

Mechanistic Insights into the Mutational Landscape of the Main Protease/3CLPro and Its Impact on Long-Term COVID-19/SARS-CoV-2 Management
 , , , ,
, , , ,
Abstract
1. Introduction
2. SARS-CoV-2 Main Protease
3. 3CLPro Mutational Landscape
4. Structural Alterations of 3CLPro
4.1. Insights into How Mutations Affect the Overall Structure of 3CLPro
4.2. Examination of Changes in Key Structural Motifs and Domains
5. Consequences for Viral Replication of SARS-CoV-2
5.1. Relationship Between 3CLPro Mutations and Viral Replication
5.2. Insights into How Mutations May Influence the Overall Fitness of the Virus
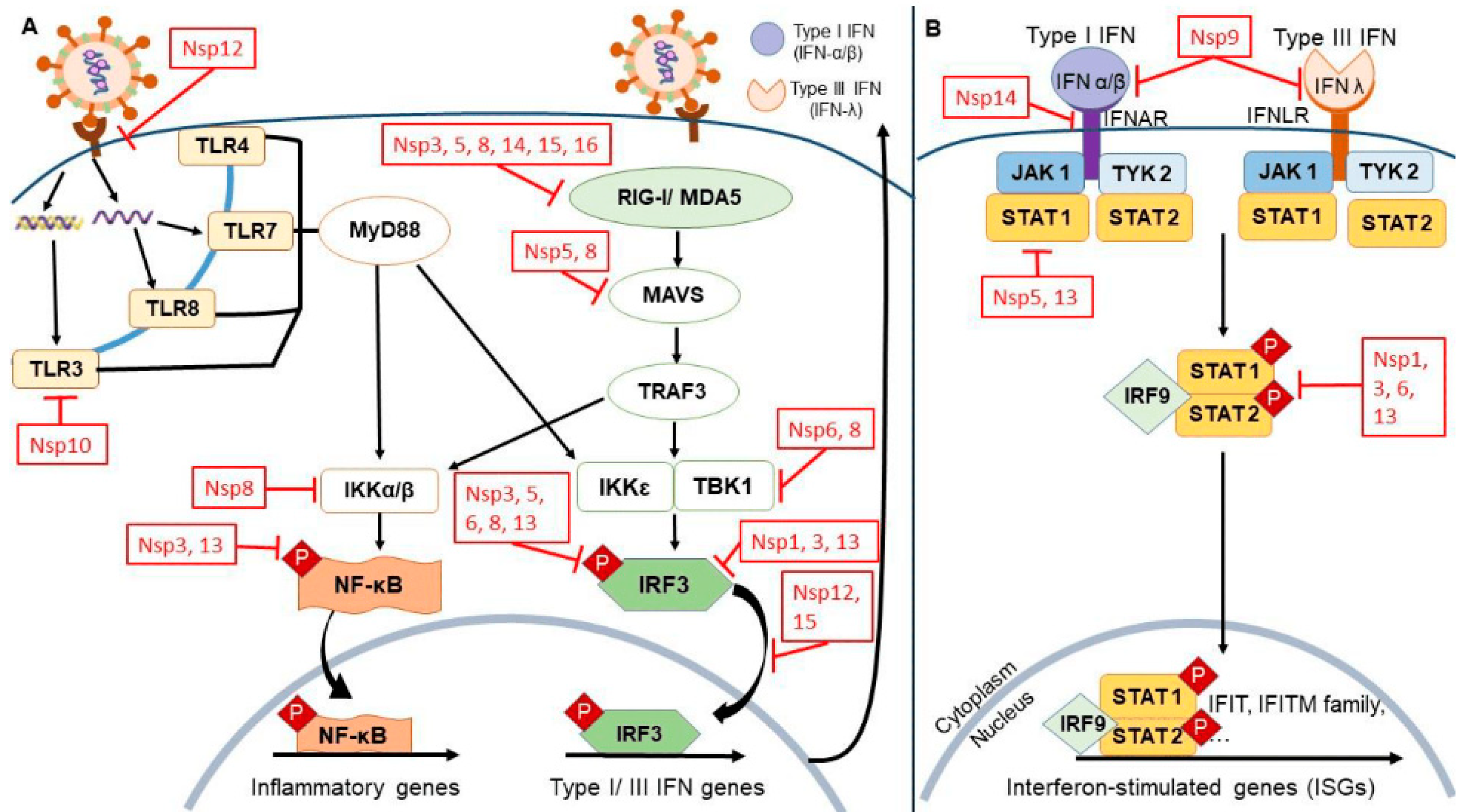
6. Host Immune Evasion in SARS-CoV-2
6.1. Exploration of Potential Mechanisms by Which 3CLPro Mutations Contribute to Immune Evasion
6.2. Discussion of the Role of Mutations in Modulating Host–Virus Interactions
7. Antiviral Drug Resistance in SARS-CoV-2
7.1. Evaluation of How Mutations in 3CLPro May Confer Resistance to Existing Antiviral Drugs
7.2. Implications for Drug Development Strategies
8. Experimental Approaches for SARS-CoV-2 3CLPro Mutations
Overview of the Experimental Techniques Used to Study the Consequences of 3CLPro Mutations
9. Future Perspectives on Tackling Similar Pandemics
10. Conclusions and the Importance of Ongoing Research in the Context of SARS-CoV-2 MPro Mutations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, M.T.; Islam, A.; Hassan, M.I. Potential drug targets of SARS-CoV-2: From genomics to therapeutics. Int. J. Biol. Macromol. 2021, 177, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Reja, A.; Pal, S.; Das, D. Systems chemistry of peptide-assemblies for biochemical transformations. Chem. Soc. Rev. 2022, 51, 3047–3070. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.S.; Hochberg, G.K.; Thornton, J.W. Simple mechanisms for the evolution of protein complexity. Protein Sci. 2022, 31, e4449. [Google Scholar] [CrossRef]
- Lubin, J.H.; Zardecki, C.; Dolan, E.M.; Lu, C.; Shen, Z.; Dutta, S.; Westbrook, J.D.; Hudson, B.P.; Goodsell, D.S.; Williams, J.K. Evolution of the SARS-CoV-2 proteome in three dimensions (3D) during the first 6 months of the COVID-19 pandemic. Proteins Struct. Funct. Bioinform. 2022, 90, 1054–1080. [Google Scholar] [CrossRef]
- Said, N.; Hilal, T.; Sunday, N.D.; Khatri, A.; Bürger, J.; Mielke, T.; Belogurov, G.A.; Loll, B.; Sen, R.; Artsimovitch, I. Steps toward translocation-independent RNA polymerase inactivation by terminator ATPase ρ. Science 2021, 371, eabd1673. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Rabeh, W.M. Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. J. Biol. Chem. 2022, 298, 102023. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, F.; Sementa, D.; Jain, A.; Kumar, M.; Tayarani-Najjaran, M.; Kroiss, D.; Ulijn, R.V. Peptide-based supramolecular systems chemistry. Chem. Rev. 2021, 121, 13869–13914. [Google Scholar] [CrossRef]
- Havranek, B.; Demissie, R.; Lee, H.; Lan, S.; Zhang, H.; Sarafianos, S.; Ayitou, A.; Islam, S. Discovery of Nirmatrelvir Resistance Mutations in SARS-CoV-2 3CLpro: A Computational-Experimental Approach. J. Chem. Inf. Model. 2023, 63, 7180–7188. [Google Scholar] [CrossRef]
- Ramos-Guzmán, C.A.; Andjelkovic, M.; Zinovjev, K.; Ruiz-Pernía, J.J.; Tuñón, I. The impact of SARS-CoV-2 3CL protease mutations on nirmatrelvir inhibitory efficiency. Computational insights into potential resistance mechanisms. Chem. Sci. 2023, 14, 2686–2697. [Google Scholar] [CrossRef]
- Jochmans, D.; Liu, C.; Donckers, K.; Stoycheva, A.; Boland, S.; Stevens, S.K.; De Vita, C.; Vanmechelen, B.; Maes, P.; Trüeb, B.; et al. The substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. MBio 2023, 14, e02815-22. [Google Scholar] [CrossRef]
- Mótyán, J.A.; Mahdi, M.; Hoffka, G.; Tőzsér, J. Potential resistance of SARS-CoV-2 main protease (Mpro) against protease inhibitors: Lessons learned from HIV-1 protease. Int. J. Mol. Sci. 2022, 23, 3507. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef]
- Iketani, S.; Mohri, H.; Culbertson, B.; Hong, S.J.; Duan, Y.; Luck, M.I.; Annavajhala, M.K.; Guo, Y.; Sheng, Z.; Uhlemann, A.-C. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 2023, 613, 558–564. [Google Scholar] [CrossRef]
- Sahoo, P.; Lenka, D.R.; Batabyal, M.; Pain, P.K.; Kumar, S.; Manna, D.; Kumar, A. Detailed Insights into the Inhibitory Mechanism of New Ebselen Derivatives against Main Protease (Mpro) of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). ACS Pharmacol. Transl. Sci. 2023, 6, 171–180. [Google Scholar] [CrossRef]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.-J.; Yang, H.; Zhang, L. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular host factors for SARS-CoV-2 infection. Nat. Microbiol. 2021, 6, 1219–1232. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, Y.; Zeng, X.; He, B.; Cheng, W. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target. Ther. 2022, 7, 26. [Google Scholar] [CrossRef]
