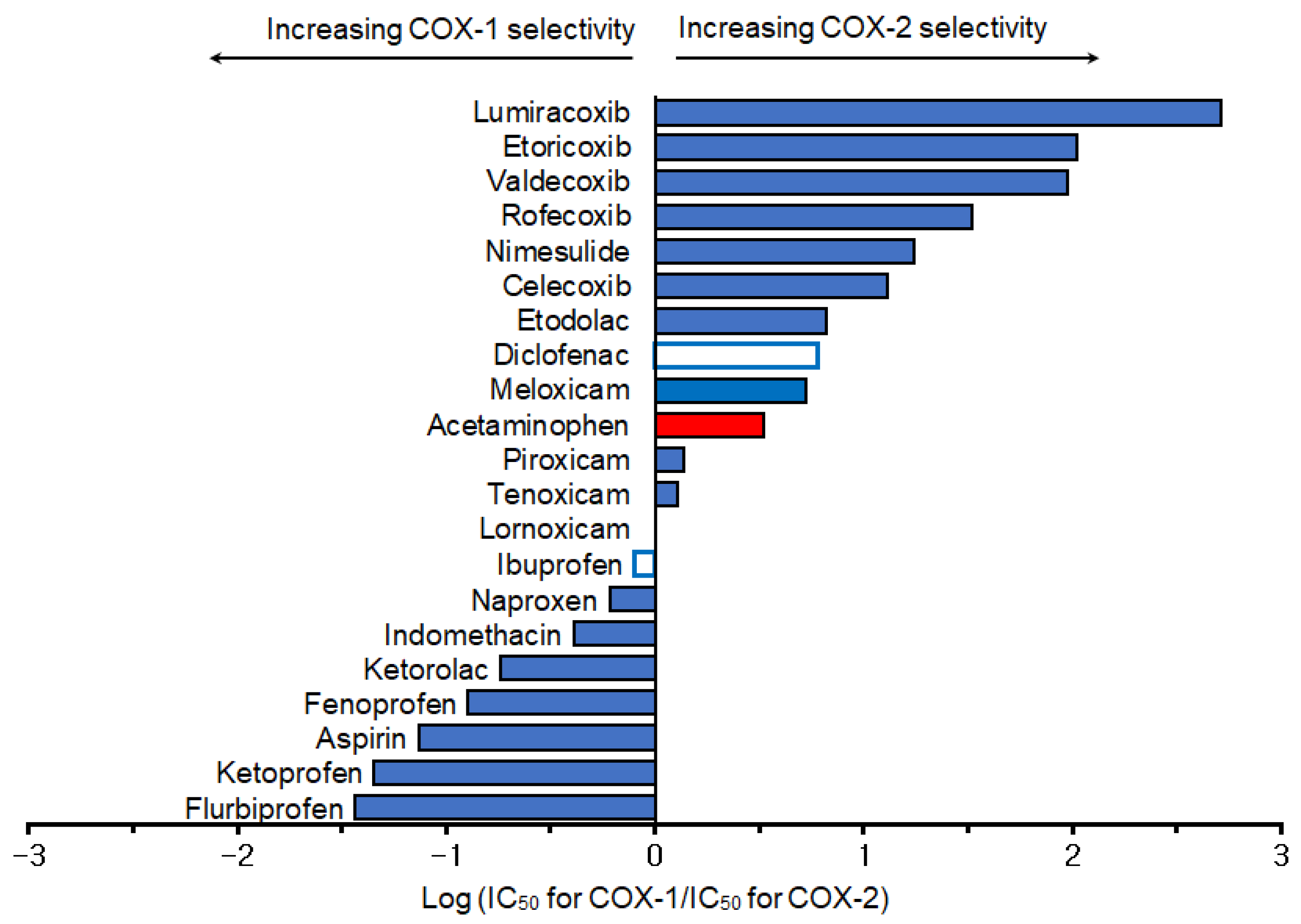
2. Selective Cyclooxygenase Inhibition
3. Anti-Inflammatory Effect
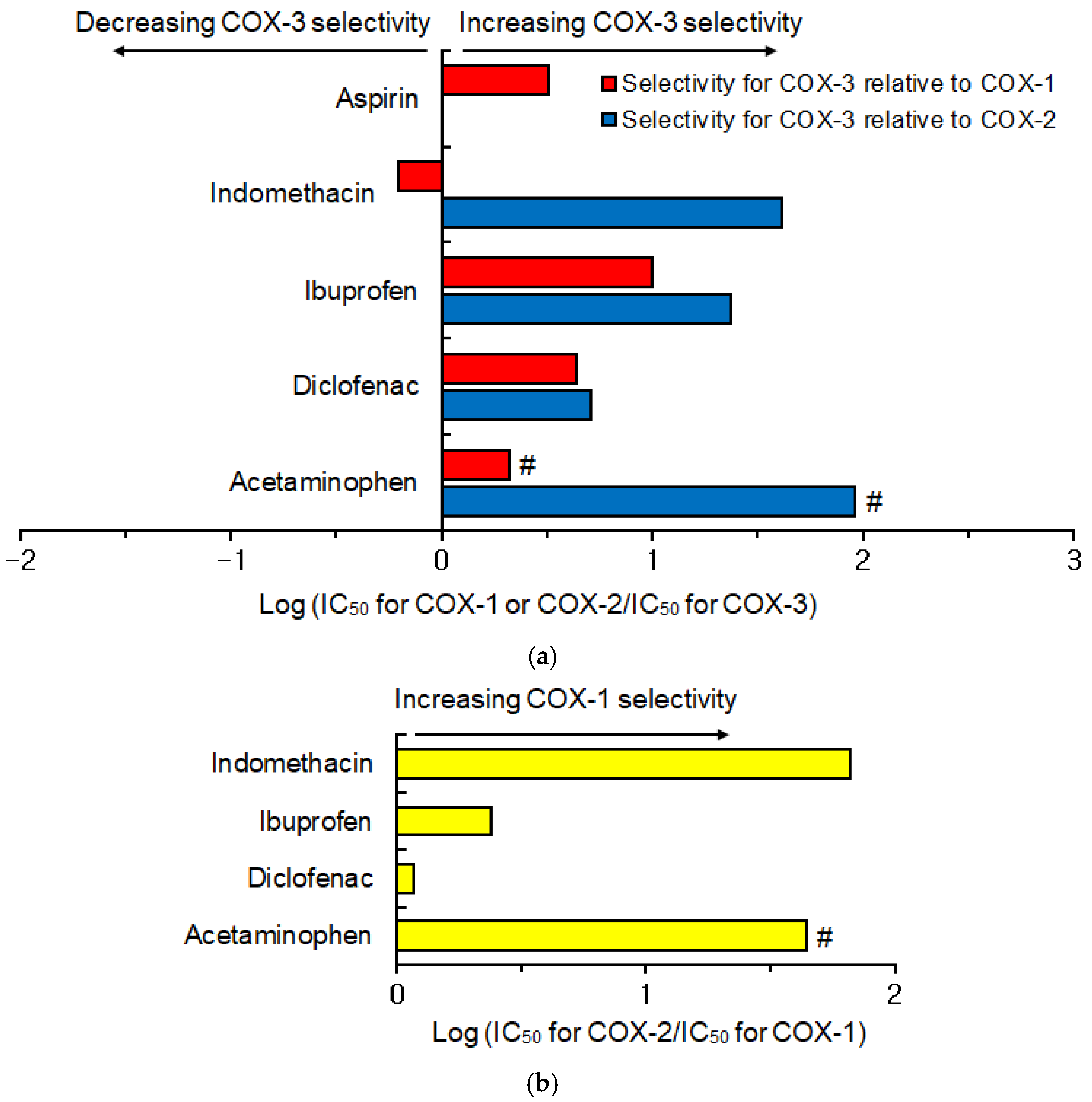
4. Hypothetical Inhibition of Cyclooxygenase-3
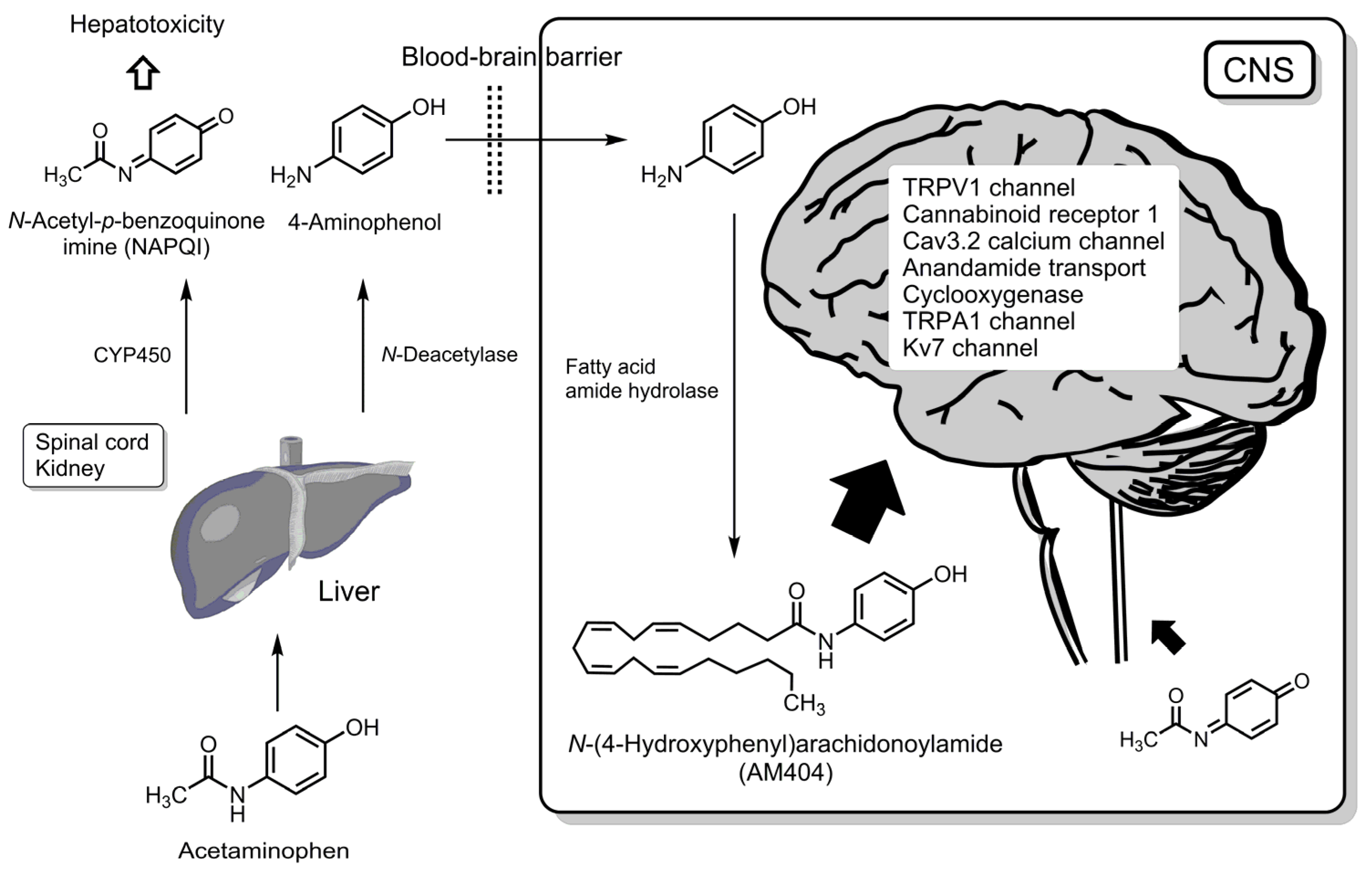
5. Conversion of Acetaminophen into Bioactive Compounds
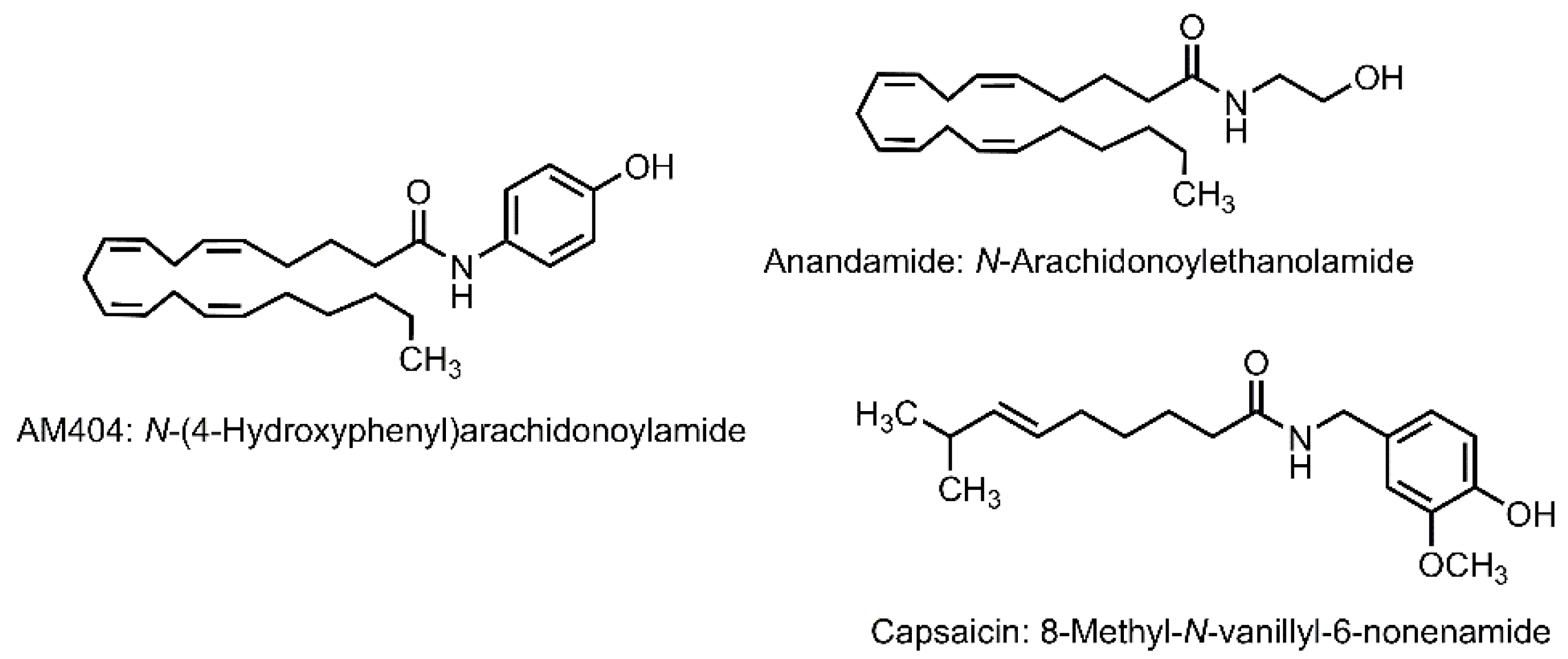
5.1. N-(4-Hydroxyphenyl)arachidonoylamide
5.1.1. Transient Receptor Potential Vanilloid 1 Channel
5.1.2. Cannabinoid Receptor
5.1.3. Calcium Channel
5.1.4. Anandamide Transport and Hydrolysis
5.1.5. Cyclooxygenase
5.1.6. Other Bioactivities of AM404
- Antibacterial Activity
- Antiviral Activity
- Anticancer Activity
5.2. N-Acetyl-p-benzoquinone Imine
5.2.1. Transient Receptor Potential Vanilloid 1 Channel
5.2.2. Transient Receptor Potential Ankyrin 1 Channel
5.2.3. Potassium Channel
6. Analgesic Strategy from the Perspective of Acetaminophen
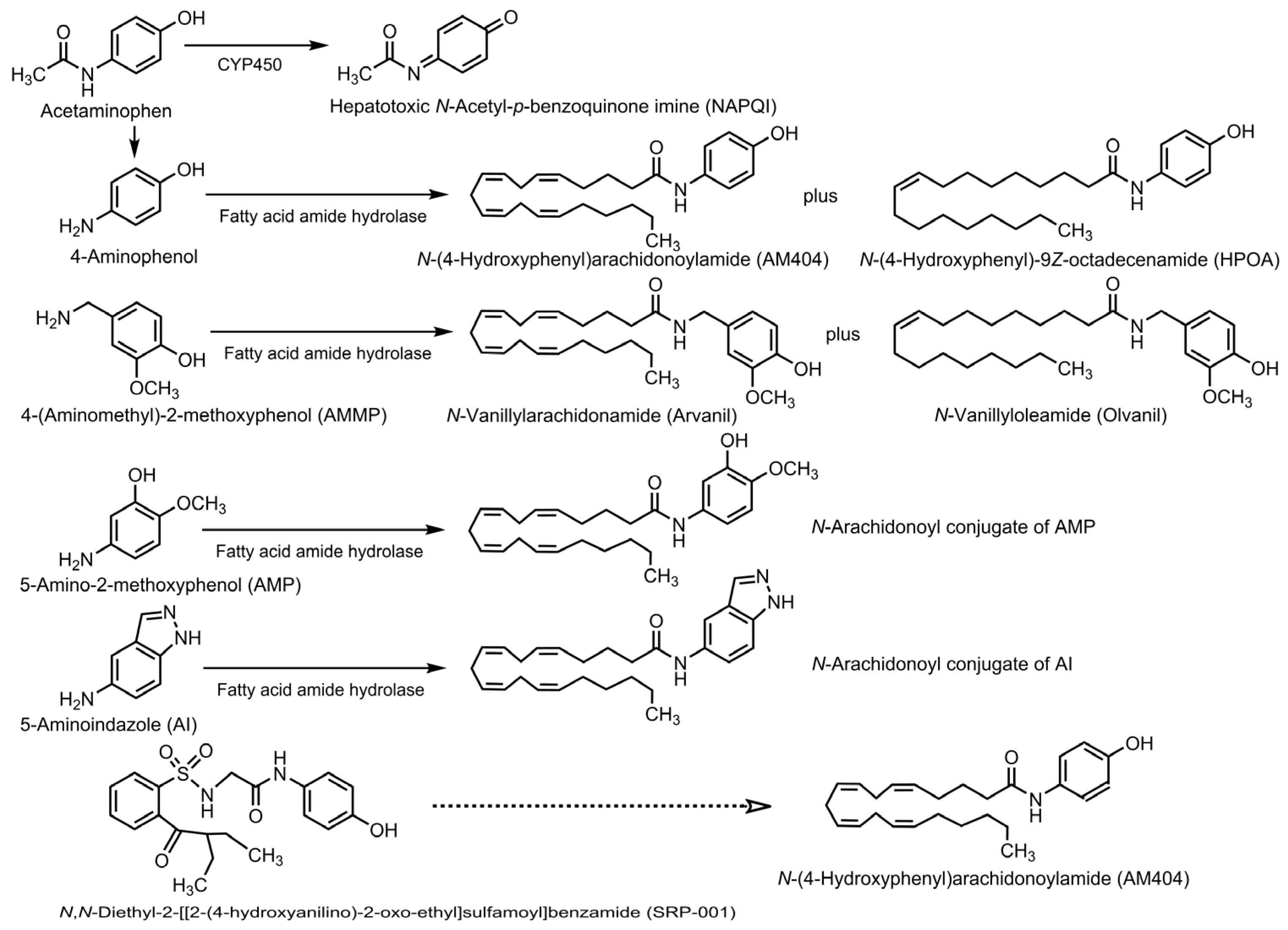
6.1. Biotransformation into AM404 or Its Analogs
6.2. Structural Modification of Acetaminophen
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AHA | 1-Acetylamino-3-hydroxyadamantane |
| AI | 5-Aminoindazole |
| AM404 | N-(4-Hydroxyphenyl)arachidonoylamide |
| AMMP | 4-(Aminomethyl)-2-methoxyphenol |
| AMP | 5-Amino-2-methoxyphenol |
| Arvanil | N-Vanillylarachidonamide |
| CB1 | Cannabinoid receptor 1 |
| CB2 | Cannabinoid receptor 2 |
| CNS | Central nervous system |
| COVID-19 | Coronavirus disease 2019 |
| COX | Cyclooxygenase |
| CYP450 | Cytochrome P450 |
| DENV | Dengue virus |
| FAAH | Fatty acid amide hydrolase |
| HAA | 1-Hydroxy-4-acetylaminoadamantane |
| HBA | N-(4-Hydroxybenzyl)acetamide |
| HMPA | N-(4-Hydroxy-3-methoxyphenyl)acetamide |
| HPOA | N-(4-Hydroxyphenyl)-9Z-octadecenamide |
| IA | N-(1H-Indazol-5-yl)acetamide |
| IC50 | 50% Inhibitory concentration |
| JNJ-10450232/NTM-006 | N-(4-Hydroxyphenyl)-5-methyl-1H-pyrazole-3-carboxamide |
| LPS | lipopolysaccharide |
| NAPQI | N-Acetyl-p-benzoquinone imine |
| NSAID | Non-steroidal anti-inflammatory drug |
| Olvanil | N-Vanillyloleamide |
| PG | Prostaglandin |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SRP-001 | N,N-Diethyl-2-[[2-(4-hydroxyanilino)-2-oxo-ethyl]sulfamoyl]benzamide |
| TRP | Transient receptor potential |
| TRPA1 | Transient receptor potential ankyrin 1 |
| TRPV1 | Transient receptor potential vanilloid 1 |
References
- Kamal, M.; Negm, W.A.; Abdelkader, A.M.; Alshehri, A.A.; El-Saber Batiha, G.; Osama, H. Most common over-the-counter medications and effects on patients. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Hider-Mlynarz, K.; Cavalié, P.; Maison, P. Trends in analgesic consumption in France over the last 10 years and comparison of patterns across Europe. Br. J. Clin. Pharmacol. 2018, 84, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Aminoshariae, A.; Khan, A. Acetaminophen: Old drug, new issues. J. Endod. 2015, 41, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Helander, E.M.; Menard, B.L.; Harmon, C.M.; Homra, B.K.; Allain, A.V.; Bordelon, G.J.; Wyche, M.Q.; Padnos, I.W.; Lavrova, A.; Kaye, A.D. Multimodal analgesia, current concepts, and acute pain considerations. Curr. Pain Headache Rep. 2017, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.; Costa, G.; Veiga, F.; Cardoso, C.; Paiva-Santos, A.C. Parenteral ready-to-use fixed-dose combinations including NSAIDs with paracetamol or metamizole for multimodal analgesia-Approved products and challenges. Pharmaceuticals 2023, 16, 1084. [Google Scholar] [CrossRef] [PubMed]
- Freo, U. Paracetamol for multimodal analgesia. Pain Manag. 2022, 12, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.P. Rational multimodal analgesia for perioperative pain management. Curr. Pain Headache Rep. 2023, 27, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Valdenassi, L.; Bjørklund, G.; Chirumbolo, S.; Lysiuk, R.; Lenchyk, L.; Doşa, M.D.; Fazio, S. COVID-19 medical and pharmacological management in the European countries compared to Italy: An overview. Int. J. Environ. Res. Public Health 2022, 19, 4262. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.; McCarberg, B.H.; Grange, L.; Kolosov, A.; Haveric, A.L.; Zucal, V.; Petruschke, R.; Bissonnette, S. The use of non-steroidal anti-inflammatory drugs (NSAIDs) in COVID-19. NPJ Prim. Care Respir. Med. 2022, 32, 35. [Google Scholar] [CrossRef] [PubMed]
- Lapi, F.; Marconi, E.; Grattagliano, I.; Rossi, A.; Fornasari, D.; Magni, A.; Lora Aprile, P.; Cricelli, C. To clarify the safety profile of paracetamol for home-care patients with COVID-19: A real-world cohort study, with nested case-control analysis, in primary care. Intern. Emerg. Med. 2022, 17, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, B.L.; Ferreira, D.P.; Borges, S.F.; Ferreira, A.M.; Holanda, F.H.; Ucella-Filho, J.G.M.; Cruz, R.A.S.; Birolli, W.G.; Luque, R.; Ferreira, I.M. Diclofenac, ibuprofen, and paracetamol biodegradation: Overconsumed non-steroidal anti-inflammatories drugs at COVID-19 pandemic. Front. Microbiol. 2023, 14, 1207664. [Google Scholar] [CrossRef] [PubMed]
