Abstract
Hydrocephalus is caused by an overproduction of cerebrospinal fluid (CSF), an obstruction of fluid movement, or improper reabsorption. CSF accumulation in the brain’s ventricles causes ventriculomegaly, increased intracranial pressure, inflammation, and neural cell injury. Hydrocephalus can arise from brain trauma, hemorrhage, infection, tumors, or genetic mutations. Currently, there is no cure for hydrocephalus. Treatments like shunting and endoscopic third ventriculostomies are used, but, unfortunately, these therapeutic approaches require brain surgery and have high failure rates. The choroid plexus epithelium (CPe) is thought to be the major producer of CSF in the brain. It is a polarized epithelium that regulates ion and water movement from a fenestrated capillary exudate to the ventricles. Despite decades of research, control of electrolyte movement in the CPe is still not fully understood. This review discusses important transporters on the CPe, how some of these are regulated, and which of them could be potential targets for hydrocephalus treatment. To advance the development of hydrocephalus treatments, physiologically relevant preclinical models are crucial. This review covers some of the current animal and cell culture methods used to study hydrocephalus and highlights the need to develop standardized preclinical models that are used by multiple investigators in order to replicate critical findings and resolve controversies regarding potential drug targets.
1. Hydrocephalus
Hydrocephalus was first described in the 5th century by Hippocrates, where he was thought to have attempted treatment with ventricular punctures [1,2]. In the Middle Ages, the Arabic surgeon Abulcasis wrote a discourse on medicine that included the treatment of hydrocephalus. Advancements in the understanding of hydrocephalus occurred after Vesalius refined the knowledge of the anatomy and physiology of the brain in the mid-1500s. In the late 1600s, Thomas Willis first hypothesized that the choroid plexus was the source of cerebrospinal fluid (CSF). In the early 1800s, Magendie advanced the understanding of the brain by describing the flow of CSF throughout the organ. Even with these advancements, hydrocephalus therapies remained crude. Some of the treatments included head wrapping, bloodletting, and purgatives. In 1881, Wernicke introduced ventricular puncture with external CSF drainage. Soon after, the first permanent shunt was invented in 1893, which ushered in a revolution for hydrocephalus therapy [2]. The invention of artificial values and silicone in the 1960s opened the door for modern shunt technology that is still used today [2].
Hydrocephalus is a disease characterized by changes in CSF dynamics. It can be caused by excessive production of CSF, an obstruction impacting CSF flow, or decreased reabsorption. Hydrocephalus can occur with or without a change in intracranial pressure. In cases where the pressure is low or normal, it is thought that there is an expansion of the skull and/or compensatory contraction of cortical tissue and encephalomalacia [3,4]. While the roles of the glymphatic system, hydrodynamic changes, and vascular pathology [4,5] are all important considerations, these are not considered in this review. We have focused our attention on emerging therapeutic targets and novel avenues for drug development related to the choroid plexus.
Hydrocephalus can be categorized into two groups: communicating and non-communicating (Figure 1). When the CSF flow is unobstructed yet fails to absorb, or there is an overproduction of CSF, it is categorized as communicating hydrocephalus. Non-communicating hydrocephalus is characterized by a blockage of CSF flow due to various causes [6]. The increased CSF in the brain and changes in intracranial pressure can cause headaches, vision loss, vomiting, and nausea, and, if it is left untreated, it can be fatal [6]. Hydrocephalus is treatable; however, current treatments are suboptimal and a burden to not only people with hydrocephalus but their families and the healthcare system as well. A cross-sectional study looking at healthcare data from 2019 in the United States determined that across all patient demographics, the cost of shunting was around USD 2.06 billion [7]. This does not include any other costs associated with hydrocephalus care [7].
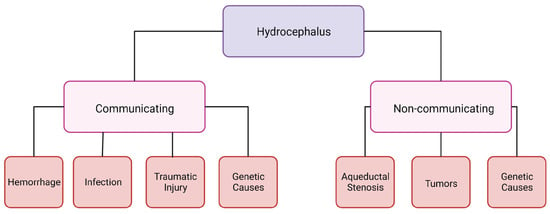
Figure 1.
Schematic of hydrocephalus categorization. Created with BioRender.com accessed on 26 January 2024.
Hydrocephalus is typically an acquired disease. Worldwide, the most common types of hydrocephalus are post-hemorrhagic and post-infectious hydrocephalus [8,9]. Pediatric post-hemorrhagic hydrocephalus occurs most commonly in preterm neonates born before 37 weeks of gestational age, especially in those that weigh less than 1500 g [10]. In adults, this condition is commonly associated with intraventricular hemorrhage. This can be caused by traumatic brain injury, hypertension, stroke, or aneurysm rupture [11]. Post-infectious hydrocephalus (PIH) is more common in developing countries, specifically Africa, Latin America, and Southeast Asia [9]. Tuberculosis and meningitis are common sources of infection in these regions due to increased risk of peripartum infection in neonatal critical care environments with limited resources. However, the sources of PIH are challenging to monitor due to limited access to advanced microbiological diagnostics. PIH is also a growing problem in resource rich medical environments and high-income countries. Indeed, a large spectrum of bacteria can be responsible including Toxoplasma gondii, Escherichia coli, Neisseria meningitidis, Streptococcus pneumoniae, Streptococcus agalactiae, and Listeria monocytogenes [12]. Prenatal or postnatal contraction of these bacteria, as well as other external pathogens, such as viruses (CMV), fungi, and protozoa, leads to infection and sepsis in the central nervous system that induces PIH.
There are also genetic mutations that can lead to either communicating or non-communicating hydrocephalus. In most cases, these genetic mutations cause congenital hydrocephalus. X-linked hydrocephalus makes up 5% of hydrocephalus cases at birth [13]. One form of X-linked hydrocephalus is caused by a mutation in L1, which affects the cell adhesion protein L1 cell adhesion molecule (L1CAM) [13]. Males with L1 syndrome have a phenotypic spectrum from mild to severe, but in all cases they are born with hydrocephalus, malformations in the corpus callosum, and intellectual disabilities [14]. Another genetic disorder, called syndromic craniosynostosis, is associated with hydrocephalus due to mutations in fibroblast growth factor receptor 2 (FGFR2) or fibroblast growth factor receptor 3 (FGFR3), leading to primary cerebral maldevelopment, brain atrophy, and structural obstruction of CSF outflow [13].
Neurofibromatosis 1 and 2 are autosomal dominant diseases that cause tumors in the central and peripheral nervous systems [13]. Tumors can lead to blockages in the ventricles and cause non-communicating hydrocephalus. Tuberous sclerosis is also an autosomal dominant disorder that causes subependymal giant cell astrocytomas that can lead to non-communicating hydrocephalus [13]. Walker–Warburg syndrome is an autosomal recessive disorder that can be associated with hydrocephalus. The disease is most commonly caused by mutations in protein O-mannosyl-transferase 1 (POMT1), but it can also be caused by defects in protein O-mannosyl-transferase 2 (POMT2), protein O-linked mannose N-acetylglucosaminyltransferase 1 (POMGNT1), acetylglucosaminyltransferase-like protein (LARGE), isoprenoid synthase domain containing (ISPD), fukutin (FKTN), or fukutin-related (FKRP) [13]. Mutations in the coiled-coil domain containing 88C (CCDC88C) cause non-syndromic autosomal recessive hydrocephalus due to defective neural tube development [15].
These are the major syndromes that are known to cause hydrocephalus, but there are many more mutations that can cause the disease. One study showed that there are almost 1500 genes that could be associated with hydrocephalus. These authors used a genome-wide association study (GWAS) on CSF and blood samples from patients with hydrocephalus [16]. This study, and studies like it, are laying the groundwork to discover more genetic causes of hydrocephalus.
A meta-analysis of hydrocephalus cases worldwide determined that per 100,000 individuals, there are 88 cases of hydrocephalus in patients under the age of 18 [17]. A child may be diagnosed with the disease for various reasons like preterm birth resulting in intraventricular hemorrhage, infection, some medications taken during pregnancy, genetic syndromes, aqueductal stenosis, or tumors [18,19]. Globally, the most common cause of pediatric hydrocephalus is infection, while in the United States, the most common cause is post-hemorrhagic hydrocephalus of prematurity [19]. It is estimated that 383,000 children will be born with hydrocephalus per year, worldwide [9].
Hydrocephalus is the least common in middle-aged adults. In the above-mentioned meta-analysis of worldwide cases of hydrocephalus, the authors reported that in adults aged from 19 to 64 years, there are 11 cases of hydrocephalus for every 100,000 individuals [17]. The causes of hydrocephalus in adults have some overlap with the causes in children, like infection, trauma, and hemorrhage.
The elderly are the most severely affected group. The worldwide meta-analysis found that in people over the age of 64, there are 175 cases per 100,000 people [17]. The most common type of hydrocephalus diagnosed in the elderly population is idiopathic normal pressure hydrocephalus (iNPH). In fact, one study estimated that 5.9% of individuals 80 years of age or older have iNPH [20]. This disease can be easily overlooked because the symptoms of iNPH are similar to the symptoms of aging in general, including urinary incontinence, gait disturbances, and cognitive dysfunction (which can progress to dementia) [20].
2. Hydrocephalus Treatments
Currently, the insertion of a shunt is the most common treatment for hydrocephalus (Figure 2, Table 1). In the simplest form, shunts consist of a ventricular catheter that is surgically implanted into one of the lateral ventricles of the brain, a valve that regulates the CSF flow out of the brain, and an additional catheter that empties the CSF into a body cavity. CSF is shunted into the peritoneal cavity, the pleura, and the atrium of the heart. Ventriculoperitoneal shunts (VPSs) are by far the most common, and CSF is diverted to the peritoneal space [21].
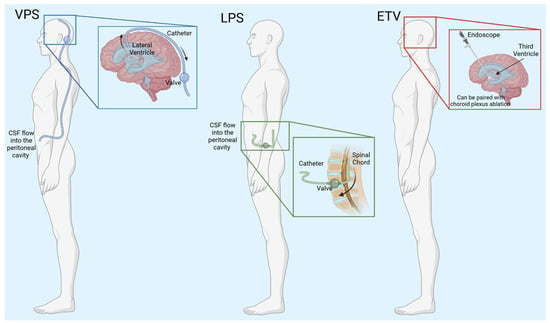
Figure 2.
Schematic of hydrocephalus treatments. VPS—ventriculoperitoneal shunt, CSF—cerebrospinal fluid, LPS—lumboperitoneal shunt, ETV—endoscopic third ventriculostomy. Created with BioRender.com accessed on 7 March 2024.

Table 1.
Benefits and complications associated with current hydrocephalus treatments.