- Rangraze, I.; Jhancy, M.; Khan, S. Recent Advances in Understanding COVID-19 Pathophysiology and Therapy: A Review. Preprints 2023. [Google Scholar] [CrossRef]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 main protease for treatment of COVID-19: Covalent inhibitors structure–activity relationship insights and evolution perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Quantum Chemical Computation of Omicron Mutations Near Cleavage Sites of the Spike Protein. Microorganisms 2022, 10, 1999. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kang, C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Perera, L.; Tillekeratne, L.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816. [Google Scholar] [CrossRef]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: State of the art and future opportunities. J. Med. Chem. 2020, 65, 2716–2746. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Roe, M.K.; Junod, N.A.; Young, A.R.; Beachboard, D.C.; Stobart, C.C. Targeting novel structural and functional features of coronavirus protease nsp5 (3CL(pro), M(pro)) in the age of COVID-19. J. Gen. Virol. 2021, 102, 001558. [Google Scholar] [CrossRef]
- Anirudhan, V.; Lee, H.; Cheng, H.; Cooper, L.; Rong, L. Targeting SARS-CoV-2 viral proteases as a therapeutic strategy to treat COVID-19. J. Med. Virol. 2021, 93, 2722–2734. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): An update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef]
- Wong, N.A.; Saier Jr, M.H. The SARS-coronavirus infection cycle: A survey of viral membrane proteins, their functional interactions and pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. [Google Scholar] [CrossRef]
- Santos, I.d.A.; Grosche, V.R.; Bergamini, F.R.G.; Sabino-Silva, R.; Jardim, A.C.G. Antivirals against coronaviruses: Candidate drugs for SARS-CoV-2 treatment? Front. Microbiol. 2020, 11, 1818. [Google Scholar] [CrossRef]
- de Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol. 2021, 95, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Konwar, M.; Sarma, D. Advances in developing small molecule SARS 3CLpro inhibitors as potential remedy for corona virus infection. Tetrahedron 2021, 77, 131761. [Google Scholar] [CrossRef]
- ul Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Lv, Z.; Cano, K.E.; Jia, L.; Drag, M.; Huang, T.T.; Olsen, S.K. Targeting SARS-CoV-2 Proteases for COVID-19 Antiviral Development. Front. Chem. 2021, 9, 819165. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Rath, S.L.; Tripathi, T. Accelerating COVID-19 research using molecular dynamics simulation. J. Phys. Chem. B 2021, 125, 9078–9091. [Google Scholar] [CrossRef]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50. [Google Scholar] [CrossRef]
- Mushebenge, A.G.A.; Ugbaja, S.C.; Mbatha, N.A.; Khan, R.B.; Kumalo, H.M. Assessing the Potential Contribution of In Silico Studies in Discovering Drug Candidates That Interact with Various SARS-CoV-2 Receptors. Int. J. Mol. Sci. 2023, 24, 15518. [Google Scholar] [CrossRef]
- Novak, J.; Rimac, H.; Kandagalla, S.; Pathak, P.; Naumovich, V.; Grishina, M.; Potemkin, V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J. Biomol. Struct. Dyn. 2021, 40, 9347–9360. [Google Scholar] [CrossRef]
- Peng, Y.; Alexov, E.; Basu, S. Structural Perspective on Revealing and Altering Molecular Functions of Genetic Variants Linked with Diseases. Int. J. Mol. Sci. 2019, 20, 548. [Google Scholar] [CrossRef]
- Sheik Amamuddy, O.; Verkhivker, G.M.; Tastan Bishop, Ö. Impact of Early Pandemic Stage Mutations on Molecular Dynamics of SARS-CoV-2 M(pro). J. Chem. Inf. Model. 2020, 60, 5080–5102. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Arutyunova, E.; Lu, J.; Khan, M.B.; Rut, W.; Zmudzinski, M.; Shahbaz, S.; Iyyathurai, J.; Moussa, E.W.; Turner, Z.; et al. SARS-CoV-2 M(pro) Protease Variants of Concern Display Altered Viral Substrate and Cell Host Target Galectin-8 Processing but Retain Sensitivity toward Antivirals. ACS Cent. Sci. 2023, 9, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Sacco, M.D.; Hu, Y.; Gongora, M.V.; Meilleur, F.; Kemp, M.T.; Zhang, X.; Wang, J.; Chen, Y. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 2022, 32, 498–500. [Google Scholar] [CrossRef]
- Chan, H.H.; Oliveira, A.S.F.; Schofield, C.J.; Mulholland, A.J.; Duarte, F. Dynamical nonequilibrium molecular dynamics simulations identify allosteric sites and positions associated with drug resistance in the SARS-CoV-2 main protease. Jacs Au 2023, 3, 1767–1774. [Google Scholar] [CrossRef]
- Flynn, J.M.; Samant, N.; Schneider-Nachum, G.; Barkan, D.T.; Yilmaz, N.K.; Schiffer, C.A.; Moquin, S.A.; Dovala, D.; Bolon, D.N. Comprehensive fitness landscape of SARS-CoV-2 Mpro reveals insights into viral resistance mechanisms. eLife 2022, 11, e77433. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Ni, D.; Chai, Z.; Wang, Y.; Li, M.; Yu, Z.; Liu, Y.; Lu, S.; Zhang, J. Along the allostery stream: Recent advances in computational methods for allosteric drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1585. [Google Scholar] [CrossRef]
- Heilmann, E.; Costacurta, F.; Moghadasi, S.A.; Ye, C.; Pavan, M.; Bassani, D.; Volland, A.; Ascher, C.; Weiss, A.K.H.; Bante, D. SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci. Transl. Med. 2022, 15, eabq7360. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Tran, N.; Dasari, S.; Barwell, S.A.E.; McLeod, M.J.; Kalyaanamoorthy, S.; Holyoak, T.; Ganesan, A. The H163A mutation unravels an oxidized conformation of the SARS-CoV-2 main protease. Nat. Commun. 2023, 14, 5625. [Google Scholar] [CrossRef]
- Tucci, A.R.; da Rosa, R.M.; Rosa, A.S.; Augusto Chaves, O.; Ferreira, V.N.S.; Oliveira, T.K.F.; Coutinho Souza, D.D.; Borba, N.R.R.; Dornelles, L.; Rocha, N.S. Antiviral Effect of 5′-Arylchalcogeno-3-aminothymidine Derivatives in SARS-CoV-2 Infection. Molecules 2023, 28, 6696. [Google Scholar] [CrossRef]