- García-Azorín, D.; García-Ruiz, C.; Sierra-Mencía, Á.; González-Osorio, Y.; Recio-García, A.; González-Celestino, A.; García-Iglesias, C.; Planchuelo-Gómez, Á.; Íñiguez, A.E.; Guerrero-Peral, Á.L. Acute and preventive treatment of COVID-19-related headache: A series of 100 patients. Life 2024, 14, 910. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.S.; Yu, Y.; Chen, Y.; Fedele, G.; Martins, L.M. Paracetamol is associated with a lower risk of COVID-19 infection and decreased ACE2 protein expression: A retrospective analysis. COVID 2021, 1, 218–229. [Google Scholar] [CrossRef]
- Barrière, D.A.; Boumezbeur, F.; Dalmann, R.; Cadeddu, R.; Richard, D.; Pinguet, J.; Daulhac, L.; Sarret, P.; Whittingstall, K.; Keller, M.; et al. Paracetamol is a centrally acting analgesic using mechanisms located in the periaqueductal grey. Br. J. Pharmacol. 2020, 177, 1773–1792. [Google Scholar] [CrossRef] [PubMed]
- Mizogami, M.; Tsuchiya, H. Acetaminophen has lipid composition-dependent membrane interactivity that could be related to nephrotoxicity but not to analgesic activity and hepatotoxicity. Med. Princ. Pract. 2022, 31, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Albuhadily, A.K.; Abd El-Maksoud, E.M.; Shokr, M.M.; Alexiou, A.; Papadakis, M.; Batiha, G.E. Paracetamol: The potential therapeutic pathways defining its clinical use. Inflammopharmacology 2025, 33, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Eschalier, A.; Daulhac, L. Chapter 10, Paracetamol: Update on its Analgesic Mechanism of Action. In Pain Relief—From Analgesics to Alternative Therapies; Maldonado, C., Ed.; IntechOpen: London, UK, 2017; Available online: http://dx.doi.org/10.5772/63264 (accessed on 28 January 2025).
- Ohashi, N.; Kohno, T. Analgesic effect of acetaminophen: A review of known and novel mechanisms of action. Front. Pharmacol. 2020, 11, 580289. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, S.S. Paracetamol (acetaminophen): A familiar drug with an unexplained mechanism of action. Temperature 2021, 8, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Högestätt, E.D.; Jönsson, B.A.; Ermund, A.; Andersson, D.A.; Björk, H.; Alexander, J.P.; Cravatt, B.F.; Basbaum, A.I.; Zygmunt, P.M. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J. Biol. Chem. 2005, 280, 31405–31412. [Google Scholar] [CrossRef] [PubMed]
- Derry, C.J.; Derry, S.; Moore, R.A. Single dose oral ibuprofen plus paracetamol (acetaminophen) for acute postoperative pain. Cochrane Database Syst. Rev. 2013, 2013, CD010210. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Tate, S.; Woolf, C.J. Transient receptor potential channels: Targeting pain at the source. Nat. Rev. Drug Discov. 2009, 8, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Burian, M.; Geisslinger, G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol. Ther. 2005, 107, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Cheremina, O.; Brune, K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J. 2008, 22, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Knaus, E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): Cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008, 11, 81s–110s. [Google Scholar] [CrossRef] [PubMed]
- Huntjens, D.R.; Danhof, M.; Della Pasqua, O.E. Pharmacokinetic-pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology 2005, 44, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.C.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Ford-Hutchinson, A.W.; Forrest, M.J.; Gauthier, J.Y.; et al. Rofecoxib [Vioxx, MK-0966; 4-(4′-methylsulfonylphenyl)-3-phenyl-2-(5H)-furanone]: A potent and orally active cyclooxygenase-2 inhibitor. Pharmacological and biochemical profiles. J. Pharmacol. Exp. Ther. 1999, 290, 551–560. [Google Scholar] [CrossRef] [PubMed]
- de Leval, X.; Delarge, J.; Devel, P.; Neven, P.; Michaux, C.; Masereel, B.; Pirotte, B.; David, J.L.; Henrotin, Y.; Dogne, J.M. Evaluation of classical NSAIDs and COX-2 selective inhibitors on purified ovine enzymes and human whole blood. Prostaglandins Leukot. Essent. Fat. Acids 2001, 64, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Fellier, H.; Christoph, T.; Grarup, J.; Stimmeder, D. The analgesic NSAID lornoxicam inhibits cyclooxygenase (COX)-1/-2, inducible nitric oxide synthase (iNOS), and the formation of interleukin (IL)-6 in vitro. Inflamm. Res. 1999, 48, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Pairet, M.; van Ryn, J.; Schierok, H.; Mauz, A.; Trummlitz, G.; Engelhardt, G. Differential inhibition of cyclooxygenases-1 and -2 by meloxicam and its 4′-isomer. Inflamm. Res. 1998, 47, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Esh, C.J.; Chrismas, B.C.R.; Mauger, A.R.; Taylor, L. Pharmacological hypotheses: Is acetaminophen selective in its cyclooxygenase inhibition? Pharmacol. Res. Perspect. 