VPSs are used to treat many types of hydrocephalus, but the shunts have varying results. When used to treat iNPH, they can successfully relieve symptoms, such as gait disturbance, urinary incontinence, and cognitive dysfunction, in 60–80% of patients [22]. However, in children, shunts fail with a frequency of 50% within the first two years [23]. Even with advances in shunt technology, the failure rate remains the same. An analysis of literature found that pediatric shunt failure rates did not improve in the thirty years from 1970 to 2000 [24]. Indeed, shunts can fail for multiple reasons such as mechanical failure, catheter migration, infection, or blockages caused by cell overgrowth [23,24]. Every shunt failure necessitates another surgery which increases the chance of infection or death for the patient [23].
Another type of shunt is the lumboperitoneal shunt (LPS). The shunt is placed into the lumbar region of the spinal cord to remove excess CSF to treat communicating hydrocephalus. LPS is beneficial because it is not necessary to have brain surgery and greatly decreases the chance of brain hemorrhage [25]. A meta-analysis performed in 2023 showed that LPS have a lower complication rate compared to VPS. Out of 3654 patients, there was a 12.98% complication rate with the patients with LPSs and 23.8% complication rate in the patients with VPSs [25]. Since their inception, LPS techniques have been improving. A new two-stage LPS procedure was safer for the patient and still outperformed VPS. Unlike a typical LPS procedure where the patient remains in one position throughout the surgery, this new technique breaks the surgery into two phases in which the patient goes from the prone position to the supine position [26]. In this study, only 1 out of 92 LPS needed revision, while 26 out of 192 VPS surgeries needed revision, and 11 surgeries had infections [26]. On the downside, LPS cannot be used as a treatment for non-communicating hydrocephalus.
Another option for treating hydrocephalus in some patients is an endoscopic third ventriculostomy (ETV). This involves neurosurgery to create an opening in the floor of the third ventricle, which allows CSF drainage into the subarachnoid space [27]. This approach may circumvent shunt dependence and can be especially beneficial in selected patients when access to emergent neurosurgery for shunt revision and/or failure is not available [28]. Indeed, it can be an alternative treatment but is highly dependent on patient age and the etiology of hydrocephalus. For example, ETV is not successful in the majority of infants younger than 1 year [29]. Kulkarni et al. created a predictive model of ETV success rates in children based on 618 ETV surgeries and discovered that if the child is under one year of age, there is only a 30% success rate for ETV [30]. Patients 10 years or older have higher success rates [30].
Choroid plexus cauterization (CPC) can also be combined with ETV for the treatment of hydrocephalus. In these cases, specific ages of patients with specific forms of hydrocephalus are more likely to benefit from ETV + CPC [31,32]. Specifically, among centers participating in the Hydrocephalus Clinical Research Network (HCRN), ETV + CPC success rates were 48% at 6 months and 44% at 2 years from 2006 to 2015 [31]. Those most likely to benefit were infants with myelomeningocele, infants with aqueductal stenosis older than 1 month corrected age, and infants older than 6 months who suffered IVH secondary to preterm birth [31]. In a study that compared ETV + CPC with VPS outcomes in children under 12 months of age, the Kaplan–Meier survival analysis showed that the estimated need for reoperation was lower in the ETV + CPC group [33]. The study estimated that 59% of the patients would avoid shunt dependency for a length of 11 years [33]. However, the long-term effects of ablating the CP, which does not regenerate, have not been studied. Notably, even after successful treatment of post-infectious hydrocephalus of infants living in sub-Saharan Africa, early post-treatment brain growth is delayed through the second year [34].
Non-surgical therapies designed to prevent and/or treat existing hydrocephalus have been explored. Unfortunately, none have proved efficacious or fully translated from preclinical studies thus far. Past pharmacologic agents were primarily diuretics and were designed to restore osmotic and fluid homeostasis. A previous review provides a historical synopsis of these trials [6]. While acetazolamide and furosemide have been shown to be important for fluid management in specific clinical scenarios, these approaches have not proven beneficial to treat or prevent existing hydrocephalus. Similarly, oral cardiac glycosides, which inhibit Na + K + ATPase [35], have failed to translate despite the putative ability to decrease CSF production.
Emerging therapies have been focused on the modulation of CSF dynamics as a whole and immunomodulation in the context of neurorepair and protection. Specifically, approaches that target systemic and neuroinflammation secondary to CSF stagnation after IVH, ventriculomegaly and associated white matter and cortical health, and elevated intracranial pressure have been designed. An example of this is a cocktail of erythropoietin and melatonin (EPO + MLT). EPO alone has been shown to help improve cognitive function in many forms of brain injury [36]. In preclinical studies of PHHP, EPO + MLT were able to reduce ventriculomegaly and macrocephaly [37]. Interestingly, the combination treatment restored previously damaged motile cilia in the brain ventricles. The rats that underwent treatment also showed improved neurodevelopment compared to control animals [37]. In another study, EPO + MLT was used to restore gait in adult rats that had brain injury secondary to chorioamnionitis [38]. Notably, a phase I clinical trial of EPO + MLT for preterm infants with severe intraventricular hemorrhage is currently underway [39].
3. Induced Animal Models
Testing potential pharmaceutical interventions requires well-characterized preclinical models. Animal models have been used to study multiple forms of hydrocephalus (Table 2).
Table 2.
Current animal models used for studying hydrocephalus.
Post-traumatic hydrocephalus caused by traumatic brain injury (TBI) can be modeled in animals. Zhao et al. used a fluid percussion device to induce TBI in adult rats [40]. After the initial injury, they injected the animals with FeCl3. This modeled intracranial hemorrhage, which is common in patients with TBI. The rats developed acute hydrocephalus within a 24 h period [40].
Models of post-hemorrhagic hydrocephalus have also been developed, including in rodents, pigs, and dogs [10]. Similar to what occurs clinically, blood is injected into the cerebral ventricles in these models [10]. In other experiments, blood components, like red blood cells, iron, or hemoglobin, have also been effective [10]. These techniques are not guaranteed to produce hydrocephalus every time. For example, Pang et al. attempted to make 10 post-hemorrhagic models of hydrocephalus in dogs, but only 8 of those caused ventriculomegaly after 3 months [41]. The techniques can be performed on young animals to model intraventricular hemorrhage secondary to preterm birth. In a study by Robinson et al., lysed red blood cell components were injected bilaterally in cerebral ventricles in postnatal day 1 rat pups after in utero exposure to intra-amniotic and placental inflammation (chorioamnionitis) [37]. An alternative method of inducing a germinal matrix hemorrhage that avoids intraventricular injection is the technique of intraperitoneal injection of glycerol in premature rabbit pups [42]. This method produced post-hemorrhagic ventriculomegaly in 42% of the pups.
Hydrocephalus can also be induced chemically, and one popular reagent is kaolin, which has been used in experiments since the 1950s [43,44]. One benefit of using kaolin is that it is inexpensive and relatively reliable in inducing hydrocephalus. In a recent experiment with juvenile pigs, the animals were injected with kaolin into the intracisternal region to produce bilateral ventriculomegaly [45]. While the severity of hydrocephalus varied among the animals, it proved to be a reliable model that was used to test shunting techniques [45]. Kaolin has been successfully used in rodents, dogs, cats, lambs, and primates [46,47,48,49,50]. One drawback of using kaolin to induce hydrocephalus is that it may kill some animals immediately after injection [46]. Depending on the animal model being used, it can take some time and effort to determine the best non-lethal concentration of the drug. Another downside of chemically induced hydrocephalus is that it does not model any naturally occurring form of the disease. Non-specific, caustic chemicals will not just ablate the choroid plexus epithelium (CPe), but also damage the ventricle lining.
6-aminonicotinamide (6-AN)-related hydrocephalus rats are used as another chemically induced model. 6-aminonicotinamide can be injected into the brain to induce symptoms similar to those described in Dandy–Walker syndrome [51]. 6-AN can also be injected into a pregnant animal to induce hydrocephalus in the fetus(es) [52]. Dandy–Walker syndrome causes malformations in the brain, particularly the cerebellum [53]. Almost 80% of patients with Dandy–Walker syndrome develop hydrocephalus [53]. 6-aminonicotinamide treatment mimics this syndrome by inducing cerebellum hypoplasia and other brain malformations [52]. When rats are treated with 6-aminonicotinamide, ventriculomegaly is visible after 72 h [54]. This is a useful model to replicate human disease; however, this is another chemically induced method that will cause non-specific damage to the brain.
4. Genetic Animal Models
The Wpk rat, also known as the Tmem67−/− rat, models pediatric communicating hydrocephalus [55,56]. The rats have a single nucleotide mutation in the TMEM67 protein which is essential for the development of primary cilia [55]. This model is orthologous to the human disease Meckel–Gruber syndrome type 3 [55]. The homozygous phenotype is characterized by severe hydrocephalus and polycystic kidney disease [55,56]. Using this genetic model has some limitations because the animals do not live very long. The homozygous phenotype is so severe that the animals typically die 18–21 days after birth [56]. These animals have been used successfully to test the efficacy of transient receptor potential vanilloid-type 4 (TRPV4) antagonists [70] and serum glucocorticoid-induced kinase 1 (SGK1) inhibitors [57].
Hydrocephalus Texas rats (H-Tx) are a good model for studying non-communicating hydrocephalus in rodents. The rats have defective cortical development and have aqueductal stenosis with detectable hydrocephalus around day 18 [71]. The lateral ventricles of the brain are enlarged, and, in some animals, the third ventricle is also enlarged [72]. It was noted that the aqueducts of the brain were abnormal due to a thickening of the midbrain [72].
LEW/Jms rats have a mutation that leads to the development of hydrocephalus immediately after birth [58]. It was noticed that more male than female rats developed symptoms, so the genetic cause may be sex-linked [58]. This model could be used to study human diseases like X-linked hydrocephalus. The L1CAM mouse is very similar to the LEW/Jms model. This mouse model lacks the L1 gene, which encodes L1CAM. This protein is a cell adhesion molecule that is important for axon growth, neuronal migration, and synaptic plasticity [59]. Mice without L1 have improper brain development and the disease disproportionately affects males [59]. This model can be used to study X-linked hydrocephalus and L1 syndrome.
The E2F5 mouse line is a homozygous knockout of the E2F5 transcription factor, which is important for the choroid plexus maturation and cilia function [60]. The knockout line has a cilia malfunction that leads to the hydrocephalus phenotype, which develops gradually [61]. The affected animals have normal size and appearance compared to their wildtype counterparts until they are 3–4 weeks of age [62]. This mutation is lethal, with the mutant mice living an average of 6 weeks [62].