- Hu, Y.; Lewandowski, E.M.; Tan, H.; Zhang, X.; Morgan, R.T.; Zhang, X.; Jacobs, L.M.; Butler, S.G.; Gongora, M.V.; Choy, J. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Cent. Sci. 2023, 9, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R. Computational studies of protein–drug binding affinity changes upon mutations in the drug target. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1563. [Google Scholar] [CrossRef]
- Yin, J.; Li, C.; Ye, C.; Ruan, Z.; Liang, Y.; Li, Y.; Wu, J.; Luo, Z. Advances in the development of therapeutic strategies against COVID-19 and perspectives in the drug design for emerging SARS-CoV-2 variants. Comput. Struct. Biotechnol. J. 2022, 20, 824–837. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Abdelrahman, A.H.; Hegazy, M.-E.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Yadav, S.; Banerjee, S.; Fakayode, S.O.; Parvathareddy, J.; Reichard, W.; Surendranathan, S.; Mahmud, F.; Whatcott, R.; Thammathong, J. Drug repurposing to identify nilotinib as a potential SARS-CoV-2 main protease inhibitor: Insights from a computational and in vitro study. J. Chem. Inf. Model. 2021, 61, 5469–5483. [Google Scholar] [CrossRef]
- Vincenzi, M.; Mercurio, F.A.; Leone, M. Looking for SARS-CoV-2 Therapeutics through Computational Approaches. Curr. Med. Chem. 2023, 30, 3158–3214. [Google Scholar] [CrossRef]
- Qiao, Z.; Wei, N.; Jin, L.; Zhang, H.; Luo, J.; Zhang, Y.; Wang, K. The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorganic Chem. 2021, 117, 105455. [Google Scholar] [CrossRef] [PubMed]
- Safiabadi Tali, S.H.; LeBlanc, J.J.; Sadiq, Z.; Oyewunmi, O.D.; Camargo, C.; Nikpour, B.; Armanfard, N.; Sagan, S.M.; Jahanshahi-Anbuhi, S. Tools and techniques for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)/COVID-19 detection. Clin. Microbiol. Rev. 2021, 34, 10–1128. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, E. Investigation of changes in protein stability and substrate affinity of 3CL-protease of SARS-CoV-2 caused by mutations. Genet. Mol. Biol. 2022, 45, e20210404. [Google Scholar] [CrossRef]
- Mahtarin, R.; Islam, S.; Islam, M.J.; Ullah, M.O.; Ali, M.A.; Halim, M.A. Structure and dynamics of membrane protein in SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 4725–4738. [Google Scholar] [CrossRef]
- Akbulut, E. Mutations in Main Protease of SARS CoV-2 Decreased Boceprevir Affinity. Braz. Arch. Biol. Technol. 2022, 64, e21200803. [Google Scholar] [CrossRef]
- Rehman, M.U.; Ali, A.; Ansar, R.; Arafah, A.; Imtiyaz, Z.; Wani, T.A.; Zargar, S.; Ganie, S.A. In Silico molecular docking and dynamic analysis of natural compounds against major non-structural proteins of SARS-COV-2. J. Biomol. Struct. Dyn. 2023, 41, 9072–9088. [Google Scholar] [CrossRef]
- Hasan, M.; Parvez, M.S.A.; Azim, K.F.; Imran, M.A.S.; Raihan, T.; Gulshan, A.; Muhit, S.; Akhand, R.N.; Ahmed, S.S.U.; Uddin, M.B. Main protease inhibitors and drug surface hotspots for the treatment of COVID-19: A drug repurposing and molecular docking approach. Biomed. Pharmacother. 2021, 140, 111742. [Google Scholar] [CrossRef]
- Al Adem, K.; Ferreira, J.C.; Fadl, S.; Mustafa, M.; Rabeh, W.M. Key allosteric and active site residues of SARS-CoV-2 3CLpro are promising drug targets. Biochem. J. 2023, 480, 791–813. [Google Scholar] [CrossRef]
- Kidera, A.; Moritsugu, K.; Ekimoto, T.; Ikeguchi, M. Allosteric regulation of 3CL protease of SARS-CoV-2 and SARS-CoV observed in the crystal structure ensemble. J. Mol. Biol. 2021, 433, 167324. [Google Scholar] [CrossRef]
- Ferreira, G.M.; Kronenberger, T.; Tonduru, A.K.; Hirata, R.D.C.; Hirata, M.H.; Poso, A. SARS-COV-2 Mpro conformational changes induced by covalently bound ligands. J. Biomol. Struct. Dyn. 2022, 40, 12347–12357. [Google Scholar] [CrossRef]
- Tekpinar, M.; Yildirim, A. Impact of dimerization and N3 binding on molecular dynamics of SARS-CoV and SARS-CoV-2 main proteases. J. Biomol. Struct. Dyn. 2022, 40, 6243–6254. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Philot, E.; Pantaleão, S.; Torres-Bonfim, N.; Kliousoff, A.; Quiroz, R.; Perahia, D.; Simões, R.; Magro, A.; Scott, A. Unveiling mutation effects on the structural dynamics of the main protease from SARS-CoV-2 with hybrid simulation methods. J. Mol. Graph. Model. 2023, 121, 108443. [Google Scholar] [CrossRef]
- Diessner, E.M.; Takahashi, G.R.; Cross, T.J.; Martin, R.W.; Butts, C.T. Mutation effects on structure and dynamics: Adaptive evolution of the SARS-CoV-2 main protease. Biochemistry 2023, 62, 747–758. [Google Scholar] [CrossRef]
- Ganesh, B.; Rajakumar, T.; Malathi, M.; Manikandan, N.; Nagaraj, J.; Santhakumar, A.; Elangovan, A.; Malik, Y.S. Epidemiology and pathobiology of SARS-CoV-2 (COVID-19) in comparison with SARS, MERS: An updated overview of current knowledge and future perspectives. Clin. Epidemiol. Glob. Health 2021, 10, 100694. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y. Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med 2021, 2, 99–112.e117. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; Nahed, A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B. 1.1. 529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef]
- Kumari, M.; Lu, R.-M.; Li, M.-C.; Huang, J.-L.; Hsu, F.-F.; Ko, S.-H.; Ke, F.-Y.; Su, S.-C.; Liang, K.-H.; Yuan, J.P.-Y. A critical overview of current progress for COVID-19: Development of vaccines, antiviral drugs, and therapeutic antibodies. J. Biomed. Sci. 2022, 29, 68. [Google Scholar] [CrossRef]