2021, 9, e00835. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, S.S.; Colville-Nash, P.R.; Willoughby, D.A.; Botting, R.M. The involvement of a cyclooxygenase 1 gene-derived protein in the antinociceptive action of paracetamol in mice. Eur. J. Pharmacol. 2006, 538, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, S.S.; Botting, R.M. Iloprost-induced nociception: Determination of the site of anti-nociceptive action of cyclooxygenase inhibitors and the involvement of cyclooxygenase products in central mechanisms of nociception. Methods Mol. Biol. 2010, 644, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Bromm, B.; Forth, W.; Richter, E.; Scharein, E. Effects of acetaminophen and antipyrine on non-inflammatory pain and EEG activity. Pain 1992, 50, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Barrière, D.A.; Ermund, A.; Jönsson, B.A.; Eschalier, A.; Zygmunt, P.M.; Högestätt, E.D. TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS ONE 2010, 5, e12748. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.J. Paracetamol (Acetaminophen): Mechanisms of action. Paediatr. Anaesth. 2008, 18, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Machado, G.C.; Maher, C.G.; Ferreira, P.H.; Pinheiro, M.B.; Lin, C.W.; Day, R.O.; McLachlan, A.J.; Ferreira, M.L. Efficacy and safety of paracetamol for spinal pain and osteoarthritis: Systematic review and meta-analysis of randomised placebo controlled trials. BMJ 2015, 350, h1225. [Google Scholar] [CrossRef] [PubMed]
- Bjørnsson, G.A.; Haanaes, H.R.; Skoglund, L.A. A randomized, double-blind crossover trial of paracetamol 1000 mg four times daily vs ibuprofen 600 mg: Effect on swelling and other postoperative events after third molar surgery. Br. J. Clin. Pharmacol. 2003, 55, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.N.; Ataoglu, H.; Yildirim, G.; Kiresi, D. Comparison of the efficacy of low doses of methylprednisolone, acetaminophen, and dexketoprofen trometamol on the swelling developed after the removal of impacted third molar. Med. Oral Patol. Oral Cir. Bucal. 2015, 20, e627–e632. [Google Scholar] [CrossRef] [PubMed]
- Brandt, K.D.; Mazzuca, S.A.; Buckwalter, K.A. Acetaminophen, like conventional NSAIDs, may reduce synovitis in osteoarthritic knees. Rheumatology 2006, 45, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Meirer, K.; Steinhilber, D.; Proschak, E. Inhibitors of the arachidonic acid cascade: Interfering with multiple pathways. Basic Clin. Pharmacol. Toxicol. 2014, 114, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Brune, K. Paracetamol and cyclooxygenase inhibition: Is there a cause for concern? Ann. Rheum. Dis. 2012, 71, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Zhang, C.; Lei, Q.; Hu, M.M.; Feng, J.; Shu, H.B.; Liu, Y.; Zhang, X.Z. Hydrogen peroxide detection with high specificity in living cells and inflamed tissues. Regen. Biomater. 2016, 3, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Boutaud, O.; Aronoff, D.M.; Richardson, J.H.; Marnett, L.J.; Oates, J.A. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proc. Natl. Acad. Sci. USA 2002, 99, 7130–7135. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, H.; Brahim, J.S.; Rowan, J.; Lee, G.; Dionne, R.A. Acetaminophen selectively suppresses peripheral prostaglandin E2 release and increases COX-2 gene expression in a clinical model of acute inflammation. Pain 2007, 129, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Scott, K.F. Mechanism of action of paracetamol. Am. J. Ther. 2005, 12, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID development: Evolution of diclofenac products using pharmaceutical technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef] [PubMed]
- Flower, R.J.; Vane, J.R. Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamidophenol). Nature 1972, 240, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Schluesener, H.J.; Meyermann, R.; Serhan, C.N. COX-3 the enzyme and the concept: Steps towards highly specialized pathways and precision therapeutics? Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Zhang, S.P.; Reitz, T.L.; Mei, J.M.; Flores, C.M. Cloning, expression, and functional characterization of human cyclooxygenase-1 splicing variants: Evidence for intron 1 retention. J. Pharmacol. Exp. Ther. 2005, 315, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Kis, B.; Snipes, J.A.; Busija, D.W. Acetaminophen and the cyclooxygenase-3 puzzle: Sorting out facts, fictions, and uncertainties. J. Pharmacol. Exp. Ther. 2005, 315, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Snipes, J.A.; Kis, B.; Shelness, G.S.; Hewett, J.A.; Busija, D.W. Cloning and characterization of cyclooxygenase-1b (putative cyclooxygenase-3) in rat. J. Pharmacol. Exp. Ther. 2005, 313, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Good, R.L.; Roupe, K.A.; Yáñez, J.A. Cyclooxygenase-3: Axiom, dogma, anomaly, enigma or splice error?—Not as easy as 1, 2, 3. J. Pharm. Pharm. Sci. 2004, 7, 217–226, Erratum in: J. Pharm. Pharm. Sci. 2006, 9, 433. [Google Scholar]