Growth arrest specific 8 (Gas8) is another genetic mouse model used for hydrocephalus. Gas8 is important for hedgehog signaling and regulating cilia movement [63]. This gene also has some clinical significance. There are at least two human patients with missense mutations in Gas8 that lead to hydrocephalus: A391V and E199K [63]. Preliminary results indicate that, dependent on the background, Gas8 mice develop hydrocephalus between postnatal days 12 and 16, making it a good model for pediatric hydrocephalus [64].
The coiled-coil domain containing 39 (CCDC39) mouse model causes neonatal hydrocephalus with abnormal motile cilia development [65]. The mice have short, thin cilia that cannot beat properly because of the loss of CCDC39. Interestingly, they also have situs inversus. They have normal brain development at birth but then progressively develop hydrocephalus until it becomes fatal at 3 weeks of age. This model mimics the timing of human congenital hydrocephalus [65].
Autosomal recessive mutation hydrocephalus-3 (Hy-3) mouse is a genetic mouse model that produces communicating hydrocephalus [66]. The mice have a mutation in the hydin protein, which is a homolog to the human protein caldesmon. This protein is important for the cellular cytoskeleton. The mice develop hydrocephalus shortly after they are born. Is it thought that they develop communicating hydrocephalus due to loss of cilia [66].
Hydrocephalus with the hop gait mouse (Hyh) model has a lethal recessive mutation on chromosome 7 that leads to hydrocephalus [67]. Homozygous mice have characteristic domed heads of hydrocephalus and develop a unique hopping gait approximately two weeks after birth. The mice die between 4 and 10 weeks of age [67]. This model can be used for genetic hydrocephalus as well as non-communicating hydrocephalus because there is a blockage of CSF movement in the fourth ventricle [67]. A similar model known as hydrocephalus associated with polydactyly mice (Hpy) has a recessive mutation that causes hydrocephalus. The mice have hopping gait, sterility in male mice, and scoliosis [68]. The mice develop communicating hydrocephalus around day 6 after birth, and most die around day 14 [68]. However, the specific cause of hydrocephalus is unknown.
Mf1 mice model congenital hydrocephalus. Mesoderm/mesenchyme forkhead 1 (Mf1) is a gene that encodes winged helix proteins that are DNA-binding proteins [75]. Mice with the Mf1 knockout die perinatally from hemorrhagic hydrocephalus. They also have issues in other parts of the body where Mf1 is expressed. There are eye abnormalities and skeletal defects [75]. Similar to Mf1 mice, hemorrhagic hydrocephalus mice (Hhy) develop hydrocephalus due to a recessive genetic mutation [51]. The symptoms of hydrocephalus occur between 3.5 and 19 weeks of age, with the majority of mice developing hydrocephalus by 15 weeks of age [73]. These models are useful for hemorrhagic hydrocephalus because there is no need to induce an injury or inject blood components into the animals to obtain the desired results.
Rho family GTPase 3 (Rnd3) is important for regulating cell migration and proliferation. When this protein is absent, in mice, the cerebral aqueduct is blocked, resulting in non-communicating hydrocephalus [74]. Mice with this mutation are described as having extreme dilation on the third ventricles but not the fourth, thick ependymal cell layers, and upregulated Notch signaling [74].
The Msx1 mouse models have malformations of the brain that result in the collapse of the cerebral aqueduct [51]. The symptoms of hydrocephalus start to develop before birth due to the deletion of msh homeobox 1 (Msx1). The function of the Msx1 protein is not fully understood, but it is thought to be involved with the development of the fasciculus retroflexus [76]. The mice are characterized with brain abnormalities, such as reduced corpus callosum, cerebral cortex, caudate putamen, and septum. Some of the homozygous mutant mice have ventriculomegaly in the third ventricles. The main cause of hydrocephalus in this animal model is thought to be ependymal detachment in the developing embryos [76].
The SUMS/NP mouse is a genetic model of congenital hydrocephalus that starts to develop 14 days after gestation [69]. The hydrocephalic phenotype is not noticeable until 4–5 days after birth, when there is a significant increase in the lateral ventricles. These mice also have absent or reduced cerebral aqueducts. This mouse model does not live very long; they typically die soon after they are weaned [69].
With the ease of use of CRISPR, genetic manipulation of the CPe is possible. There are also many genetic knockout lines that can be created to study hydrocephalus in vivo, and a recent excellent review by Jang and Lehtinen covers many of them [77]. Viral vectors are another method to genetically manipulate the CPe. Viral vectors can change gene expression within 24–48 h without the need for breeding [77]. They can be used in many stages of life in animals, including in utero [77].
Animal models are useful for understanding the physiology of hydrocephalus and testing potential drug treatments, but they do have some drawbacks. Animal models do not perfectly model the human brain. The size of the ventricles varies across species, and some animal CPe cells may have different ion channels compared to humans. For example, it has been reported that mice CPe cells have a sodium transporter known as the epithelial Na+ channel (ENaC), while humans cells do not [78]. The animal models may not represent the symptoms of hydrocephalus perfectly and they may not respond to drug treatments in the same way as humans. Large animal models that more closely mimic the human brain are very costly. Using cell culture models in conjunction with animal models can help reduce confounding factors and provide depth to the experimental reliability. Arguably, the primary limiting factor in the field of hydrocephalus research is the sheer number of animal models and the lack of comparisons of models between laboratory groups. There are no standard models in the field, and this is a detriment to comparison of research results.
5. The Choroid Plexus Epithelium
One major target in the search to find treatment options for hydrocephalus has been the functionality of the choroid plexus. The CPe is primarily responsible for the production of CSF [78]. The CPe is a monolayer of epithelial cells that resides in all four ventricles of the brain and receives nutrients and substrates from a fenestrated capillary network it surrounds. Epithelial cells contain tight junctions, desmosomes, and adherent junctions, thus forming the CSF–blood barrier [79,80]. The cells are cuboidal in shape and are ciliated [79,80]. Interestingly, in the CPe, many of the crucial transport proteins are polarized in a manner that is different from most other epithelial cells. Typically, ion transporters like Na+-K+-ATPase, sodium potassium chloride co-transporter 1 (NKCC1), and Na/H exchanger 1 (NHE1) are located in the basolateral membrane [79,81]. On the CPe, these are located apically facing the CSF [79,81]. There are also some transporters found on the basolateral side of the CPe that are normally found on the apical side. These include anion exchanger 2 (AE2), K+-Cl− co-transporter (KCC3), and Na+-HCO3− co-transporter 1 (NBCn1) [79].
It is estimated that the adult human brain contains about 150 mL of CSF, with 125 mL located in the cranial and spinal subarachnoid spaces and the remaining 25 mL in the ventricles [82]. The CPe is thought to secrete around 80% of the total CSF, while the remaining 20% is produced by leakage across the blood–brain barrier [79]. A healthy human adult produces between 400 and 600 mL of CSF per day [82]. The CPe produces CSF by controlling transepithelial electrolyte and water flow, which has been reported to be independent of the trans-epithelial osmotic gradient [83]. Control of electrolyte and water movement in the CPe is still not fully understood.
Along with producing CSF, the CPe has many other functions. One function is removing waste products from the CSF, including β-amyloid, phosphatidylinositol, and uric acid [81]. The CPe is also important for immune function. It is a bridge where immune cells can cross into the CSF from the blood [81]. Additionally, it acts as a physical barrier so that some pathogens cannot enter the CSF [81]. The CPe is also an important entry point for drug delivery to the brain [81].
6. Cell Culture Models
Primary cultures derived from the CPe have been obtained from many species of animals, including amphibians [84], mice [85], rats [86], rabbits [87], pigs [88], sheep [89], and cows [90]. While these studies have provided much useful information, the downside to primary cell culture is that it can be difficult to obtain pure cultures and that the cultures have short viability [91]. The CPe must be removed soon after the animal is sacrificed, and the cells need to be purified. Additionally, there is usually low cell abundance, so many animals are required for cell culture [91].
CPe cell cultures can also be obtained from biotech companies. The human choroid plexus epithelial cell line (HCPEpiC) can be purchased from different companies and has been referenced in publications. For example, this cell line has been used to study the inflammatory response [92] and the effects of chemotherapy on CPe cells [93].
As an alternative to primary cultures, there are some continuous CPe cell lines (Figure 3). The porcine choroid plexus–Riems (PCP-R) cell line has been used to model the CPe [94]. It has been used to identify ion channels on the CPe using Ussing-style electrophysiology and to study changes in barrier function. Recent studies have, however, indicated that the PCP-R is mispolarized, meaning that the ion channels are on the opposite side of the cell when compared to the native epithelium [95]. Therefore, this cell line is not ideal for studying CSF production. However, this cell line is still useful for other experiments. For example, the PCP-R has been used to test what xenobiotic drugs can cross the blood–CSF barrier [88]. Similarly, it has been used to study immune cell movement across the blood–CSF barrier [96]. The cells also contain tight junctions like Claudin-1 and Claudin-2 [94]. This cell line has high transepithelial electrical resistance (TEER), which is typical of mammalian choroid plexus cells [94,97]. On average, the TEER was approximately 1000 Ω cm2 when the resistance was measured with Ussing-style electrophysiology [95].
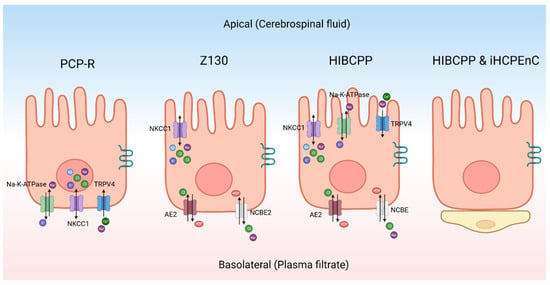
Figure 3.
Schematic of different choroid plexus models. The transporters and channels indicated in each cell are not meant to be comprehensive but to indicate those proteins that have been conclusively identified. PCP-R—porcine choroid plexus–Riems, NKCC1—sodium potassium chloride co-transporter 1, TRPV4—transient receptor potential vanilloid-type 4, NCBE2—sodium-driven chloride bicarbonate exchanger 2, HIBCPP—human choroid plexus papilloma, AE2—anion exchanger 2, NCBE—sodium-driven chloride bicarbonate exchanger, iHCPEnC—immortalized human choroid plexus endothelial cell. Created with BioRender.com.
The Z130 cell line is an immortalized rat CPe cell line that was created by transfecting simian virus 40 (SV40) large T into primary cultures [98]. When compared to primary rat cell cultures, the morphology was the same and it was able to mimic the blood–cerebrospinal fluid barrier [98]. The polarity of the cells are the same as primary rat cultures [98,99]. Z130 cells have tight junctions but can only maintain a maximum TEER value around 100 Ω cm2 [100]. Z130 cells have been used to study how heavy metals cross the CPe along with the toxic effects of these metals [101].