- Alzyoud, L.; Ghattas, M.A.; Atatreh, N. Allosteric binding sites of the SARS-CoV-2 main protease: Potential targets for broad-spectrum anti-coronavirus agents. Drug Des. Dev. Ther. 2022, 16, 2463–2478. [Google Scholar] [CrossRef]
- Liu, H.; Iketani, S.; Zask, A.; Khanizeman, N.; Bednarova, E.; Forouhar, F.; Fowler, B.; Hong, S.J.; Mohri, H.; Nair, M.S.; et al. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nat. Commun. 2022, 13, 1891. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Panwar, U.; Dinesh, D.C.; Boura, E.; Singh, P.; Dubey, V.K.; Singh, S.K. Microsecond MD simulation and multiple-conformation virtual screening to identify potential anti-COVID-19 inhibitors against SARS-CoV-2 main protease. Front. Chem. 2021, 8, 595273. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Wang, Z.; Wang, P.; Ren, W.; Yu, Y.; Yu, Y.; Yuan, B.; Song, J.; Zhang, X.; Zhang, Y. SARS-CoV-2 main protease cleaves MAGED2 to antagonize host antiviral defense. Mbio 2023, 14, e01373-23. [Google Scholar] [CrossRef]
- Khataniar, A.; Pathak, U.; Rajkhowa, S.; Jha, A.N. A comprehensive review of drug repurposing strategies against known drug targets of COVID-19. Covid 2022, 2, 148–167. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Aruleba, R.T.; Sibuyi, N.R.S.; Klein, A.; Madiehe, A.M.; Meyer, M. Inhibitory potential of repurposed drugs against the SARS-CoV-2 main protease: A computational-aided approach. J. Biomol. Struct. Dyn. 2022, 40, 3416–3427. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An updated review of computer-aided drug design and its application to COVID-19. BioMed Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef]
- Chatzigoulas, A.; Cournia, Z. Rational design of allosteric modulators: Challenges and successes. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1529. [Google Scholar] [CrossRef]
- Reinke, P.Y.; Schubert, R.; Oberthür, D.; Galchenkova, M.; Rahmani Mashhour, A.; Günther, S.; Chretien, A.; Round, A.; Seychell, B.C.; Norton-Baker, B.; et al. SARS-CoV-2 Mpro Responds to Oxidation by Forming Disulfide and NOS/SONOS Bonds. Nat. Commun. 2024, 15, 3827. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, B.; Deng, L.; Liang, B.; Ping, J. Virus-host interaction networks as new antiviral drug targets for IAV and SARS-CoV-2. Emerg. Microbes Infect. 2022, 11, 1371–1389. [Google Scholar] [CrossRef]
- Aniana, A.; Nashed, N.; Ghirlando, R.; Coates, L.; Kneller, D.; Kovalevsky, A.; Louis, J. Insights into the mechanism of SARS-CoV-2 main protease autocatalytic maturation from model precursors. Commun. Biol. 2023, 6, 1159. [Google Scholar] [CrossRef]
- Kovalevsky, A.; Coates, L.; Kneller, D.W.; Ghirlando, R.; Aniana, A.; Nashed, N.T.; Louis, J.M. Unmasking the conformational stability and inhibitor binding to SARS-CoV-2 main protease active site mutants and miniprecursor. J. Mol. Biol. 2022, 434, 167876. [Google Scholar] [CrossRef] [PubMed]
- Mushebenge, A.G.; Ugbaja, S.C.; Mtambo, S.E.; Ntombela, T.; Metu, J.I.; Babayemi, O.; Chima, J.I.; Appiah-Kubi, P.; Odugbemi, A.I.; Ntuli, M.L.; et al. Unveiling the Inhibitory Potentials of Peptidomimetic Azanitriles and Pyridyl Esters Towards SARS-CoV-2 Main Protease: A Molecular Modelling Investigation. Molecules 2023, 28, 2641. [Google Scholar] [CrossRef]
- Duerr, R.; Crosse, K.M.; Valero-Jimenez, A.M.; Dittmann, M. SARS-CoV-2 portrayed against HIV: Contrary viral strategies in similar disguise. Microorganisms 2021, 9, 1389. [Google Scholar] [CrossRef] [PubMed]
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020, 21, 3099. [Google Scholar] [CrossRef]
- Nguyen, K.V. Problems associated with antiviral drugs and vaccines development for COVID-19: Approach to intervention using expression vectors via GPI anchor. Nucleosides Nucleotides Nucleic Acids 2021, 40, 665–706. [Google Scholar] [CrossRef]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm 2022, 3, e151. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorganic Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gammeltoft, K.A.; Ryberg, L.A.; Pham, L.V.; Tjørnelund, H.D.; Binderup, A.; Duarte Hernandez, C.R.; Fernandez-Antunez, C.; Offersgaard, A.; Fahnøe, U. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 2022, 8, eadd7197. [Google Scholar] [CrossRef]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef]
- Bloom, J.D.; Neher, R.A. Fitness effects of mutations to SARS-CoV-2 proteins. Virus Evol. 2023, 9, vead055. [Google Scholar] [CrossRef]
- Iketani, S.; Hong, S.J.; Sheng, J.; Bahari, F.; Culbertson, B.; Atanaki, F.F.; Aditham, A.K.; Kratz, A.F.; Luck, M.I.; Tian, R. Functional map of SARS-CoV-2 3CL protease reveals tolerant and immutable sites. Cell Host Microbe 2022, 30, 1354–1362.e1356. [Google Scholar] [CrossRef] [PubMed]
- Goyal, B.; Goyal, D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Fischer, C.; Feys, J.R. SARS-CoV-2 Mpro inhibitors: Achieved diversity, developing resistance and future strategies. Future Pharmacol. 2023, 3, 80–107. [Google Scholar] [CrossRef]
- Kim, C.-H. SARS-CoV-2 evolutionary adaptation toward host entry and recognition of receptor O-Acetyl sialylation in virus–host interaction. Int. J. Mol. Sci. 2020, 21, 4549. [Google Scholar] [CrossRef]
- Swaraj, S.; Malpani, T.; Tripathi, S. Antagonism and Evasion of Cellular Innate Immunity by SARS-CoV-2. In Uncovering The Science of COVID-19; World Scientific: Singapore, 2023; pp. 233–258. [Google Scholar]