- Dalmann, R.; Daulhac, L.; Antri, M.; Eschalier, A.; Mallet, C. Supra-spinal FAAH is required for the analgesic action of paracetamol in an inflammatory context. Neuropharmacology 2015, 91, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, S.; Shiraishi, S.; Miyano, K.; Sudo, Y.; Toda, A.; Mogi, M.; Hara, M.; Yokoyama, A.; Kawasaki, Y.; Taniguchi, M.; et al. Metabolism of AM404 from acetaminophen at human therapeutic dosages in the rat brain. Anesth. Pain Med. 2016, 6, e32873. [Google Scholar] [CrossRef] [PubMed]
- Fegley, D.; Kathuria, S.; Mercier, R.; Li, C.; Goutopoulos, A.; Makriyannis, A.; Piomelli, D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc. Natl. Acad. Sci. USA 2004, 101, 8756–8761. [Google Scholar] [CrossRef] [PubMed]
- Langford, R.A.; Hogg, M.; Bjorksten, A.R.; Williams, D.L.; Leslie, K.; Jamsen, K.; Kirkpatrick, C. Comparative plasma and cerebrospinal fluid pharmacokinetics of paracetamol after intravenous and oral administration. Anesth. Analg. 2016, 123, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, N.; Uta, D.; Sasaki, M.; Ohashi, M.; Kamiya, Y.; Kohno, T. Acetaminophen metabolite N-acylphenolamine induces analgesia via transient receptor potential vanilloid 1 receptors expressed on the primary afferent terminals of C-fibers in the spinal dorsal horn. Anesthesiology 2017, 127, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.V.; Long, J.H.; Shah, S.; Rahman, J.; Perrett, D.; Ayoub, S.S.; Mehta, V. First evidence of the conversion of paracetamol to AM404 in human cerebrospinal fluid. J. Pain Res. 2017, 10, 2703–2709. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, W.; Wu, N.; He, X.; Wang, J.; Feng, Z.; Xie, X.Q. An insight into paracetamol and its metabolites using molecular docking and molecular dynamics simulation. J. Mol. Model. 2018, 24, 243. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Desmeules, J.; Pegahi, R.; Eschalier, A. An updated review on the metabolite (AM404)-mediated central mechanism of action of paracetamol (acetaminophen): Experimental evidence and potential clinical impact. J. Pain Res. 2023, 16, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Wiesenberg-Boettcher, I.; Pfeilschifter, J.; Schweizer, A.; Sallmann, A.; Wenk, P. Pharmacological properties of five diclofenac metabolites identified in human plasma. Agents Actions 1991, 34, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Hashimoto, R.; Shibata, M.; Shimada, H.; Iwaki, M. Involvement of reactive metabolites of diclofenac in cytotoxicity in sandwich-cultured rat hepatocytes. Int. J. Toxicol. 2017, 36, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Amaya-Rodriguez, C.A.; Carvajal-Zamorano, K.; Bustos, D.; Alegría-Arcos, M.; Castillo, K. A journey from molecule to physiology and in silico tools for drug discovery targeting the transient receptor potential vanilloid type 1 (TRPV1) channel. Front. Pharmacol. 2024, 14, 1251061. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Jo, Y.Y.; Kim, Y.H.; Park, C.K. Current insights and therapeutic strategies for targeting TRPV1 in neuropathic pain management. Life Sci. 2024, 355, 122954. [Google Scholar] [CrossRef] [PubMed]
- Maione, S.; Radanova, L.; De Gregorio, D.; Luongo, L.; De Petrocellis, L.; Di Marzo, V.; Imming, P. Effects of metabolites of the analgesic agent dipyrone (metamizol) on rostral ventromedial medulla cell activity in mice. Eur. J. Pharmacol. 2015, 748, 115–122. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, D.; McLaughlin, R.J.; Posa, L.; Ochoa-Sanchez, R.; Enns, J.; Lopez-Canul, M.; Aboud, M.; Maione, S.; Comai, S.; Gobbi, G. Cannabidiol modulates serotonergic transmission and reverses both allodynia and anxiety-like behavior in a model of neuropathic pain. Pain 2019, 160, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Chuang, H.; Movahed, P.; Julius, D.; Högestätt, E.D. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur. J. Pharmacol. 2000, 396, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Stueber, T.; Meyer, S.; Jangra, A.; Hage, A.; Eberhardt, M.; Leffler, A. Activation of the capsaicin-receptor TRPV1 by the acetaminophen metabolite N-arachidonoylaminophenol results in cytotoxicity. Life Sci. 2018, 194, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Nozadze, I.; Tsiklauri, N.; Gurtskaia, G.; Tsagareli, M.G. NSAIDs attenuate hyperalgesia induced by TRP channel activation. Data Brief 2016, 6, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Tsagareli, M.G.; Nozadze, I.; Tsiklauri, N.; Gurtskaia, G. Non-steroidal anti-inflammatory drugs attenuate agonist-evoked activation of transient receptor potential channels. Biomed. Pharmacother. 2018, 97, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Mlost, J.; Kostrzewa, M.; Malek, N.; Starowicz, K. Molecular understanding of the activation of CB1 and blockade of TRPV1 receptors: Implications for novel treatment strategies in osteoarthritis. Int. J. Mol. Sci. 2018, 19, 342. [Google Scholar] [CrossRef] [PubMed]