The human choroid plexus papilloma (HIBCPP) is an immortalized cell line that was derived from a malignant papilloma in the choroid plexus of a 29-year-old Japanese woman [102]. It has been characterized to have structures similar to the CPe-like villus structures located apically [97]. The important transport proteins are correctly polarized with reference to the native epithelium. Notably, TRPV4 and NKCC1 are located apically [97], while the basolateral side of the cell contains the sodium-driven chloride bicarbonate exchanger (NCBE) and AE2 [97]. Both sides of the cell have aquaporin channels [97]. This cell line has a moderately high resistance when it is grown in a monolayer with TEER measurements over 400 Ω cm2 [97]. This cell line can be used for many techniques, including Ussing-style electrophysiology. The downside to this cell line is that it is relatively difficult to maintain in culture.
Another interesting model is the co-culture of the immortalized human CP endothelial cell (iHCPEnC) line with HIBCPP cells. Cell types are cultivated on opposite sides of the same permeable support to produce a combined CP model [103]. The addition of the endothelial cells increases the tightness of the epithelial cells and more closely mimics the conditions found in the brain [103]. Using tracer voltohmmeter technology, the authors measured the TEER of the combined cell culture which reached a max of 700 Ω cm2 [103].
CP organoids can be derived from stem cells. Interestingly, the organoids can produce CSF-like fluid. When the fluid from the organoid was compared with CSF proteins from human patients, the same proteins were present [104]. This provides an important experimental tool that allows investigators to explore the transporters and subcellular signaling pathways that are responsible for CSF production and composition. Organoids derived from human cells provide a species specificity that is key to understanding CP-related diseases in humans. Another group made a CP organoid that had a similar morphology and expressed the genes LIM homeobox transcription factor 1 alpha (LMX1A), orthodenticle homeobox 2 (OTX2), and aquaporin 1 (AQP1), which are also expressed in the CP [93]. Again, these findings provide a model that can be used for the study of the regulatory processes important for CP function. There are some limitations for organoids. They are expensive and time-consuming. There are also different protocols that produce organoids of different quality and composition [93]. These inconsistencies make it difficult to compare studies from different research groups. However, this technology is developing rapidly and will likely provide an important addition to preclinical models in the near future.
7. Electrolyte Transporters, Channels, and Pumps: Potential Roles in Fluid/Electrolyte Homeostasis
The main driving force for the transepithelial ion flux is Na+ [84,105]. In one potential scenario, Na+ enters the CPe from the bloodstream by the NCBE and Na+-HCO3− co-transporter 1 (NBCn1) channels. Subsequently Na+-K+-ATPase pumps Na+ into the CSF [79,81,84]. The Na+-K+-ATPase is also important for K+ homeostasis. It removes excess K+ from the CSF by pumping the ions into the cell [106]. It has been suggested that the potassium voltage-gated channel subfamily Q member 1 (KCNQ1) and potassium voltage-gated channel shaker-related subfamily member 3 (KCNA3) can join with potassium voltage-gated channel subfamily E regulatory subunit 2 (KCNE2) to form K+ channels on the basolateral side of cell to excrete K+ into the blood [107]. The K+ Cl− co-transporter KCC3 also moves K+ from the CSF into the bloodstream [108]. While these multiple K+ channels have been implicated, which are critical, regulated transporters and channels remain unknown.
NKCC1 is a co-transporter that regulates the movement of Na+, K+, and two Cl− ions across the CPe apical membrane [79]. The directionality of the electrolyte transport as well as the ability of the protein to transport water is controversial. For a synopsis of the controversy surrounding the directionality of the flux, the reader is referred to a point–counterpoint exchange in the Journal of Physiology [109,110,111,112]. More recently, data suggest that the direction of electrolyte movement may be heavily influenced by local ion concentrations [113] or the developmental stage [114]. It was previously reported that around 600 molecules could be moved by NKCC1 [115], but recent studies suggest that the transporter only moves around 86 water molecules [116]. The NKCC1 is not only expressed on the CPe but in many tissues where it is important for maintaining cell volume [117,118,119]. When NKCC1 is mutated or knocked out, it causes severe disease symptoms [120]. However, NKCC1 knockout rodent models do not have altered ventricle sizes, but they do have brain malformations and smaller CPe cells compared to their wildtype counterparts [120,121]. There are some rare cases where human patients are deficient in NKCC1 due to a mutation in solute carrier family 12 member 2 (SLc12a2). Since NKCC1 is found throughout the body, patients suffer from multiple serious problems, including severe intellectual disability, hearing loss, respiratory weakness, and gastrointestinal issues [120,122].
There are many groups that have investigated NKCC1’s role in CSF production, but their conclusions are conflicting. For example, when NKCC1 is pharmacologically blocked during inflammatory conditions in rats, less CSF is produced by the CPe [123]. However, when NKCC1 was overexpressed in a neonatal post-hemorrhagic hydrocephalic mouse model, CSF production diminished, and the brain ventricle size returned to normal [124]. Another study investigated the role of NKCC1 under normal conditions in a rat model and found that when NKCC1 was blocked, CSF production was not reduced [125]. One explanation for these diverse findings is that the transporter can adapt to maintain homeostasis. NKCC1 seems to be able to move ions to either side of the membrane depending on the external/internal environment [113,119]. Additional studies will be required to determine the factors involved in the control and mechanism of NKCC1 in the CPe. One way to investigate the role of NKCC1 would be to test cultured CPe cells for fluid secretion in different conditions. The recently characterized human cell line, HIBCPP, provides a unique epithelium in which fluid secretion can be easily determined [57]. Culturing the CPe in vitro removes any confounding factors and can help determine the role of NKCC1 in CSF production.
Pharmacologically blocking NKCC1 has been investigated in preclinical models. However, before this can be translated into the clinical sphere, better NKCC1 antagonists need to be developed. Bumetanide is a common clinical inhibitor, but this drug has very low brain penetration [126]. Therefore, it needs to be infused directly into the ventricles for efficacy in decreasing CSF production [123]. Additionally, this drug affects both NKCC1 and NKCC2 and therefore has substantial renal effects, causing excessive diuresis [126]. Some other symptoms include low K+ and Cl− in the blood, metabolic alkalosis, and elevated levels of uric acid in the blood. There has been some work carried out to create better NKCC1 inhibitors, and a recent review written by Savardi et al. covers a few of them [126].
TRPV4 is a channel that is osmo-, shear-, temperature-, and pressure-sensitive [127,128,129,130,131,132]. Activation results in a Ca2+ and Na+ influx into the CPe and, secondarily, causes a substantial change in transepithelial permeability and an electrolyte flux that appears to involve multiple transporters [97]. This channel may play a role in CSF production in pathological states. In an experiment with Tmem67−/− rats, a genetic form of hydrocephalus was reversed with a treatment with the TRPV4 antagonists HC067047 and RN 1734 [70]. Blocking TRPV4 with an antagonist may be a promising form of treatment for communicating hydrocephalus. TRPV4 knockout mice have no obvious physiological or behavioral phenotypes under normal conditions [132,133]. Therefore, using a TRPV4 antagonist would likely be safe for patients. In fact, oral TRPV4 antagonists were tested in clinical trials as a treatment for heart failure [134]. The drug, GSK2798745, was well tolerated by healthy and ill patients alike [134,135].
There may be a relationship between TRPV4 and NKCC1. When TRPV4 transports Ca2+ into the cell, WNK (with no lysine kinase) may be stimulated. A study in human salivary glands found that WNK can be activated when there is an increase in the cellular levels of Ca2+ [136]. WNK then phosphorylates the SPS1-related proline/alanine-rich kinase (SPAK), which secondarily activates NKCC1 [106,137].
KCC3 is associated with inherited forms of hydrocephalus. When the channel is mutated, it leads to decreased K+ transport in the brain [79,138]. KCC3 dysfunction is also associated with Mendelian disease agenesis of the corpus callosum with peripheral neuropathy, which causes malformation of the corpus callosum [139]. NKCC1 is related to KCC3, with both ion channels being regulated by the WNK/SPAK pathway [140]. KCC3 is inhibited by WNK/SPAK activation. When it is phosphorylated, it moves Na+ and Cl− ions out of the cell. It is thought that KCC3 can be phosphorylated to increase cell volume, or that it can be dephosphorylated to decrease cell swelling [140,141,142].
The cystic fibrosis transmembrane conductance regulator (CFTR) is another transport protein whose role in the CPe is controversial. CFTR secretes Cl− ions from epithelial cells [143,144]. It has been reported that there is no CFTR mRNA in the CPe of rats [78,79]. However, unpublished data from our laboratory indicates the presence of CFTR in both human and porcine epithelial cells at the mRNA level and an effect of CFTR inhibitors on the transepithelial ion flux. In other tissues of the body, like the lungs, TRPV4 has been shown to be important for activating CFTR [145].
Additionally, the CPe is important for pH regulation. There are many acid–base transporter proteins like the NCBE (Na+-driven chloride bicarbonate exchanger), AE2, and NBCE2 (Na+-driven bicarbonate co-transporter) [79]. Other important transporters for pH homeostasis include acid–base transporters Cl−−/H+, sodium/hydrogen exchanger 6 (NHE6), and chloride/proton exchanger 7 (CLC-7) [80].
NCBE may be an important transporter for understanding the pathophysiology of hydrocephalus (Figure 4). The transporter is likely regulated by inflammation in the brain. When mouse CPe cells were treated with inflammatory cytokines TNFα and IL-1β, NCBE expression decreased [146]. Inhibiting carbonic anhydrase with acetazolamide prevents CSF secretion, pointing to the importance of HCO3- in CSF production [147]. There may be interplay between NCBE and multiple transporters that impact CSF secretion. When the gene responsible for NCBE, solute carrier family 4 member 10 (Slc4a10), is knocked out in mouse models, aquaporin 1 and Na+-K+-ATPase expression is drastically decreased [148]. Additionally, the WNK/SPAK pathway regulates the functional activity of NCBE [149]. It was shown in pancreatic epithelial cells that SPAK inhibits the expression of the sodium-dependent bicarbonate transporter NBCE1-B [150]. As previously discussed, the WNK/SPAK pathway regulates ion channels like TRPV4 and NKCC1 that may be important for the regulation of CSF. It has also been noted that mice with Slc4a10 mutations have small ventricles [148,151]. Humans with de novo Slc4a10 mutations have small ventricles as well as cognitive impairments and epilepsy [152,153].