- Park, A.; Iwasaki, A. Type I and type III interferons–induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Mensah, J.O.; Ampomah, G.B.; Gasu, E.N.; Adomako, A.K.; Menkah, E.S.; Borquaye, L.S. Allosteric modulation of the main protease (MPro) of SARS-CoV-2 by casticin—Insights from molecular dynamics simulations. Chem. Afr. 2022, 5, 1305–1320. [Google Scholar] [CrossRef]
- Irfan, M.; Akhtar, N.; Ahmad, M.; Shahzad, F.; Elavarasan, R.M.; Wu, H.; Yang, C. Assessing public willingness to wear face masks during the COVID-19 pandemic: Fresh insights from the theory of planned behavior. Int. J. Environ. Res. Public Health 2021, 18, 4577. [Google Scholar] [CrossRef]
- Lee, J.T.; Yang, Q.; Gribenko, A.; Perrin Jr, B.S.; Zhu, Y.; Cardin, R.; Liberator, P.A.; Anderson, A.S.; Hao, L. Genetic surveillance of SARS-CoV-2 Mpro reveals high sequence and structural conservation prior to the introduction of protease inhibitor Paxlovid. Mbio 2022, 13, e00869-22. [Google Scholar] [CrossRef]
- Reis, S.; Metzendorf, M.-I.; Kuehn, R.; Popp, M.; Gagyor, I.; Kranke, P.; Meybohm, P.; Skoetz, N.; Weibel, S. Nirmatrelvir combined with ritonavir for preventing and treating COVID-19. Cochrane Database Syst. Rev. 2022. [Google Scholar] [CrossRef]
- Sacchi, A.; Giannessi, F.; Sabatini, A.; Percario, Z.A.; Affabris, E. SARS-CoV-2 Evasion of the Interferon System: Can We Restore Its Effectiveness? Int. J. Mol. Sci. 2023, 24, 9353. [Google Scholar] [CrossRef]
- Lotke, R.; Petersen, M.; Sauter, D. Restriction of Viral Glycoprotein Maturation by Cellular Protease Inhibitors. Viruses 2024, 16, 332. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, X.; Luo, H.; Qian, R.; Yang, Y.; Yu, H.; Huang, J.; Shi, P.-Y.; Hu, Q. Resistance mechanisms of SARS-CoV-2 3CLpro to the non-covalent inhibitor WU-04. Cell Discov. 2024, 10, 40. [Google Scholar] [CrossRef]
- Tan, B.; Joyce, R.; Tan, H.; Hu, Y.; Wang, J. SARS-CoV-2 main protease drug design, assay development, and drug resistance studies. Acc. Chem. Res. 2022, 56, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Feng, Y.; Li, W.; Liu, T.; Lv, X.; Tong, X.; Xi, G.; Ye, X.; Li, X. Development of Novel Antiviral Agents That Induce the Degradation of the Main Protease of Human-Infecting Coronaviruses. Eur. J. Med. Chem. 2024, 275, 116629. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorganic Med. Chem. 2021, 29, 115860. [Google Scholar] [CrossRef] [PubMed]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Baghi, H.B.; Nahand, J.S.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A new approach to COVID-19 therapy? Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef]
- Magwaza, N.N.; Mushebenge, A.G.-A.; Ugbaja, S.C.; Mbatha, N.A.; Khan, R.B.; Kumalo, H.M. Mechanistic Insights into Targeting SARS-CoV-2 Papain-like Protease in the Evolution and Management of COVID-19. BioChem 2024, 4, 268–299. [Google Scholar] [CrossRef]
- Mushebenge, A.G.-A.; Ugbaja, S.C.; Mbatha, N.A.; Khan, R.B.; Kumalo, H.M. Unravelling Insights into the Evolution and Management of SARS-CoV-2. BioMedInformatics 2024, 4, 385–409. [Google Scholar] [CrossRef]
- Chen, J.-T. Ethnopharmacology and Drug Discovery for COVID-19: Anti-SARS-CoV-2 Agents from Herbal Medicines and Natural Products; Springer Nature: Berlin/Heidelberg, Germany, 2023. [Google Scholar]
- Heilmann, E.; Costacurta, F.; Geley, S.; Mogadashi, S.A.; Volland, A.; Rupp, B.; Harris, R.S.; von Laer, D. A VSV-based assay quantifies coronavirus Mpro/3CLpro/Nsp5 main protease activity and chemical inhibition. Commun. Biol. 2022, 5, 391. [Google Scholar] [CrossRef]
- Heilmann, E.; Costacurta, F.; Volland, A.; von Laer, D. SARS-CoV-2 3CLpro mutations confer resistance to Paxlovid (nirmatrelvir/ritonavir) in a VSV-based, non-gain-of-function system. bioRxiv 2022. [Google Scholar] [CrossRef]
- Moghadasi, S.A.; Heilmann, E.; Khalil, A.M.; Nnabuife, C.; Kearns, F.L.; Ye, C.; Moraes, S.N.; Costacurta, F.; Esler, M.A.; Aihara, H.; et al. Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. Sci. Adv. 2023, 9, eade8778. [Google Scholar] [CrossRef]
- Rella, S.A.; Kulikova, Y.A.; Dermitzakis, E.T.; Kondrashov, F.A. Rates of SARS-CoV-2 transmission and vaccination impact the fate of vaccine-resistant strains. Sci. Rep. 2021, 11, 15729. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Pozzi, C.; Vanet, A.; Francesconi, V.; Tagliazucchi, L.; Tassone, G.; Venturelli, A.; Spyrakis, F.; Mazzorana, M.; Costi, M.P.; Tonelli, M. Antitarget, Anti-SARS-CoV-2 Leads, Drugs, and the Drug Discovery–Genetics Alliance Perspective. J. Med. Chem. 2023, 66, 3664–3702. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nirmatrelvir plus ritonavir: First approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef]
- Parigger, L.; Krassnigg, A.; Schopper, T.; Singh, A.; Tappler, K.; Köchl, K.; Hetmann, M.; Gruber, K.; Steinkellner, G.; Gruber, C.C. Recent changes in the mutational dynamics of the SARS-CoV-2 main protease substantiate the danger of emerging resistance to antiviral drugs. Front. Med. 2022, 9, 1061142. [Google Scholar] [CrossRef]
- Cobey, S.; Larremore, D.B.; Grad, Y.H.; Lipsitch, M. Concerns about SARS-CoV-2 evolution should not hold back efforts to expand vaccination. Nat. Rev. Immunol. 2021, 21, 330–335. [Google Scholar] [CrossRef]