- Rotpenpian, N.; Arayapisit, T.; Roumwong, A.; Pakaprot, N.; Tantisira, M.; Wanasuntronwong, A. A standardized extract of Centella asiatica (ECa 233) prevents temporomandibular joint osteoarthritis by modulating the expression of local inflammatory mediators in mice. J. Appl. Oral Sci. 2021, 29, e20210329. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.M.; Reilly, C.A.; Deering-Rice, C.E.; Brewster, C.; Brewster, C. Inhibition of FAAH, TRPV1, and COX2 by NSAID-serotonin conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 5695–5698. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, D.; Qiu, Q.; Deng, X.; Lin, H.; Qian, H.; Huang, W. Evaluation of anti-inflammatory and analgesic effects of synthesized derivatives of ibuprofen. Chem. Biol. Drug Des. 2015, 85, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Milligan, A.L.; Szabo-Pardi, T.A.; Burton, M.D. Cannabinoid receptor type 1 and its role as an analgesic: An opioid alternative? J. Dual Diagn. 2020, 16, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Ottani, A.; Leone, S.; Sandrini, M.; Ferrari, A.; Bertolini, A. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur. J. Pharmacol. 2006, 531, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C.; Daulhac, L.; Bonnefont, J.; Ledent, C.; Etienne, M.; Chapuy, E.; Libert, F.; Eschalier, A. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain 2008, 139, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Hama, A.T.; Sagen, J. Cannabinoid receptor-mediated antinociception with acetaminophen drug combinations in rats with neuropathic spinal cord injury pain. Neuropharmacology 2010, 58, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; De Léan, A.; Beaulieu, P. Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain 2006, 121, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.; Romero, T.R.; Guzzo, L.S.; Duarte, I.D. Participation of cannabinoid receptors in peripheral nociception induced by some NSAIDs. Braz. J. Med. Biol. Res. 2012, 45, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Tamaddonfard, E.; Erfanparast, A.; Salighedar, R.; Tamaddonfard, S. Medial prefrontal cortex diclofenac-induced antinociception is mediated through GPR55, cannabinoid CB1, and mu-opioid receptors of this area and periaqueductal gray. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Alloui, A.; Nargeot, J.; Lory, P.; Eschalier, A.; Bourinet, E.; Chemin, J. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J. Neurosci. 2009, 29, 13106–13114. [Google Scholar] [CrossRef] [PubMed]
- Kerckhove, N.; Mallet, C.; François, A.; Boudes, M.; Chemin, J.; Voets, T.; Bourinet, E.; Alloui, A.; Eschalier, A. Cav3.2 calcium channels: The key protagonist in the supraspinal effect of paracetamol. Pain 2014, 155, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Stella, N.; Calignano, A.; Lin, S.Y.; Makriyannis, A.; Piomelli, D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 1997, 277, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Rodriguez de Fonseca, F.; Nava, F.; Loubet-Lescoulié, P.; Piomelli, D. Elevated circulating levels of anandamide after administration of the transport inhibitor, AM404. Eur. J. Pharmacol. 2000, 408, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.T.; Abumrad, N.A.; Fatade, F.; Kaczocha, M.; Studholme, K.M.; Deutsch, D.G. Evidence against the presence of an anandamide transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 4269–4274. [Google Scholar] [CrossRef] [PubMed]
- Vandevoorde, S.; Fowler, C.J. Inhibition of fatty acid amide hydrolase and monoacylglycerol lipase by the anandamide uptake inhibitor VDM11: Evidence that VDM11 acts as an FAAH substrate. Br. J. Pharmacol. 2005, 145, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J.; Björklund, E.; Lichtman, A.H.; Naidu, P.S.; Congiu, C.; Onnis, V. Inhibitory properties of ibuprofen and its amide analogues towards the hydrolysis and cyclooxygenation of the endocannabinoid anandamide. J. Enzym. Inhib. Med. Chem. 2013, 28, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; LoVerme, J.; De Léan, A.; Piomelli, D.; Beaulieu, P. Synergistic antinociceptive effects of anandamide, an endocannabinoid, and nonsteroidal anti-inflammatory drugs in peripheral tissue: A role for endogenous fatty-acid ethanolamides? Eur. J. Pharmacol. 2006, 550, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Păunescu, H.; Coman, O.A.; Coman, L.; Ghiţă, I.; Georgescu, S.R.; Drăghia, F.; Fulga, I. Cannabinoid system and cyclooxygenases inhibitors. J. Med. Life 2011, 4, 11–20. [Google Scholar] [PubMed]