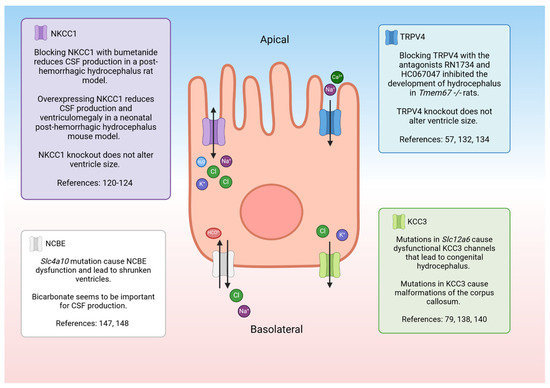
Figure 4.
Schematic of a choroid plexus cell with the ion transporters that may be involved with hydrocephalus. NKCC1—sodium potassium chloride co-transporter 1, CSF—cerebrospinal fluid, TRPV4—transient receptor potential vanilloid-type 4, KCC3—potassium chloride co-transporter 3. Created with BioRender.com accessed 7 March 2024 [57,79,120,121,122,123,124,132,134,138,140,147,148].
Even though the CP is a small tissue, it is essential for the production of CSF. CPe cells are uniquely polarized so that they can produce large amounts of CSF to maintain homeostasis. The CPe is able to complete this task by precisely controlling the flow of electrolytes and water with the help of a complex system of ion channels, transporters, pumps, and their intracellular regulators. Some of these electrolyte channels, specifically TRPV4, NKCC1, NCBE, and KCC3, seem to be involved in CSF production and ventricle size. These channels as well as the biochemical pathways that regulate their activity are prime targets to explore in the search for potential pharmaceutical interventions to treat hydrocephalus. However, the lack of consensus as to which electrolyte transporters and channels are the primary drivers of CSF production and composition has a limiting effect on drug development. An increased understanding of the pathways that regulate CSF production is also critical for a full elucidation of potential overlap in regulation of various electrolyte movements. The standardization of model systems as well as collaboration between laboratories with apparently conflicting results is important for forward progress in this field.
8. Conclusions
Hydrocephalus is a complex disease with many origins. There have been myriad attempts to find better treatments for hydrocephalus, but, currently, invasive shunt surgeries or ETVs remain the standard of care. To facilitate the development of pharmacological interventions that can be used on an as-needed basis, it is key to increase our understanding of how CSF production and composition are regulated. An important component of this research is to develop and characterize animal and cell culture models that are widely used within the research community. Currently, each laboratory or group of collaborating investigators appears to have their own animal model(s), and there is little consistency in how hydrocephalus is induced and little agreement as to which genetic models best mirror human pathology. A similar situation exists with regard to the cultured cell models. There is no consensus as to which characteristics are most important and should be de rigueur in any cultured lines. Both animal and cell culture models are necessary as both can be used in an iterative fashion, particularly in the realm of drug testing. If there were agreements with regard to standardized models, the replication of results may facilitate the resolution of existing controversies and would, undoubtedly, facilitate pharmaceutical development from targets to drugs.
Author Contributions
Writing—original draft preparation (including figures), V.N.; writing—review and editing, B.L.B.-Y., L.L.J. and V.N.; funding acquisition, B.L.B.-Y. All authors have read and agreed to the published version of the manuscript.
Funding
The research in the authors’ laboratories was funded by grants from the United States Department of Defense Office of the Congressionally Directed Medical Research Programs (CDMRP): Expansion Award, W81XWH-22-PRMRP-EA (BBY) and Focused Program Award, W81XWH-22-PRMRP-FPA (BBY, LJ).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank T. Cummins, T. Belecky-Adams, and H. Damkier for proofreading and commenting on the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lifshutz, J.I.; Johnson, W.D. History of hydrocephalus and its treatments. Neurosurg. Focus 2001, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Aschoff, A.; Kremer, P.; Hashemi, B.; Kunze, S. The scientific history of hydrocephalus and its treatment. Neurosurg. Rev. 1999, 22, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Mokri, B. The Monro–Kellie hypothesis: Applications in CSF volume depletion. Neurology 2001, 56, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Greitz, D. Radiological assessment of hydrocephalus: New theories and implications for therapy. Neurosurg. Rev. 2004, 27, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, ra111–ra147. [Google Scholar] [CrossRef] [PubMed]
- Hochstetler, A.; Raskin, J.; Blazer-Yost, B.L. Hydrocephalus: Historical analysis and considerations for treatment. Eur. J. Med. Res. 2022, 27, 168. [Google Scholar] [CrossRef] [PubMed]
- Koschnitzky, J.E.; Yap, E.; Zhang, Y.; Chau, M.J.; Yerneni, K.; Somera, A.L.; Luciano, M.; Moghekar, A. Inpatient healthcare burden and variables influencing hydrocephalus-related admissions across the lifespan. J. Neurosurg. 2022, 139, 502–511. [Google Scholar] [CrossRef]
- Karimy, J.K.; Reeves, B.C.; Damisah, E.; Duy, P.Q.; Antwi, P.; David, W.; Wang, K.; Schiff, S.J.; Limbrick, D.D., Jr.; Alper, S.L. Inflammation in acquired hydrocephalus: Pathogenic mechanisms and therapeutic targets. Nat. Rev. Neurol. 2020, 16, 285–296. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Mekary, R.; Glancz, L.J.; Yunusa, I.; Baticulon, R.E.; Fieggen, G.; Wellons, J.C.; Park, K.B.; Warf, B.C. Global hydrocephalus epidemiology and incidence: Systematic review and meta-analysis. J. Neurosurg. 2018, 130, 1065–1079. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, Z.; Tan, Q.; Guo, J.; Tang, J.; Tan, L.; Feng, H.; Chen, Z. Post-hemorrhagic hydrocephalus: Recent advances and new therapeutic insights. J. Neurol. Sci. 2017, 375, 220–230. [Google Scholar] [CrossRef]
- Cioca, A.; Gheban, D.; Perju-Dumbrava, D.; Chiroban, O.; Mera, M. Sudden death from ruptured choroid plexus arteriovenous malformation. Am. J. Forensic Med. Pathol. 2014, 35, 100–102. [Google Scholar] [CrossRef]
- Thigpen, M.C.; Whitney, C.G.; Messonnier, N.E.; Zell, E.R.; Lynfield, R.; Hadler, J.L.; Harrison, L.H.; Farley, M.M.; Reingold, A.; Bennett, N.M. Bacterial meningitis in the United States, 1998–2007. N. Engl. J. Med. 2011, 364, 2016–2025. [Google Scholar] [CrossRef]
- Varagur, K.; Sanka, S.A.; Strahle, J.M. Syndromic hydrocephalus. Neurosurg. Clin. 2022, 33, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, C.; Vos, Y.J. L1 Syndrome; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Marguet, F.; Vezain, M.; Marcorelles, P.; Audebert-Bellanger, S.; Cassinari, K.; Drouot, N.; Chambon, P.; Gonzalez, B.J.; Horowitz, A.; Laquerriere, A. Neuropathological hallmarks of fetal hydrocephalus linked to CCDC88C pathogenic variants. Acta Neuropathol. Commun. 2021, 9, 104. [Google Scholar] [CrossRef]
- Hale, A.T.; Bastarache, L.; Morales, D.M.; Wellons, J.C.; Limbrick, D.D.; Gamazon, E.R. Multi-omic analysis elucidates the genetic basis of hydrocephalus. Cell Rep. 2021, 35, 109085. [Google Scholar] [CrossRef]
- Isaacs, A.M.; Riva-Cambrin, J.; Yavin, D.; Hockley, A.; Pringsheim, T.M.; Jette, N.; Lethebe, B.C.; Lowerison, M.; Dronyk, J.; Hamilton, M.G. Age-specific global epidemiology of hydrocephalus: Systematic review, metanalysis and global birth surveillance. PLoS ONE 2018, 13, e0204926. [Google Scholar] [CrossRef] [PubMed]
- Tully, H.M.; Dobyns, W.B. Infantile hydrocephalus: A review of epidemiology, classification and causes. Eur. J. Med. Genet. 2014, 57, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Wright, Z.; Larrew, T.W.; Eskandari, R. Pediatric hydrocephalus: Current state of diagnosis and treatment. Pediatr. Rev. 2016, 37, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Rosell, M.; Kockum, K.; Lilja-Lund, O.; Söderström, L.; Laurell, K. Prevalence of idiopathic normal pressure hydrocephalus: A prospective, population-based study. PLoS ONE 2019, 14, e0217705. [Google Scholar] [CrossRef]
- Fowler, J.B.; De Jesus, O.; Mesfin, F.B. Ventriculoperitoneal Shunt; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Akyol, M.E.; Cetin, E. Effects of shunt types used in idiopathic normal pressure hydrocephalus on patients’ clinical outcomes. Ann. Med. Res. 2023, 30, 146–281. [Google Scholar] [CrossRef]
- Shannon, C.N.; Carr, K.R.; Tomycz, L.; Wellons, J.C.; Tulipan, N. Time to first shunt failure in pediatric patients over 1 year old: A 10-year retrospective study. Pediatr. Neurosurg. 2015, 49, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Guo, W. Have we made progress in preventing shunt failure? A critical analysis. J. Neurosurg. Pediatr. 2008, 1, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.J.; Chiang, W.C.; Huang, H.Y.; Lin, S.Z.; Tsai, S.T. Effectiveness and safety of ventriculoperitoneal shunt versus lumboperitoneal shunt for communicating hydrocephalus: A systematic review and meta-analysis with trial sequential analysis. CNS Neurosci. Ther. 2023, 29, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-H.; Chang, C.-S.; Sung, W.-W.; Liu, J.-T. Lumboperitoneal shunt: A new modified surgical technique and a comparison of the complications with ventriculoperitoneal shunt in a single center. Medicina 2019, 55, 643. [Google Scholar] [CrossRef] [PubMed]
- Yadav, Y.R.; Parihar, V.; Pande, S.; Namdev, H.; Agarwal, M. Endoscopic third ventriculostomy. J. Neurosci. Rural Pract. 2012, 3, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.W.; Bahuleyan, B.; Robinson, S.; Cohen, A.R. The role of endoscopic third ventriculostomy in the treatment of hydrocephalus. J. Neurosurg. Pediatr. 2013, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Marano, P.J.; Stone, S.S.; Mugamba, J.; Ssenyonga, P.; Warf, E.B.; Warf, B.C. Reopening of an obstructed third ventriculostomy: Long-term success and factors affecting outcome in 215 infants. J. Neurosurg. Pediatr. 2015, 15, 399–405. [Google Scholar] [CrossRef]
- Kulkarni, A.V.; Drake, J.M.; Mallucci, C.L.; Sgouros, S.; Roth, J.; Constantini, S.; Canadian Pediatric Neurosurgery Study Group. Endoscopic third ventriculostomy in the treatment of childhood hydrocephalus. J. Pediatr. 2009, 155, 254–259.e1. [Google Scholar] [CrossRef]
- Riva-Cambrin, J.; Kestle, J.R.; Rozzelle, C.J.; Naftel, R.P.; Alvey, J.S.; Reeder, R.W.; Holubkov, R.; Browd, S.R.; Cochrane, D.D.; Limbrick, D.D. Predictors of success for combined endoscopic third ventriculostomy and choroid plexus cauterization in a North American setting: A Hydrocephalus Clinical Research Network study. J. Neurosurg. Pediatr. 2019, 24, 128–138. [Google Scholar] [CrossRef]
- Zaben, M.; Manivannan, S.; Sharouf, F.; Hammad, A.; Patel, C.; Bhatti, I.; Leach, P. The efficacy of endoscopic third ventriculostomy in children 1 year of age or younger: A systematic review and meta-analysis. Eur. J. Paediatr. Neurol. 2020, 26, 7–14. [Google Scholar] [CrossRef]
- Warf, B.C.; Weber, D.S.; Day, E.L.; Riordan, C.P.; Staffa, S.J.; Baird, L.C.; Fehnel, K.P.; Stone, S.S. Endoscopic third ventriculostomy with choroid plexus cauterization: Predictors of long-term success and comparison with shunt placement for primary treatment of infant hydrocephalus. J. Neurosurg. Pediatr. 2023, 32, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Schiff, S.J.; Kulkarni, A.V.; Mbabazi-Kabachelor, E.; Mugamba, J.; Ssenyonga, P.; Donnelly, R.; Levenbach, J.; Monga, V.; Peterson, M.; Cherukuri, V. Brain growth after surgical treatment for infant postinfectious hydrocephalus in Sub-Saharan Africa: 2-year results of a randomized trial. J. Neurosurg. Pediatr. 2021, 28, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Bass, N.H.; Fällström, S.; Lundborg, P. Digoxin-induced arrest of the cerebrospinal fluid circulation in the infant rat: Implications for medical treatment of hydrocephalus during early postnatal life. Pediatr. Res. 1979, 13, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Winer, J.L.; Kitase, Y.; Brigman, J.L.; Jantzie, L.L. Neonatal administration of erythropoietin attenuates cognitive deficits in adult rats following placental insufficiency. J. Neurosci. Res. 2022, 100, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Conteh, F.S.; Oppong, A.Y.; Yellowhair, T.R.; Newville, J.C.; Demerdash, N.E.; Shrock, C.L.; Maxwell, J.R.; Jett, S.; Northington, F.J. Extended combined neonatal treatment with erythropoietin plus melatonin prevents posthemorrhagic hydrocephalus of prematurity in rats. Front. Cell. Neurosci. 2018, 12, 322. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Muthukumar, S.; Kitase, Y.; Vasan, V.; Fouda, M.A.; Hamimi, S.; Burkhardt, C.; Burton, V.J.; Gerner, G.; Scafidi, J. Infantile Cocktail of Erythropoietin and Melatonin Restores Gait in Adult Rats with Preterm Brain Injury. Dev. Neurosci. 2022, 44, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Safety of Erythropoietin and Melatonin for Very Preterm Infants with Intraventricular Hemorrhage (SCEMPI). Available online: https://clinicaltrials.gov/study/NCT05617833?term=robinson&intr=erythropoietin&rank=1 (accessed on 14 January 2024).