- Weng, Y.L.; Naik, S.R.; Dingelstad, N.; Lugo, M.R.; Kalyaanamoorthy, S.; Ganesan, A. Molecular dynamics and in silico mutagenesis on the reversible inhibitor-bound SARS-CoV-2 main protease complexes reveal the role of lateral pocket in enhancing the ligand affinity. Sci. Rep. 2021, 11, 7429. [Google Scholar] [CrossRef]
- Gao, K.; Wang, R.; Chen, J.; Cheng, L.; Frishcosy, J.; Huzumi, Y.; Qiu, Y.; Schluckbier, T.; Wei, X.; Wei, G.-W. Methodology-centered review of molecular modeling, simulation, and prediction of SARS-CoV-2. Chem. Rev. 2022, 122, 11287–11368. [Google Scholar] [CrossRef]
- Westberg, M.; Su, Y.; Zou, X.; Huang, P.; Rustagi, A.; Garhyan, J.; Patel, P.B.; Fernandez, D.; Wu, Y.; Hao, C. An orally bioavailable SARS-CoV-2 main protease inhibitor exhibits improved affinity and reduced sensitivity to mutations. Sci. Transl. Med. 2024, 16, eadi0979. [Google Scholar] [CrossRef]
- Hattori, S.-i.; Bulut, H.; Hayashi, H.; Kishimoto, N.; Takamune, N.; Hasegawa, K.; Furusawa, Y.; Yamayoshi, S.; Murayama, K.; Tamamura, H. Structural and virologic mechanism of the emergence of resistance to Mpro inhibitors in SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2024, 121, e2404175121. [Google Scholar] [CrossRef]
- Kaniyala Melanthota, S.; Banik, S.; Chakraborty, I.; Pallen, S.; Gopal, D.; Chakrabarti, S.; Mazumder, N. Elucidating the microscopic and computational techniques to study the structure and pathology of SARS-CoVs. Microsc. Res. Tech. 2020, 83, 1623–1638. [Google Scholar] [CrossRef]
- Chan, C.C.-Y.; Guo, Q.; Chan, J.F.-W.; Tang, K.; Cai, J.-P.; Chik, K.K.-H.; Huang, Y.; Dai, M.; Qin, B.; Ong, C.P. Identification of novel small-molecule inhibitors of SARS-CoV-2 by chemical genetics. Acta Pharm. Sin. B 2024, 14, 4028–4044. [Google Scholar] [CrossRef]
- Abe, K.; Kabe, Y.; Uchiyama, S.; Iwasaki, Y.W.; Ishizu, H.; Uwamino, Y.; Takenouchi, T.; Uno, S.; Ishii, M.; Maruno, T. Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19. Sci. Rep. 2022, 12, 1299. [Google Scholar] [CrossRef]
- James, V.K.; Godula, R.N.; Perez, J.M.; Beckham, J.T.; Butalewicz, J.P.; Sipe, S.N.; Huibregtse, J.M.; Brodbelt, J.S. Native mass spectrometry reveals binding interactions of SARS-CoV-2 PLpro with inhibitors and cellular targets. ACS Infect. Dis. 2024, 10, 3597–3606. [Google Scholar] [CrossRef]
- Vlachou, A.; Nchioua, R.; Regensburger, K.; Kirchhoff, F.; Kmiec, D. A Gaussia luciferase reporter assay for the evaluation of coronavirus Nsp5/3CLpro activity. Sci. Rep. 2024, 14, 20697. [Google Scholar] [CrossRef]
- Funk, L.-M.; Poschmann, G.; Rabe von Pappenheim, F.; Chari, A.; Stegmann, K.M.; Dickmanns, A.; Wensien, M.; Eulig, N.; Paknia, E.; Heyne, G. Multiple redox switches of the SARS-CoV-2 main protease in vitro provide opportunities for drug design. Nat. Commun. 2024, 15, 411. [Google Scholar] [CrossRef]
- Moghadasi, S.A.; Esler, M.A.; Otsuka, Y.; Becker, J.T.; Moraes, S.N.; Anderson, C.B.; Chamakuri, S.; Belica, C.; Wick, C.; Harki, D.A. Gain-of-signal assays for probing inhibition of SARS-CoV-2 Mpro/3CLpro in living cells. MBio 2022, 13, e00784-22. [Google Scholar] [CrossRef]
- Cetin, Y.; Aydinlik, S.; Gungor, A.; Kan, T.; Avsar, T.; Durdagi, S. Review on in silico methods, high-throughput screening techniques, and cell culture based in vitro assays for SARS-CoV-2. Curr. Med. Chem. 2022, 29, 5925–5948. [Google Scholar] [CrossRef]
- Azzeri, A.; Mohamed, N.A.; Wan Rosli, S.H.; Abdul Samat, M.N.; Rashid, Z.Z.; Mohamad Jamali, M.A.; Md Zoqratt, M.Z.H.; Mohammad Nasir, M.A.; Ranjit Singh, H.K.; Azmi, L. Unravelling the link between SARS-CoV-2 mutation frequencies, patient comorbidities, and structural dynamics. PLoS ONE 2024, 19, e0291892. [Google Scholar] [CrossRef]
- Zeng, L.; Li, D.; Tong, W.; Shi, T.; Ning, B. Biochemical features and mutations of key proteins in SARS-CoV-2 and their impacts on RNA therapeutics. Biochem. Pharmacol. 2021, 189, 114424. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, J.; Wang, P.; Zhang, Z. The mechanisms of immune response and evasion by the main SARS-CoV-2 variants. Iscience 2022, 25, 105044. [Google Scholar] [CrossRef]
- Vilar, S.; Isom, D.G. One year of SARS-CoV-2: How much has the virus changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef]
- Mascellino, M.T.; Di Timoteo, F.; De Angelis, M.; Oliva, A. Overview of the main anti-SARS-CoV-2 vaccines: Mechanism of action, efficacy and safety. Infect. Drug Resist. 2021, 14, 3459–3476. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef]
- Sargsyan, K.; Mazmanian, K.; Lim, C. A strategy for evaluating potential antiviral resistance to small molecule drugs and application to SARS-CoV-2. Sci. Rep. 2023, 13, 502. [Google Scholar] [CrossRef]
- Nikhra, V. Living with COVID-19: The Nemesis, the Hubris, and the Elpis. Dynamics 2021, 3, 012. [Google Scholar]
- Chtita, S.; Belhassan, A.; Aouidate, A.; Belaidi, S.; Bouachrine, M.; Lakhlifi, T. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking and virtual screening. Comb. Chem. High Throughput Screen. 2021, 24, 441–454. [Google Scholar] [CrossRef] [PubMed]
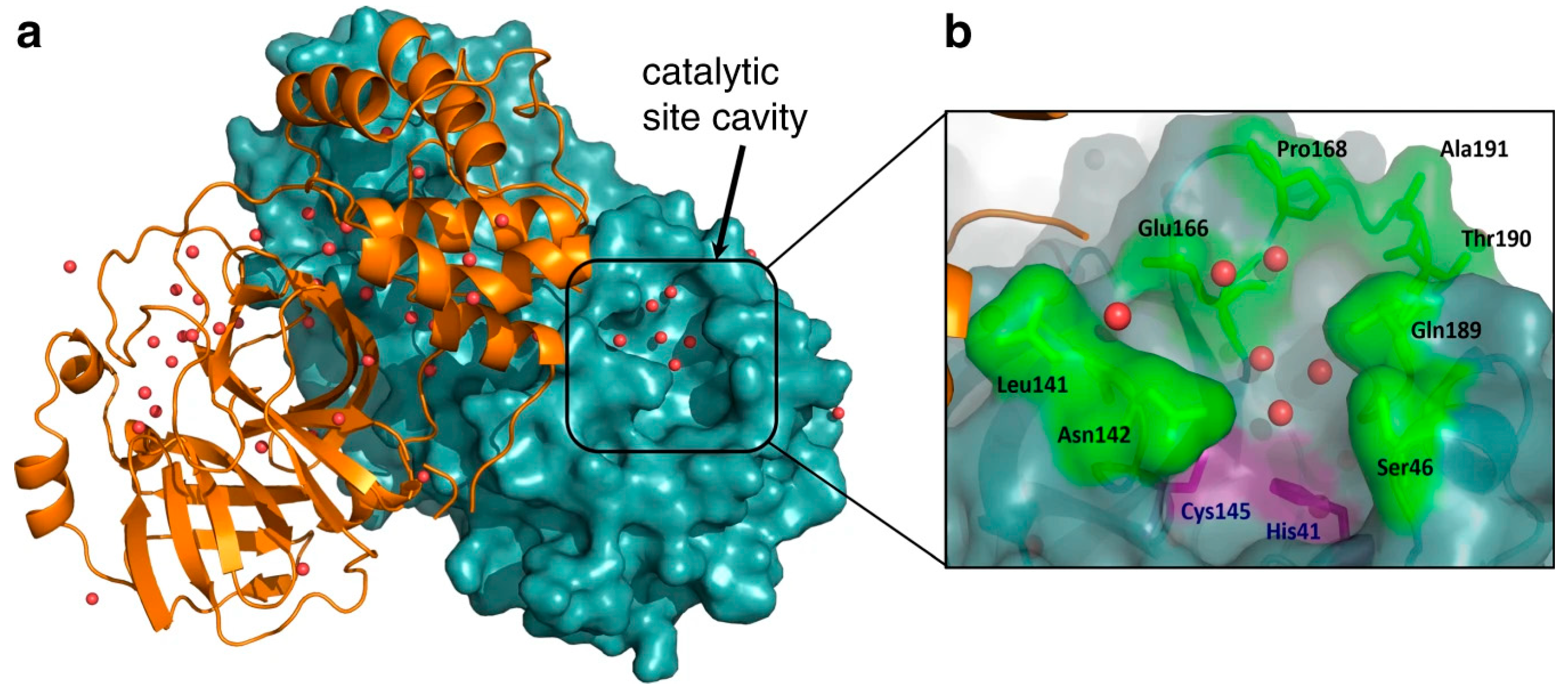

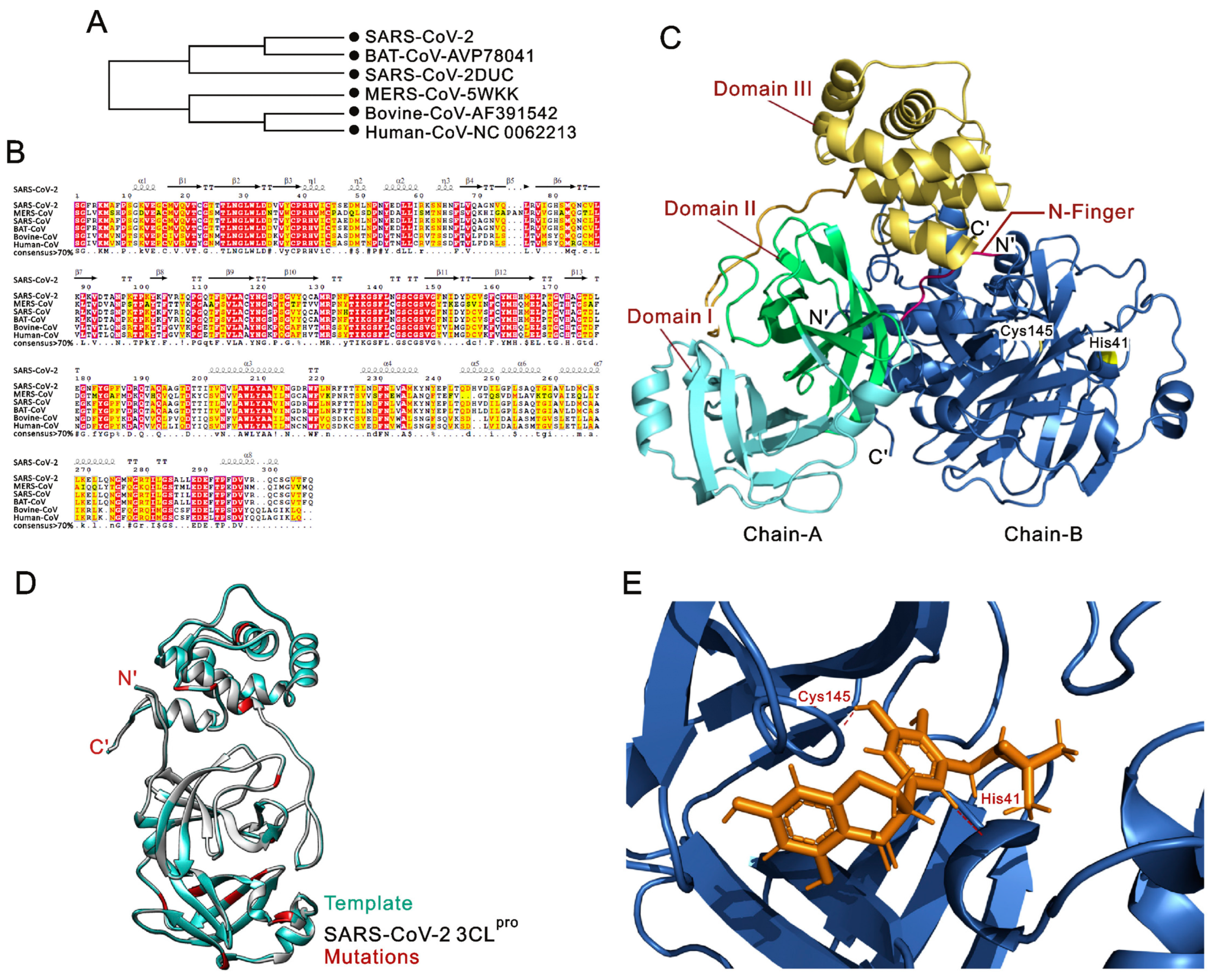
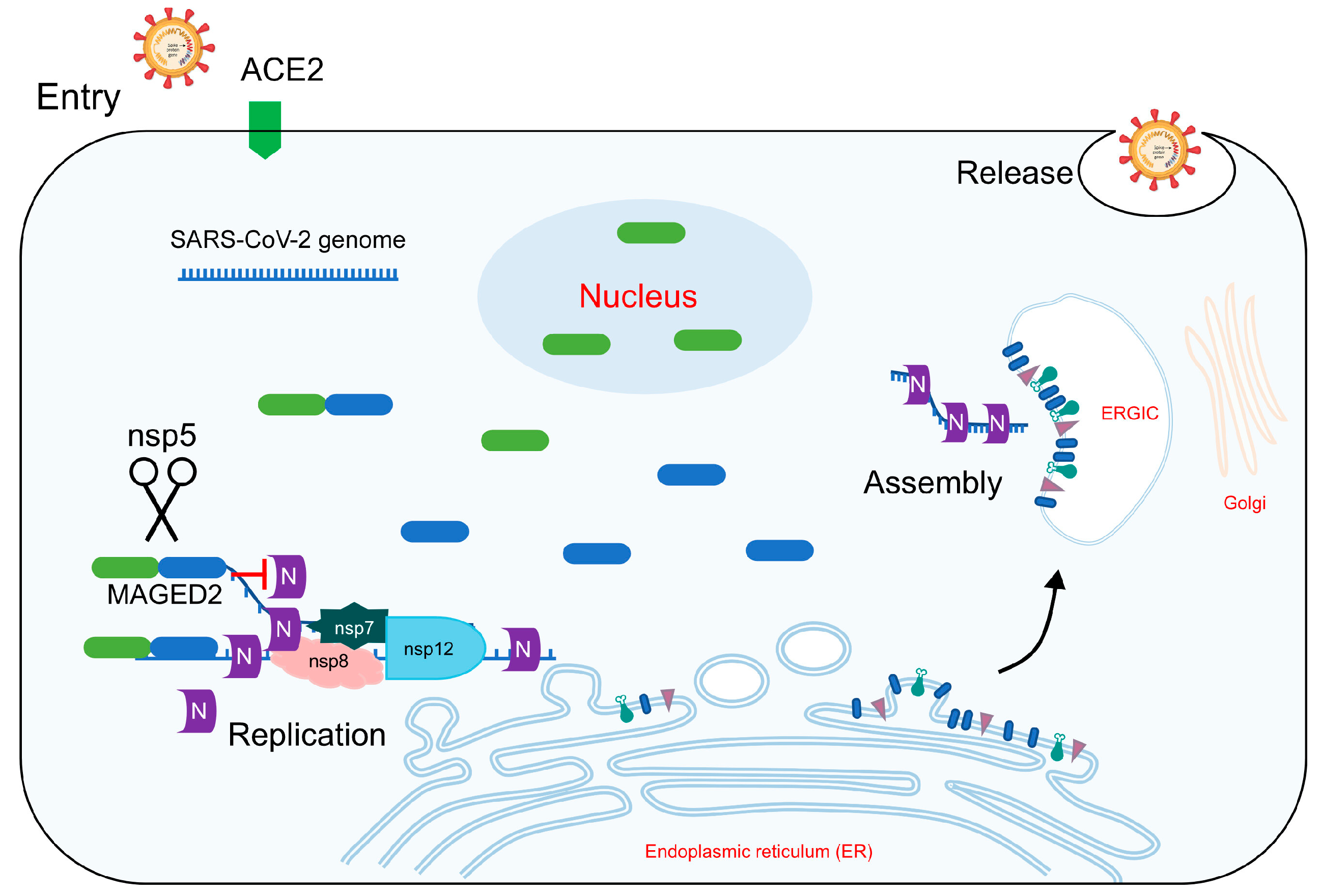
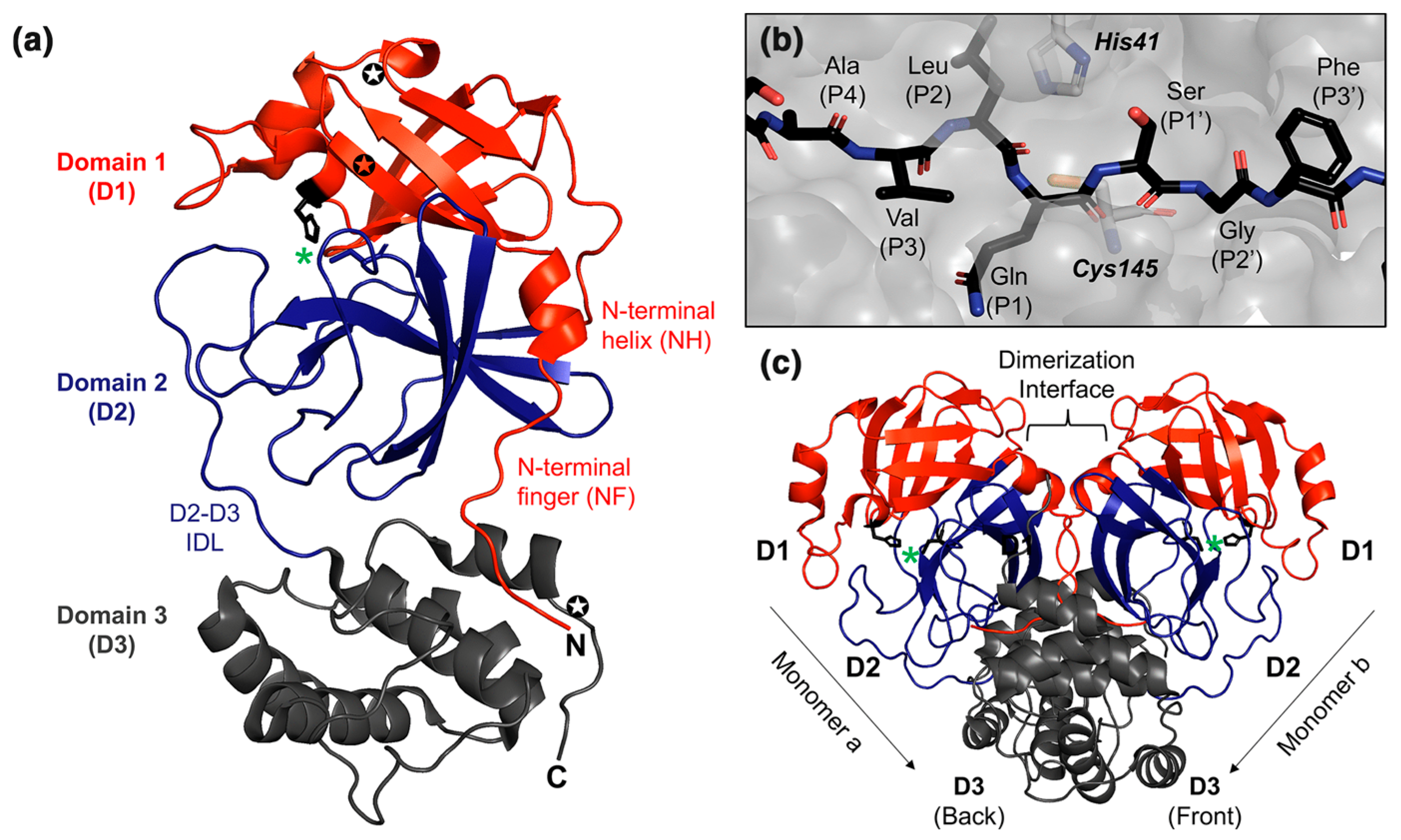

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Alterations | Description | Potential Consequences | Research Implications | Ref. |