- Turman, M.V.; Kingsley, P.J.; Marnett, L.J. Characterization of an AM404 analogue, N-(3-hydroxyphenyl)arachidonoylamide, as a substrate and inactivator of prostaglandin endoperoxide synthase. Biochemistry 2009, 48, 12233–12241. [Google Scholar] [CrossRef] [PubMed]
- Saliba, S.W.; Marcotegui, A.R.; Fortwängler, E.; Ditrich, J.; Perazzo, J.C.; Muñoz, E.; de Oliveira, A.C.P.; Fiebich, B.L. AM404, paracetamol metabolite, prevents prostaglandin synthesis in activated microglia by inhibiting COX activity. J. Neuroinflamm. 2017, 14, 246, Erratum in: J. Neuroinflamm. 2018, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Treatments of COVID-19-associated taste and saliva aecretory disorders. Dent. J. 2023, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Gerits, E.; Van der Massen, I.; Vandamme, K.; De Cremer, K.; De Brucker, K.; Thevissen, K.; Cammue, B.P.A.; Beullens, S.; Fauvart, M.; Verstraeten, N.; et al. In vitro activity of the antiasthmatic drug zafirlukast against the oral pathogens Porphyromonas gingivalis and Streptococcus mutans. FEMS Microbiol. Lett. 2017, 364, fnx005. [Google Scholar] [CrossRef] [PubMed]
- Gerits, E.; Defraine, V.; Vandamme, K.; De Cremer, K.; De Brucker, K.; Thevissen, K.; Cammue, B.P.; Beullens, S.; Fauvart, M.; Verstraeten, N.; et al. Repurposing toremifene for treatment of oral bacterial infections. Antimicrob. Agents Chemother. 2017, 61, e01846-16. [Google Scholar] [CrossRef] [PubMed]
- Gerits, E.; Spincemaille, P.; De Cremer, K.; De Brucker, K.; Beullens, S.; Thevissen, K.; Cammue, B.P.A.; Vandamme, K.; Fauvart, M.; Verstraeten, N.; et al. Repurposing AM404 for the treatment of oral infections by Porphyromonas gingivalis. Clin. Exp. Dent. Res. 2017, 3, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Al-Janabi, A.A. In vitro antibacterial activity of ibuprofen and acetaminophen. J. Glob. Infect. Dis. 2010, 2, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Leão, C.; Borges, A.; Simões, M. NSAIDs as a drug repurposing strategy for biofilm control. Antibiotics 2020, 9, 591. [Google Scholar] [CrossRef] [PubMed]
- Paes Leme, R.C.; da Silva, R.B. Antimicrobial activity of non-steroidal anti-inflammatory drugs on biofilm: Current evidence and potential for drug repurposing. Front. Microbiol. 2021, 12, 707629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qu, X.; Tang, H.; Wang, Y.; Yang, H.; Yuan, W.; Yue, B. Diclofenac resensitizes methicillin-resistant Staphylococcus aureus to β-lactams and prevents implant infections. Adv. Sci. 2021, 8, 2100681. [Google Scholar] [CrossRef] [PubMed]
- Mizogami, M.; Tsuchiya, H. Lipid composition-, medium pH-, and drug-concentration-dependent membrane interactions of ibuprofen, diclofenac, and celecoxib: Hypothetical association with their analgesic and gastrointestinal toxic effects. Future Pharmacol. 2024, 4, 437–448. [Google Scholar] [CrossRef]
- Pina-Vaz, C.; Sansonetty, F.; Rodrigues, A.G.; Martinez-DE-Oliveira, J.; Fonseca, A.F.; Mårdh, P.A. Antifungal activity of ibuprofen alone and in combination with fluconazole against Candida species. J. Med. Microbiol. 2000, 49, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.F.; Wu, Y.S.; Poh, C.L. Molecular mechanisms of antiviral agents against dengue virus. Viruses 2023, 15, 705. [Google Scholar] [CrossRef] [PubMed]
- van Cleef, K.W.; Overheul, G.J.; Thomassen, M.C.; Marjakangas, J.M.; van Rij, R.P. Escape mutations in NS4B render dengue virus insensitive to the antiviral activity of the paracetamol metabolite AM404. Antimicrob. Agents Chemother. 2016, 60, 2554–2557. [Google Scholar] [CrossRef] [PubMed]
- Gerges, R.H. NSAIDs: A double edged sword in viral infections. Int. J. Med. Rev. 2022, 9, 288–297. [Google Scholar] [CrossRef]
- Paul, A.; Raj, V.S.; Vibhuti, A. In silico and in vitro evaluation of generic medicines against DENV-2. J. Adv. Pharmacog. 2022, 2, 64–73. [Google Scholar]
- Ahmed, M.; Jinks, N.; Babaei-Jadidi, R.; Kashfi, H.; Castellanos-Uribe, M.; May, S.T.; Mukherjee, A.; Nateri, A.S. Repurposing antibacterial AM404 as a potential anticancer drug for targeting colorectal cancer stem-like cells. Cancers 2019, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.J.; Soler-Torronteras, R.; Lara-Chica, M.; García, V.; Fiebich, B.L.; Muñoz, E.; Calzado, M.A. AM404 inhibits NFAT and NF-κB signaling pathways and impairs migration and invasiveness of neuroblastoma cells. Eur. J. Pharmacol. 2015, 746, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.I.; DuBois, R.N. NSAIDs and cancer prevention: Targets downstream of COX-2. Annu. Rev. Med. 2007, 58, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin. Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.M.; Xavier, C.P.R.; Vasconcelos, M.H.; Palmeira, A. Repurposing some of the well-known non-steroid anti-inflammatory drugs (NSAIDs) for cancer treatment. Curr. Top. Med. Chem. 2023, 23, 1171–1195. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Seliger, C.; Jachnik, B.; Welz, T.; Leukel, P.; Vollmann-Zwerenz, A.; Bogdahn, U.; Kreutz, M.; Grauer, O.M.; Hau, P. Ibuprofen and diclofenac restrict migration and proliferation of human glioma cells by distinct molecular mechanisms. PLoS ONE 2015, 10, e0140613. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Marinov, L.; Georgieva, A.; Voynikov, Y.; Toshkova, R.; Nikolova, I.; Malchev, M. Cytotoxic and antiproliferative effects of the nonsteroidal anti-inflammatory drug diclofenac in human tumour cell lines. Biotechnol. Biotechnol. Equip. 2021, 35, 1118–1126. [Google Scholar] [CrossRef]
- Moyer, A.M.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.J.; Pelleymounter, L.L.; Kalari, K.R.; Ji, Y.; Chai, Y.; Nordgren, K.K.; Weinshilboum, R.M. Acetaminophen-NAPQI hepatotoxicity: A cell line model system genome-wide association study. Toxicol. Sci. 2011, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Gentry, C.; Alenmyr, L.; Killander, D.; Lewis, S.E.; Andersson, A.; Bucher, B.; Galzi, J.L.; Sterner, O.; Bevan, S.; et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ9-tetrahydrocannabiorcol. Nat. Commun. 2011, 2, 551. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.A.; Kennon-McGill, S.; Parker, L.D.; James, L.P.; Fantegrossi, W.E.; McGill, M.R. NAPQI is absent in the mouse brain after Sub-hepatotoxic and hepatotoxic doses of acetaminophen. Toxicol. Sci. 2025, 205, 274–278, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dannesboe, J.; Bastrup, J.A.; Nielsen, K.H.; Munck, P.; Thomsen, M.B.; Hawkins, C.L.; Jepps, T.A. Paracetamol metabolism by endothelial cells—Potential mechanism underlying intravenous paracetamol-induced hypotension. Pharmacol. Res. 2025, 211, 107540. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, M.J.; Schillers, F.; Eberhardt, E.M.; Risser, L.; de la Roche, J.; Herzog, C.; Echtermeyer, F.; Leffler, A. Reactive metabolites of acetaminophen activate and sensitize the capsaicin receptor TRPV1. Sci. Rep. 2017, 7, 12775. [Google Scholar] [CrossRef] [PubMed]
- Iannone, L.F.; Nassini, R.; Patacchini, R.; Geppetti, P.; De Logu, F. Neuronal and non-neuronal TRPA1 as therapeutic targets for pain and headache relief. Temperature 2022, 10, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Gentry, C.; Andersson, D.A.; Bevan, S. TRPA1 mediates the hypothermic action of acetaminophen. Sci. Rep. 2015, 5, 12771. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, J.; Zhu, Y.; Wang, C.; Xiao, R.; Herz, J.M.; Wood, J.D.; Zhu, M.X. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflug. Arch. 2010, 459, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Hu, F.; Yang, J.; Guo, X.; Hou, X.; Ju, C.; Wang, K. Activation of neuronal voltage-gated potassium Kv7/KCNQ/M-current by a novel channel opener SCR2682 for alleviation of chronic pain. J. Pharmacol. Exp. Ther. 2021, 377, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Salzer, I.; Kronschläger, M.T.; Boehm, S. The paracetamol metabolite N-acetyl-p-benzoquinone imine reduces excitability in first- and second-order neurons of the pain pathway through actions on KV7 channels. Pain 2019, 160, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Iwata, M.; Tsuchimori, N.; Matsumoto, T. Activation of peripheral KCNQ channels attenuates inflammatory pain. Mol. Pain 2014, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Stampf, J.L.; Ciotu, C.I.; Heber, S.; Boehm, S.; Fischer, M.J.M.; Salzer, I. Analgesic action of acetaminophen via Kv7 channels. Int. J. Mol. Sci. 2022, 24, 650. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, H.; Xu, Y.; Xu, X.; Xu, Y.; Huang, H.; Lv, X.; Liao, C.; Ye, J.; Li, H. Underlying mechanisms and treatment of acetaminophen-induced liver injury (Review). Mol. Med. Rep. 2025, 31, 106. [Google Scholar] [CrossRef] [PubMed]
- Żur, J.; Piński, A.; Marchlewicz, A.; Hupert-Kocurek, K.; Wojcieszyńska, D.; Guzik, U. Organic micropollutants paracetamol and ibuprofen-toxicity, biodegradation, and genetic background of their utilization by bacteria. Environ. Sci. Pollut. Res. Int. 2018, 25, 21498–21524. [Google Scholar] [CrossRef] [PubMed]
- Parolini, M. Toxicity of the non-steroidal anti-inflammatory drugs (NSAIDs) acetylsalicylic acid, paracetamol, diclofenac, ibuprofen and naproxen towards freshwater invertebrates: A review. Sci. Total Environ. 2020, 740, 140043. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.F.; Pan, P.; Li, T.; Song, X.; Xu, Y.; Zhou, N.Y. The universal accumulation of p-aminophenol during the microbial degradation of analgesic and antipyretic acetaminophen in WWTPs: A novel metagenomic perspective. Microbiome 2025, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Barrière, D.A.; Mallet, C.; Blomgren, A.; Simonsen, C.; Daulhac, L.; Libert, F.; Chapuy, E.; Etienne, M.; Högestätt, E.D.; Zygmunt, P.M.; et al. Fatty acid amide hydrolase-dependent generation of antinociceptive drug metabolites acting on TRPV1 in the brain. PLoS ONE 2013, 8, e70690. [Google Scholar] [CrossRef] [PubMed]