- Zhao, J.; Chen, Z.; Xi, G.; Keep, R.F.; Hua, Y. Deferoxamine attenuates acute hydrocephalus after traumatic brain injury in rats. Transl. Stroke Res. 2014, 5, 586–594. [Google Scholar] [CrossRef]
- Pang, D.; Sclabassi, R.J.; Horton, J.A. Lysis of intraventricular blood clot with urokinase in a canine model: Part 1. Canine intraventricular blood cast model. Neurosurgery 1986, 19, 540–546. [Google Scholar] [CrossRef]
- Chua, C.O.; Chahboune, H.; Braun, A.; Dummula, K.; Chua, C.E.; Yu, J.; Ungvari, Z.; Sherbany, A.A.; Hyder, F.; Ballabh, P. Consequences of intraventricular hemorrhage in a rabbit pup model. Stroke 2009, 40, 3369–3377. [Google Scholar] [CrossRef]
- Kiseleva, Z.N.; Volzhina, N.S. Experimental hydrocephalus in young rats. Arkhiv Patol. 1957, 19, 44–52. [Google Scholar]
- Pudenz, R.H. Experimental and clinical observations on the shunting of cerebrospinal fluid into the circulatory system. Neurosurgery 1958, 5, 98–115. [Google Scholar] [CrossRef] [PubMed]
- McAllister, J.P.; Talcott, M.R.; Isaacs, A.M.; Zwick, S.H.; Garcia-Bonilla, M.; Castaneyra-Ruiz, L.; Hartman, A.L.; Dilger, R.N.; Fleming, S.A.; Golden, R.K. A novel model of acquired hydrocephalus for evaluation of neurosurgical treatments. Fluids Barriers CNS 2021, 18, 49. [Google Scholar] [CrossRef] [PubMed]
- da Silva Lopes, L.; Slobodian, I.; Del Bigio, M.R. Characterization of juvenile and young adult mice following induction of hydrocephalus with kaolin. Exp. Neurol. 2009, 219, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Darder, J.; Barbera, J.; Cerda-Nicolas, M.; Segura, D.; Broseta, J.; Barcia-Salorio, J.L. Sequential morphological and functional changes in kaolin-induced hydrocephalus. J. Neurosurg. 1984, 61, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, D.K.; Harrison, M.R.; Berger, M.S.; Chinn, D.H.; Halks-Miller, M.; Edwards, M.S. Correction of congenital hydrocephalus in utero I. The model: Intracisternal kaolin produces hydrocephalus in fetal lambs and rhesus monkeys. J. Pediatr. Surg. 1983, 18, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Marlin, A.; Wald, A.; Hochwald, G.; Malhan, C. Kaolin-induced hydrocephalus impairs CSF secretion by the choroid plexus. Neurology 1978, 28, 945. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Y.; Cui, W.; Cao, Y.; Zhao, L.; Wang, H.; Liu, X.; Fan, S.; Huang, K.; Tong, A. Inhibition of neuronal necroptosis mediated by RIP1/RIP3/MLKL provides neuroprotective effects on kaolin-induced hydrocephalus in mice. Cell Prolif. 2021, 54, e13108. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Guo, J.; Yu, C.; Yang, J. Molecular mechanisms and risk factors for the pathogenesis of hydrocephalus. Front. Genet. 2022, 12, 777926. [Google Scholar] [CrossRef]
- Yamada, H.; Oi, S.; Tamaki, N.; Matsumoto, S.; Taomoto, K. Embryopathoetiology of Congenital Hydrocephalus in Experimental Models: A Comparative Morphological Study in Two Different Models. In Hydrocephalus: Pathogenesis and Treatment; Springer: Tokyo, Japan, 1991; pp. 27–35. [Google Scholar]
- Stambolliu, E.; Ioakeim-Ioannidou, M.; Kontokostas, K.; Dakoutrou, M.; Kousoulis, A.A. The most common comorbidities in Dandy-Walker syndrome patients: A systematic review of case reports. J. Child Neurol. 2017, 32, 886–902. [Google Scholar] [CrossRef]
- Chamberlain, J. Early neurovascular abnormalities underlying 6-aminonicotinamide (6-AN)-induced congenital hydrocephalus in rats. Teratology 1970, 3, 377–387. [Google Scholar] [CrossRef]
- Gattone, V.H.; Tourkow, B.A.; Trambaugh, C.M.; Yu, A.C.; Whelan, S.; Phillips, C.L.; Harris, P.C.; Peterson, R.G. Development of multiorgan pathology in the wpk rat model of polycystic kidney disease. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. Off. Publ. Am. Assoc. Anat. 2004, 277, 384–395. [Google Scholar] [CrossRef]
- Shim, J.W.; Territo, P.R.; Simpson, S.; Watson, J.C.; Jiang, L.; Riley, A.A.; McCarthy, B.; Persohn, S.; Fulkerson, D.; Blazer-Yost, B.L. Hydrocephalus in a rat model of Meckel Gruber syndrome with a TMEM67 mutation. Sci. Rep. 2019, 9, 1069. [Google Scholar] [CrossRef] [PubMed]
- Hochstetler, A.; Smith, H.; Reed, M.; Hulme, L.; Territo, P.; Bedwell, A.; Persohn, S.; Perrotti, N.; D’Antona, L.; Musumeci, F. Inhibition of serum-and glucocorticoid-induced kinase 1 ameliorates hydrocephalus in preclinical models. Fluids Barriers CNS 2023, 20, 61. [Google Scholar] [CrossRef]
- Jones, H.C.; Carter, B.J.; Morel, L. Characteristics of hydrocephalus expression in the LEW/Jms rat strain with inherited disease. Child’s Nerv. Syst. 2003, 19, 11–18. [Google Scholar] [CrossRef]
- Itoh, K.; Fushiki, S. The role of L1 cam in murine corticogenesis, and the pathogenesis of hydrocephalus. Pathol. Int. 2015, 65, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Swetloff, A.; Ferretti, P. Changes in E2F5 intracellular localization in mouse and human choroid plexus epithelium with development. Int. J. Dev. Biol. 2004, 49, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, G.J.; Dagnino, L.; Gaubatz, S.; Xu, Y.; Bronson, R.T.; Warren, H.B.; Livingston, D.M. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998, 12, 1092–1098. [Google Scholar] [CrossRef]
- Lewis, W.R.; Malarkey, E.B.; Tritschler, D.; Bower, R.; Pasek, R.C.; Porath, J.D.; Birket, S.E.; Saunier, S.; Antignac, C.; Knowles, M.R. Mutation of growth arrest specific 8 reveals a role in motile cilia function and human disease. PLoS Genet. 2016, 12, e1006220. [Google Scholar] [CrossRef]
- Hochstetler, A.E.; Whitehouse, L.; Antonellis, P.; Berbari, N.F.; Blazer-Yost, B.L. Characterizing the Expression of TRPV4 in the Choroid Plexus Epithelia as a Prospective Component in the Development of Hydrocephalus in the Gas8GT Juvenile Mouse Model. FASEB J. 2018, 32, C1823–C1842. [Google Scholar] [CrossRef]
- Abdelhamed, Z.; Vuong, S.M.; Hill, L.; Shula, C.; Timms, A.; Beier, D.; Campbell, K.; Mangano, F.T.; Stottmann, R.W.; Goto, J. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development 2018, 145, dev154500. [Google Scholar] [CrossRef] [PubMed]
- Davy, B.E.; Robinson, M.L. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003, 12, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Bronson, R.T.; Lane, P.W. Hydrocephalus with hop gait (hyh): A new mutation on chromosome 7 in the mouse. Dev. Brain Res. 1990, 54, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Di Curzio, D.L. Animal models of hydrocephalus. Open J. Mod. Neurosurg. 2017, 8, 57–71. [Google Scholar] [CrossRef]
- Jones, H.; Dack, S.; Ellis, C. Morphological aspects of the development of hydrocephalus in a mouse mutant (SUMS/NP). Acta Neuropathol. 1987, 72, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Hochstetler, A.E.; Smith, H.M.; Preston, D.C.; Reed, M.M.; Territo, P.R.; Shim, J.W.; Fulkerson, D.; Blazer-Yost, B.L. TRPV4 antagonists ameliorate ventriculomegaly in a rat model of hydrocephalus. JCI Insight 2020, 5, e137646. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, F.; Draper, C.E.; Bannister, C.M.; Pourghasem, M.; Owen-Lynch, P.J.; Miyan, J.A. Deficient cortical development in the hydrocephalic Texas (H-Tx) rat: A role for CSF. Brain 2002, 125, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.; Bucknall, R. Inherited prenatal hydrocephalus in the H–Tx rat: A morphological study. Neuropathol. Appl. Neurobiol. 1988, 14, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Kuwamura, M.; Kinoshita, A.; Okumoto, M.; Yamate, J.; Mori, N. Hemorrhagic hydrocephalus (hhy): A novel mutation on mouse chromosome 12. Dev. Brain Res. 2004, 152, 69–72. [Google Scholar] [CrossRef]
- Lin, X.; Liu, B.; Yang, X.; Yue, X.; Diao, L.; Wang, J.; Chang, J. Genetic deletion of Rnd3 results in aqueductal stenosis leading to hydrocephalus through up-regulation of Notch signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 8236–8241. [Google Scholar] [CrossRef]
- Kume, T.; Deng, K.-Y.; Winfrey, V.; Gould, D.B.; Walter, M.A.; Hogan, B.L. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell 1998, 93, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Fernández-Llebrez, P.; Bach, A.; Robert, B.; Soriano, E. Msx1 disruption leads to diencephalon defects and hydrocephalus. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2004, 230, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lehtinen, M.K. Experimental approaches for manipulating choroid plexus epithelial cells. Fluids Barriers CNS 2022, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Millar, I.D.; Bruce, J.I.; Brown, P.D. Ion channel diversity, channel expression and function in the choroid plexuses. Cerebrospinal Fluid Res. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Damkier, H.H.; Brown, P.D.; Praetorius, J. Cerebrospinal fluid secretion by the choroid plexus. Physiol. Rev. 2013, 93, 1847–1892. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, Z.; Chen, Y.; Liao, J.; Wang, Y.; Liu, J.; Lin, Z.; Xiao, G. Choroid plexus epithelium and its role in neurological diseases. Front. Mol. Neurosci. 2022, 15, 949231. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, J.; Wang, Y.; Wang, C.; Tan, C.; Liao, J.; Tong, L.; Xiao, G. Targeting choroid plexus epithelium as a novel therapeutic strategy for hydrocephalus. J. Neuroinflamm. 2022, 19, 156. [Google Scholar] [CrossRef] [PubMed]