| Active Site | Analysis of changes in residues crucial for substrate binding and catalysis, including mutations that may affect substrate specificity or enzymatic activity. Structural modifications in the active site can impact inhibitor binding and efficacy. | Mutations in the active site may lead to altered substrate specificity, reduced enzymatic activity, or resistance to inhibitors. | Understanding alterations in the active site provides insights into the design of specific inhibitors targeting mutant enzymes. | [68,69] |
| Substrate Binding Pocket | Examination of modifications in the pocket’s shape, size, and residues involved in substrate recognition. Alterations in the substrate binding pocket can influence substrate affinity, catalytic efficiency, and enzyme–substrate interactions. Understanding changes in this region is crucial for predicting the impact on enzymatic function and drug binding. | Changes in the substrate binding pocket may affect substrate recognition, leading to altered enzymatic activity or decreased inhibitor binding. | Knowledge of substrate binding pocket alterations aids in the design of novel inhibitors with improved binding affinity and specificity. | [6,68] |
| Catalytic Residues | Assessment of alterations in residues directly involved in catalysis, such as those participating in nucleophilic attack and formation of the enzyme–substrate complex. Changes in catalytic residues can disrupt enzymatic activity, leading to loss of function or altered kinetics. Identifying mutations in these residues provides insights into the mechanisms underlying enzyme dysfunction. | Mutations in catalytic residues can impair enzymatic activity, resulting in reduced substrate turnover or altered reaction kinetics. | Understanding the effects of catalytic residue mutations helps elucidate the molecular basis of enzyme dysfunction and guides the development of therapeutic strategies targeting these mutations. | [6,69] |
| Dimerization Interface | Investigation of changes in residues forming the dimer interface, affecting enzyme activity and stability. The dimerization interface plays a crucial role in maintaining enzyme structure and function. Mutations in this region can disrupt dimer formation, leading to monomerization or altered dimer stability. Understanding alterations in the dimer interface is essential for elucidating the impact on enzyme oligomerization and function. | Mutations at the dimerization interface may disrupt enzyme dimerization, leading to decreased enzyme stability or altered catalytic activity. | Knowledge of dimerization interface alterations informs strategies for stabilizing enzyme dimers or disrupting aberrant dimerization as therapeutic interventions. | [70,71] |
| Allosteric Sites | Analysis of modifications in sites distal from the active site, influencing enzyme activity through allosteric regulation. Changes in allosteric sites can allosterically modulate enzyme function, altering substrate binding affinity or catalytic activity. Identifying alterations in these sites provides insights into potential allosteric regulatory mechanisms and their impact on enzyme function. | Mutations in allosteric sites may affect enzyme regulation, leading to altered substrate binding or catalytic activity in response to regulatory signals. | Understanding alterations in allosteric sites elucidates mechanisms of enzyme regulation and provides opportunities for developing allosteric modulators to modulate enzyme activity. | [44,68] |
| Domain Arrangement | Examination of changes in the arrangement or conformation of structural domains, affecting enzyme function and stability. Alterations in domain arrangement can disrupt domain–domain interactions, affecting enzyme stability or catalytic efficiency. Understanding modifications in domain arrangement provides insights into structural changes that may impact enzyme function and stability. | Changes in domain arrangement may affect enzyme stability, alter domain–domain interactions, or disrupt catalytic activity. | Knowledge of domain arrangement alterations informs strategies for stabilizing enzyme structure or designing domain-specific inhibitors to target mutant enzymes. | [69,72] |
| Overall Structure | Assessment of global structural changes, including alterations in secondary structure elements and overall folding pattern. Changes in overall structure can impact enzyme stability, substrate binding, and catalytic activity. Analysing alterations in overall structure provides insights into the structural basis of enzyme dysfunction and the potential effects on enzymatic function. | Modifications in overall structure may lead to protein misfolding, decreased stability, or the loss of enzymatic function. | Understanding alterations in overall structure aids in identifying structural determinants of enzyme dysfunction and guides the development of strategies to restore enzyme function or stability. | [12,73] |
| Experimental Technique | Description | Advantages | Limitations | Ref. |
| X-ray Crystallography | Determines the three-dimensional structure of proteins, including mutant forms of 3CLPro, by analysing the diffraction pattern of X-rays passing through protein crystals. Provides detailed atomic resolution information about protein structure. | Provides high-resolution structural data, allowing the precise visualization of mutant 3CLPro conformations. | Requires protein crystallization, which can be challenging for some proteins and mutants. The technique is also time-consuming and requires access to specialized equipment and expertise. | [132,133] |