- Å Nilsson, J.L.; Mallet, C.; Shionoya, K.; Blomgren, A.; Sundin, A.P.; Grundemar, L.; Boudieu, L.; Blomqvist, A.; Eschalier, A.; Nilsson, U.J.; et al. Paracetamol analogues conjugated by FAAH induce TRPV1-mediated antinociception without causing acute liver toxicity. Eur. J. Med. Chem. 2021, 213, 113042. [Google Scholar] [CrossRef] [PubMed]
- Bazan, H.A.; Bhattacharjee, S.; Burgos, C.; Recio, J.; Abet, V.; Pahng, A.R.; Jun, B.; Heap, J.; Ledet, A.J.; Gordon, W.C.; et al. A novel pipeline of 2-(benzenesulfonamide)-N-(4-hydroxyphenyl) acetamide analgesics that lack hepatotoxicity and retain antipyresis. Eur. J. Med. Chem. 2020, 202, 112600. [Google Scholar] [CrossRef] [PubMed]
- Bazan, H.A.; Bhattacharjee, S.; Reid, M.M.; Jun, B.; Polk, C.; Strain, M.; St Pierre, L.A.; Desai, N.; Daly, P.W.; Cucinello-Ragland, J.A.; et al. Transcriptomic signature, bioactivity and safety of a non-hepatotoxic analgesic generating AM404 in the midbrain PAG region. Sci. Rep. 2024, 14, 11103. [Google Scholar] [CrossRef] [PubMed]
- Fresno, N.; Pérez-Fernández, R.; Goicoechea, C.; Alkorta, I.; Fernández-Carvajal, A.; de la Torre-Martínez, R.; Quirce, S.; Ferrer-Montiel, A.; Martín, M.I.; Goya, P.; et al. Adamantyl analogues of paracetamol as potent analgesic drugs via inhibition of TRPA1. PLoS ONE 2014, 9, e113841. [Google Scholar] [CrossRef] [PubMed]
- Sinning, C.; Watzer, B.; Coste, O.; Nüsing, R.M.; Ott, I.; Ligresti, A.; Di Marzo, V.; Imming, P. New analgesics synthetically derived from the paracetamol metabolite N-(4-hydroxyphenyl)-(5Z,8Z,11Z,14Z)-icosatetra-5,8,11,14-enamide. J. Med. Chem. 2008, 51, 7800–7805. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.M.; Carson, J.R.; Codd, E.E.; Dax, S.L.; Kuffner, E.K.; Stahle, P.L.; Neff, R.A.; Eichenbaum, G.E. The discovery and preclinical pharmacology of JNJ-10450232 (NTM-006), a centrally penetrant, non-opioid structural analog of acetaminophen with comparable analgesic and anti-pyretic properties but no evidence of hepatotoxicity. Regul. Toxicol. Pharmacol. 2025, 105830, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Gelotte, C.K.; Vakil, A.M.; Berwaerts, J.; Zimmerman, B.A.; Eichenbaum, G.E.; Flores, C.M.; Kuffner, E.K. First-in-human study to evaluate the safety, tolerability and pharmacokinetics of a novel analgesic and antipyretic drug with structural similarity to acetaminophen. Regul. Toxicol. Pharmacol. 2022, 134, 105236. [Google Scholar] [CrossRef] [PubMed]
- Gelotte, C.K.; Vakil, A.M.; Zimmerman, B.A.; Zannikos, P.; Mishra, R.; Eichenbaum, G.; Kuffner, E.K.; Flores, C.M. JNJ-10450232 (NTM-006), A novel non-opioid with structural similarities to acetaminophen, produces relatively long-lasting analgesia after a single dose in patients undergoing 3rd molar extraction. Regul. Toxicol. Pharmacol. 2023, 105480, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; De Jonghe, S.; Codd, E.E.; Weiner, S.; Gallacher, D.; Stahle, P.; Kelley, M.F.; Kuffner, E.K.; Flores, C.M.; Eichenbaum, G.E. Preclinical safety assessment of JNJ-10450232 (NTM-006), a structural analog of acetaminophen, that does not cause hepatotoxicity at supratherapeutic doses. Regul. Toxicol. Pharmacol. 2023, 105334, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.K.; Chen, J.; Lam, W.; Gong, Y.; Leclercq, L.; Silva, J.; Salter, R.; Berwaerts, J.; Gelotte, C.K.; Vakil, A.M.; et al. Metabolism and disposition of JNJ-10450232 (NTM-006) in rats, dogs, monkeys and humans. Regul. Toxicol. Pharmacol. 2023, 105379, Online ahead of print. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuchiya, H.; Mizogami, M. Old and New Analgesic Acetaminophen: Pharmacological Mechanisms Compared with Non-Steroidal Anti-Inflammatory Drugs. Future Pharmacol. 2025, 5, 40. https://doi.org/10.3390/futurepharmacol5030040
Tsuchiya H, Mizogami M. Old and New Analgesic Acetaminophen: Pharmacological Mechanisms Compared with Non-Steroidal Anti-Inflammatory Drugs. Future Pharmacology. 2025; 5(3):40. https://doi.org/10.3390/futurepharmacol5030040
Chicago/Turabian StyleTsuchiya, Hironori, and Maki Mizogami. 2025. "Old and New Analgesic Acetaminophen: Pharmacological Mechanisms Compared with Non-Steroidal Anti-Inflammatory Drugs" Future Pharmacology 5, no. 3: 40. https://doi.org/10.3390/futurepharmacol5030040
APA StyleTsuchiya, H., & Mizogami, M. (2025). Old and New Analgesic Acetaminophen: Pharmacological Mechanisms Compared with Non-Steroidal Anti-Inflammatory Drugs. Future Pharmacology, 5(3), 40. https://doi.org/10.3390/futurepharmacol5030040