- Sakka, L.; Coll, G.; Chazal, J. Anatomy and physiology of cerebrospinal fluid. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2011, 128, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Oernbo, E.K.; Steffensen, A.B.; Razzaghi Khamesi, P.; Toft-Bertelsen, T.L.; Barbuskaite, D.; Vilhardt, F.; Gerkau, N.J.; Tritsaris, K.; Simonsen, A.H.; Lolansen, S.D. Membrane transporters control cerebrospinal fluid formation independently of conventional osmosis to modulate intracranial pressure. Fluids Barriers CNS 2022, 19, 65. [Google Scholar] [CrossRef]
- Wright, E.M. Mechanisms of ion transport across the choroid plexus. J. Physiol. 1972, 226, 545–571. [Google Scholar] [CrossRef]
- Bouillé, C.; Mesnil, M.; Barriere, H.; Gabrion, J. Gap junctional intercellular communication between cultured ependymal cells, revealed by lucifer yellow CH transfer and freeze-fracture. Glia 1991, 4, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Q.; Graziano, J.H. Primary culture of choroidal epithelial cells: Characterization of an in vitro model of blood-CSF barrier. Vitr. Cell. Dev. Biol.-Anim. 1998, 34, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.E.; Sanders-Bush, E. Sodium-dependent antiporters in choroid plexus epithelial cultures from rabbit. J. Neurochem. 1993, 60, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Baehr, C.; Reichel, V.; Fricker, G. Choroid plexus epithelial monolayers—A cell culture model from porcine brain. Cerebrospinal Fluid Res. 2006, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Harter, D.; Hsu, K.; Rose, H. Immunofluorescence and cytochemical studies of visna virus in cell culture. J. Virol. 1967, 1, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Whittico, M.T.; Hui, A.C.; Giacomini, K.M. Preparation of brush border membrane vesicles from bovine choroid plexus. J. Pharmacol. Methods 1991, 25, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Monnot, A.D.; Zheng, W. Culture of choroid plexus epithelial cells and in vitro model of blood–CSF barrier. Epithel. Cell Cult. Protoc. 2013, 945, 13–29. [Google Scholar]
- Fejes, Z.; Pócsi, M.; Takai, J.; Erdei, J.; Tóth, A.; Balogh, E.; Rusznyák, Á.; Fenyvesi, F.; Nagy, A.; Kappelmayer, J. Preterm intraventricular hemorrhage-induced inflammatory response in human choroid plexus epithelial cells. Int. J. Mol. Sci. 2021, 22, 8648. [Google Scholar] [CrossRef] [PubMed]
- Guy, B.; Zhang, J.S.; Duncan, L.H.; Johnston, R.J., Jr. Human neural organoids: Models for developmental neurobiology and disease. Dev. Biol. 2021, 478, 102–121. [Google Scholar] [CrossRef]
- Schroten, M.; Hanisch, F.-G.; Quednau, N.; Stump, C.; Riebe, R.; Lenk, M.; Wolburg, H.; Tenenbaum, T.; Schwerk, C. A novel porcine in vitro model of the blood-cerebrospinal fluid barrier with strong barrier function. PLoS ONE 2012, 7, e39835. [Google Scholar] [CrossRef]
- Hochstetler, A.; Hulme, L.; Delpire, E.; Schwerk, C.; Schroten, H.; Preston, D.; Simpson, S.; Blazer-Yost, B.L. Porcine choroid plexus-riems cell line demonstrates altered polarization of transport proteins compared with the native epithelium. Am. J. Physiol.-Cell Physiol. 2022, 323, C1–C13. [Google Scholar] [CrossRef] [PubMed]
- Lauer, A.N.; März, M.; Meyer, S.; Meurer, M.; de Buhr, N.; Borkowski, J.; Weiß, C.; Schroten, H.; Schwerk, C. Optimized cultivation of porcine choroid plexus epithelial cells, a blood–cerebrospinal fluid barrier model, for studying granulocyte transmigration. Lab. Investig. 2019, 99, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Hulme, L.; Hochstetler, A.; Schwerk, C.; Schroten, H.; Ishikawa, H.; Tung, C.-Y.; Perrin, B.; Blazer-Yost, B. Characterization of TRPV4-mediated signaling pathways in an optimized human choroid plexus epithelial cell line. Am. J. Physiol.-Cell Physiol. 2022, 323, C1823–C1842. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Q. Establishment and characterization of an immortalized Z310 choroidal epithelial cell line from murine choroid plexus. Brain Res. 2002, 958, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Dziegielewska, K.M.; Ek, C.J.; Habgood, M.D.; Bauer, H.; Bauer, H.-C.; Lindsay, H.; Wakefield, M.J.; Strazielle, N.; Kratzer, I. Mechanisms that determine the internal environment of the developing brain: A transcriptomic, functional and ultrastructural approach. PLoS ONE 2016, 11, e0147680. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Zheng, W. Establishment of an in vitro brain barrier epithelial transport system for pharmacological and toxicological study. Brain Res. 2005, 1057, 37–48. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, W. Toxicology of choroid plexus: Special reference to metal-induced neurotoxicities. Microsc. Res. Tech. 2001, 52, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, I.; Ishiwat, C.; Ishiwata, E.; Sato, Y.; Kiguchi, K.; Tachibana, T.; Hashimoto, H.; Ishikawa, H. Establishment and characterization of a human malignant choroids plexus papilloma cell line (HIBCPP). Hum. Cell 2005, 18, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Schwerk, C.; Herold, R.; Stump-Guthier, C.; Lampe, M.; Fallier-Becker, P.; Weiß, C.; Sticht, C.; Ishikawa, H.; Schroten, H. Immortalized human choroid plexus endothelial cells enable an advanced endothelial-epithelial two-cell type in vitro model of the choroid plexus. Iscience 2022, 25, 104383. [Google Scholar] [CrossRef]
- Giovannucci, T.A.; Leckey, C.A.; Moncur, E.; Tariq, K.; Thorne, L.; Watkins, L.; Toma, A.; Fox, N.C.; Bateman, R.J.; Mills, K. Choroid plexus protein turnover in human choroid plexus organoids recapitulates turnover in humans measured using stable isotope labeling kinetics (SILK). Alzheimer’s Dement. 2023, 19, e074240. [Google Scholar] [CrossRef]
- Praetorius, J.; Nielsen, S. Distribution of sodium transporters and aquaporin-1 in the human choroid plexus. Am. J. Physiol. -Cell Physiol. 2006, 291, C59–C67. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N.; Toft-Bertelsen, T.L. Dual function of the choroid plexus: Cerebrospinal fluid production and control of brain ion homeostasis. Cell Calcium 2023, 116, 102797. [Google Scholar] [CrossRef] [PubMed]
- Roepke, T.K.; Kanda, V.A.; Purtell, K.; King, E.C.; Lerner, D.J.; Abbott, G.W. KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J. 2011, 25, 4264. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.; Lu, J.; Mount, D.; Delpire, E. Localization of the K+–Cl− cotransporter, KCC3, in the central and peripheral nervous systems: Expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 2001, 103, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Leefmans, F.J. CrossTalk proposal: Apical NKCC1 of choroid plexus epithelial cells works in the net inward flux mode under basal conditions, maintaining intracellular Cl− and cell volume. J. Physiol. 2020, 598, 4733–4736. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N.; Rose, C. CrossTalk opposing view: NKCC1 in the luminal membrane of choroid plexus is outwardly directed under basal conditions and contributes directly to cerebrospinal fluid secretion. J. Physiol. 2020, 598, 4737–4739. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Leefmans, F.J. Rebuttal from Francisco J. Alvarez-Leefmans. J. Physiol. 2020, 598, 4741–4742. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N.; Rose, C.R. Rebuttal from Nanna MacAulay and Christine R. Rose. J. Physiol. 2020, 598, 4743. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Gagnon, K.B. Elusive role of the Na-K-2Cl cotransporter in the choroid plexus. Am. J. Physiol.-Cell Physiol. 2019, 316, C522–C524. [Google Scholar] [CrossRef]
- Fame, R.M.; Xu, H.; Pragana, A.; Lehtinen, M. Age-appropriate potassium clearance from perinatal cerebrospinal fluid depends on choroid plexus NKCC1. Fluids Barriers CNS 2023, 20, 45. [Google Scholar] [CrossRef]