| Cryo-Electron Microscopy (Cryo-EM) | Utilizes electron microscopy to visualize biological samples, including mutant 3CLPro proteins, at cryogenic temperatures. Provides high-resolution images of protein structure, offering insights into conformational changes caused by mutations. | Enables the visualization of protein structures in near-native states, including flexible regions and large protein complexes. | Requires expensive equipment and significant expertise. Image processing and analysis can be complex, and resolution may be lower compared to X-ray crystallography for some samples. | [134,135] |
| Mass Spectrometry | Identifies and quantifies proteins, peptides, and posttranslational modifications in mutant 3CLPro samples. Enables the characterization of protein structure, stability, and interactions, as well as the detection of mutation-induced alterations. | Highly sensitive and versatile technique for analysing protein samples, including mutant forms of 3CLPro. | Requires specialized equipment and expertise. Data analysis can be complex, particularly for large proteins and complex samples. Sample preparation and handling may affect results. | [136,137] |
| Enzyme Activity Assays | Measures the catalytic activity of mutant 3CLPro enzymes by monitoring substrate turnover or product formation. Assesses the impact of mutations on enzyme function, substrate specificity, and catalytic efficiency. | Provides the direct assessment of mutant 3CLPro functionality, allowing the quantitative analysis of enzymatic activity. | May require optimization for specific mutants and conditions. The results may be influenced by assay conditions, substrate choice, and enzyme purification methods. | [52,138] |
| Circular Dichroism Spectroscopy (CD) | Analyses the secondary structure of mutant 3CLPro proteins by measuring the differential absorption of circularly polarized light. Provides information about protein folding, stability, and conformational changes induced by mutations. | Rapid and nondestructive technique for studying protein secondary structure and stability. | Limited to analysing protein secondary structure and may not provide detailed information about tertiary or quaternary structure. Requires careful interpretation of results. | [136,139] |
| Fluorescence Spectroscopy | Studies the structural and dynamic properties of mutant 3CLPro proteins by monitoring changes in fluorescence emission upon ligand binding or conformational transitions. Offers insights into protein stability, folding, and interaction dynamics. | Sensitive method for detecting changes in protein structure and dynamics. Can be used to study protein-ligand interactions and conformational changes induced by mutations. | Requires fluorescent labelling of proteins, which may affect protein function. Data interpretation can be complex, and results may be influenced by environmental factors. | [140,141] |
| Molecular Dynamics Simulations | Uses computational models to simulate the behaviour and dynamics of mutant 3CLPro proteins at the atomic level over time. Predicts protein structure, flexibility, and interactions, elucidating the effects of mutations on protein stability and function. | Allows the exploration of mutant 3CLPro behaviour and interactions at an atomic resolution, providing insights into dynamic processes. | Requires computational resources and expertise. The results may be influenced by force field parameters, simulation length, and initial protein conformation. Interpretation can be challenging. | [72,142] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushebenge, A.G.-A.; Ugbaja, S.C.; Magwaza, N.N.; Mbatha, N.A.; Muzumbukilwa, T.W.; Kadima, M.G.; Tata, F.Y.; Nxumalo, M.B.; Manimani, R.G.; Ndage, N.; et al. Mechanistic Insights into the Mutational Landscape of the Main Protease/3CLPro and Its Impact on Long-Term COVID-19/SARS-CoV-2 Management. Future Pharmacol. 2024, 4, 825-852. https://doi.org/10.3390/futurepharmacol4040044
Mushebenge AG-A, Ugbaja SC, Magwaza NN, Mbatha NA, Muzumbukilwa TW, Kadima MG, Tata FY, Nxumalo MB, Manimani RG, Ndage N, et al. Mechanistic Insights into the Mutational Landscape of the Main Protease/3CLPro and Its Impact on Long-Term COVID-19/SARS-CoV-2 Management. Future Pharmacology. 2024; 4(4):825-852. https://doi.org/10.3390/futurepharmacol4040044
Chicago/Turabian StyleMushebenge, Aganze Gloire-Aimé, Samuel Chima Ugbaja, Nonjabulo Ntombikhona Magwaza, Nonkululeko Avril Mbatha, Tambwe Willy Muzumbukilwa, Mukanda Gedeon Kadima, Fave Yohanna Tata, Mthokosizi Bongani Nxumalo, Riziki Ghislain Manimani, Ntabaza Ndage, and et al. 2024. "Mechanistic Insights into the Mutational Landscape of the Main Protease/3CLPro and Its Impact on Long-Term COVID-19/SARS-CoV-2 Management" Future Pharmacology 4, no. 4: 825-852. https://doi.org/10.3390/futurepharmacol4040044
APA StyleMushebenge, A. G.-A., Ugbaja, S. C., Magwaza, N. N., Mbatha, N. A., Muzumbukilwa, T. W., Kadima, M. G., Tata, F. Y., Nxumalo, M. B., Manimani, R. G., Ndage, N., Amuri, B. S., Byanga, K., Nlooto, M., Khan, R. B., & Kumalo, H. M. (2024). Mechanistic Insights into the Mutational Landscape of the Main Protease/3CLPro and Its Impact on Long-Term COVID-19/SARS-CoV-2 Management. Future Pharmacology, 4(4), 825-852. https://doi.org/10.3390/futurepharmacol4040044