- Zeuthen, T.; MacAulay, N. Cotransport of water by Na+–K+–2Cl− cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J. Physiol. 2012, 590, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Zhang, Y.; Liu, T.; Friedel, P.; Zhuo, W.; Somasekharan, S.; Roy, K.; Zhang, L.; Liu, Y. The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 2021, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Macvicar, B.A.; Feighan, D.; Brown, A.; Ransom, B. Intrinsic optical signals in the rat optic nerve: Role for K+ uptake via NKCC1 and swelling of astrocytes. Glia 2002, 37, 114–123. [Google Scholar] [CrossRef]
- Chou, C.-L.; Yu, M.-J.; Kassai, E.M.; Morris, R.G.; Hoffert, J.D.; Wall, S.M.; Knepper, M.A. Roles of basolateral solute uptake via NKCC1 and of myosin II in vasopressin-induced cell swelling in inner medullary collecting duct. Am. J. Physiol.-Ren. Physiol. 2008, 295, F192–F201. [Google Scholar] [CrossRef]
- Blazer-Yost, B.L. Consideration of Kinase Inhibitors for the Treatment of Hydrocephalus. Int. J. Mol. Sci. 2023, 24, 6673. [Google Scholar] [CrossRef]
- Koumangoye, R.; Bastarache, L.; Delpire, E. NKCC1: Newly found as a human disease-causing ion transporter. Function 2021, 2, zqaa028. [Google Scholar] [CrossRef] [PubMed]
- Gregoriades, J.M.; Madaris, A.; Alvarez, F.J.; Alvarez-Leefmans, F.J. Genetic and pharmacological inactivation of apical Na+-K+-2Cl− cotransporter 1 in choroid plexus epithelial cells reveals the physiological function of the cotransporter. Am. J. Physiol. Cell Physiol. 2019, 316, C525–C544. [Google Scholar] [CrossRef]
- Szymanski, J.; Minichiello, L. NKCC1 deficiency in forming hippocampal circuits triggers neurodevelopmental disorder: Role of BDNF-TrkB signaling. Brain Sci. 2022, 12, 502. [Google Scholar] [CrossRef]
- Karimy, J.K.; Zhang, J.; Kurland, D.B.; Theriault, B.C.; Duran, D.; Stokum, J.A.; Furey, C.G.; Zhou, X.; Mansuri, M.S.; Montejo, J. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med. 2017, 23, 997–1003. [Google Scholar] [CrossRef]
- Sadegh, C.; Xu, H.; Sutin, J.; Fatou, B.; Gupta, S.; Pragana, A.; Taylor, M.; Kalugin, P.N.; Zawadzki, M.E.; Alturkistani, O. Choroid plexus-targeted NKCC1 overexpression to treat post-hemorrhagic hydrocephalus. Neuron 2023, 111, 1591–1608.e4. [Google Scholar] [CrossRef]
- Bothwell, S.W.; Omileke, D.; Patabendige, A.; Spratt, N.J. CSF secretion is not altered by NKCC1 nor TRPV4 antagonism in healthy rats. Brain Sci. 2021, 11, 1117. [Google Scholar] [CrossRef] [PubMed]
- Savardi, A.; Borgogno, M.; De Vivo, M.; Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 2021, 42, 1009–1034. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K. TRPV4 ion channel as important cell sensors. J. Anesth. 2016, 30, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Choe, Y.; Martí-Renom, M.A.; Bell, A.M.; Denis, C.S.; Hudspeth, A.; Friedman, J.M.; Heller, S. Vanilloid receptor–related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T. TRPV4 calcium entry channel: A paradigm for gating diversity. Am. J. Physiol. Cell Physiol. 2004, 286, C195–C205. [Google Scholar] [CrossRef] [PubMed]
- Strotmann, R.; Harteneck, C.; Nunnenmacher, K.; Schultz, G.; Plant, T.D. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000, 2, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Darby, W.; Grace, M.; Baratchi, S.; McIntyre, P. Modulation of TRPV4 by diverse mechanisms. Int. J. Biochem. Cell Biol. 2016, 78, 217–228. [Google Scholar] [CrossRef]
- Liedtke, W.; Friedman, J.M. Abnormal osmotic regulation in trpv4-/-mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13698–13703. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Suzuki, M.; Mizuno, A.; Hara, A. Hearing impairment in TRPV4 knockout mice. Neurosci. Lett. 2005, 382, 304–308. [Google Scholar] [CrossRef]
- Goyal, N.; Skrdla, P.; Schroyer, R.; Kumar, S.; Fernando, D.; Oughton, A.; Norton, N.; Sprecher, D.L.; Cheriyan, J. Clinical pharmacokinetics, safety, and tolerability of a novel, first-in-class TRPV4 ion channel inhibitor, GSK2798745, in healthy and heart failure subjects. Am. J. Cardiovasc. Drugs 2019, 19, 335–342. [Google Scholar] [CrossRef]
- Lawhorn, B.G.; Brnardic, E.J.; Behm, D.J. TRPV4 antagonists: A patent review (2015–2020). Expert Opin. Ther. Pat. 2021, 31, 773–784. [Google Scholar] [CrossRef]
- Park, S.; Ku, S.K.; Ji, H.W.; Choi, J.-H.; Shin, D.M. Ca(2+) is a Regulator of the WNK/OSR1/NKCC Pathway in a Human Salivary Gland Cell Line. Korean J. Physiol. Pharmacol. 2015, 19, 249–255. [Google Scholar] [CrossRef][Green Version]
- Toft-Bertelsen, T.L.; Barbuskaite, D.; Heerfordt, E.K.; Lolansen, S.D.; Andreassen, S.N.; Rostgaard, N.; Olsen, M.H.; Norager, N.H.; Capion, T.; Rath, M.F. Lysophosphatidic acid as a CSF lipid in posthemorrhagic hydrocephalus that drives CSF accumulation via TRPV4-induced hyperactivation of NKCC1. Fluids Barriers CNS 2022, 19, 69. [Google Scholar] [CrossRef]
- Jin, S.C.; Furey, C.G.; Zeng, X.; Allocco, A.; Nelson-Williams, C.; Dong, W.; Karimy, J.K.; Wang, K.; Ma, S.; Delpire, E. SLC12A ion transporter mutations in sporadic and familial human congenital hydrocephalus. Mol. Genet. Genom. Med. 2019, 7, e892. [Google Scholar] [CrossRef]
- Kahle, K.T.; Flores, B.; Bharucha-Goebel, D.; Zhang, J.; Donkervoort, S.; Hegde, M.; Begum, G.; Duran, D.; Liang, B.; Sun, D. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci. Signal. 2016, 9, ra77. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Alessi, D.R. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signaling pathway. J. Cell Sci. 2008, 121, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Lauf, P.K.; Bauer, J. Direct evidence for chloride-dependent volume reduction in macrocytic sheep reticulocytes. Biochem. Biophys. Res. Commun. 1987, 144, 849–855. [Google Scholar] [CrossRef]
- Mercado, A.; Broumand, V.; Zandi-Nejad, K.; Enck, A.H.; Mount, D.B. A C-terminal domain in KCC2 confers constitutive K+-Cl-cotransport. J. Biol. Chem. 2006, 281, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Chloride impermeability in cystic fibrosis. Nature 1983, 301, 421–422. [Google Scholar] [CrossRef]
- Bear, C.E.; Li, C.; Kartner, N.; Bridges, R.J.; Jensen, T.J.; Ramjeesingh, M.; Riordan, J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992, 68, 809–818. [Google Scholar] [CrossRef]
- Genovese, M.; Borrelli, A.; Venturini, A.; Guidone, D.; Caci, E.; Viscido, G.; Gambardella, G.; di Bernardo, D.; Scudieri, P.; Galietta, L.J. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J. Physiol. 2019, 597, 5859–5878. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, L.Ø.; Friis, K.A.; Damkier, H.H. In vitro investigation of the effect of proinflammatory cytokines on mouse choroid plexus membrane transporters Ncbe and NKCC1. Fluids Barriers CNS 2023, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, J.; Damkier, H.H. Transport across the choroid plexus epithelium. Am. J. Physiol. Cell Physiol. 2017, 312, C673–C686. [Google Scholar] [CrossRef] [PubMed]
- Damkier, H.H.; Praetorius, J. Genetic ablation of Slc4a10 alters the expression pattern of transporters involved in solute movement in the mouse choroid plexus. Am. J. Physiol. Cell Physiol. 2012, 302, C1452–C1459. [Google Scholar] [CrossRef] [PubMed]
- Christensen, I.B.; Wu, Q.; Bohlbro, A.S.; Skals, M.G.; Damkier, H.H.; Hübner, C.A.; Fenton, R.A.; Praetorius, J. Genetic disruption of slc4a10 alters the capacity for cellular metabolism and vectorial ion transport in the choroid plexus epithelium. Fluids Barriers CNS 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Li, Q.; So, I.; Huang, C.-L.; Ando, H.; Mizutani, A.; Seki, G.; Mikoshiba, K.; Thomas, P.J.; Muallem, S. IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. J. Clin. Investig. 2011, 121, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Ruusuvuori, E.; Sipilä, S.T.; Haapanen, A.; Damkier, H.H.; Kurth, I.; Hentschke, M.; Schweizer, M.; Rudhard, Y.; Laatikainen, L.M. Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc. Natl. Acad. Sci. USA 2008, 105, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Gurnett, C.A.; Veile, R.; Zempel, J.; Blackburn, L.; Lovett, M.; Bowcock, A. Disruption of sodium bicarbonate transporter SLC4A10 in a patient with complex partial epilepsy and mental retardation. Arch. Neurol. 2008, 65, 550–553. [Google Scholar] [CrossRef]
- Fasham, J.; Huebner, A.K.; Liebmann, L.; Khalaf-Nazzal, R.; Maroofian, R.; Kryeziu, N.; Wortmann, S.B.; Leslie, J.S.; Ubeyratna, N.; Mancini, G.M. SLC4A10 mutation causes a neurological disorder associated with impaired GABAergic transmission. Brain 2023, 146, 4547–4561. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).