
Elexacaftor/Tezacaftor/Ivacaftor Efficacy in a Cohort of Italian Patients with CFTR Rare Mutations

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
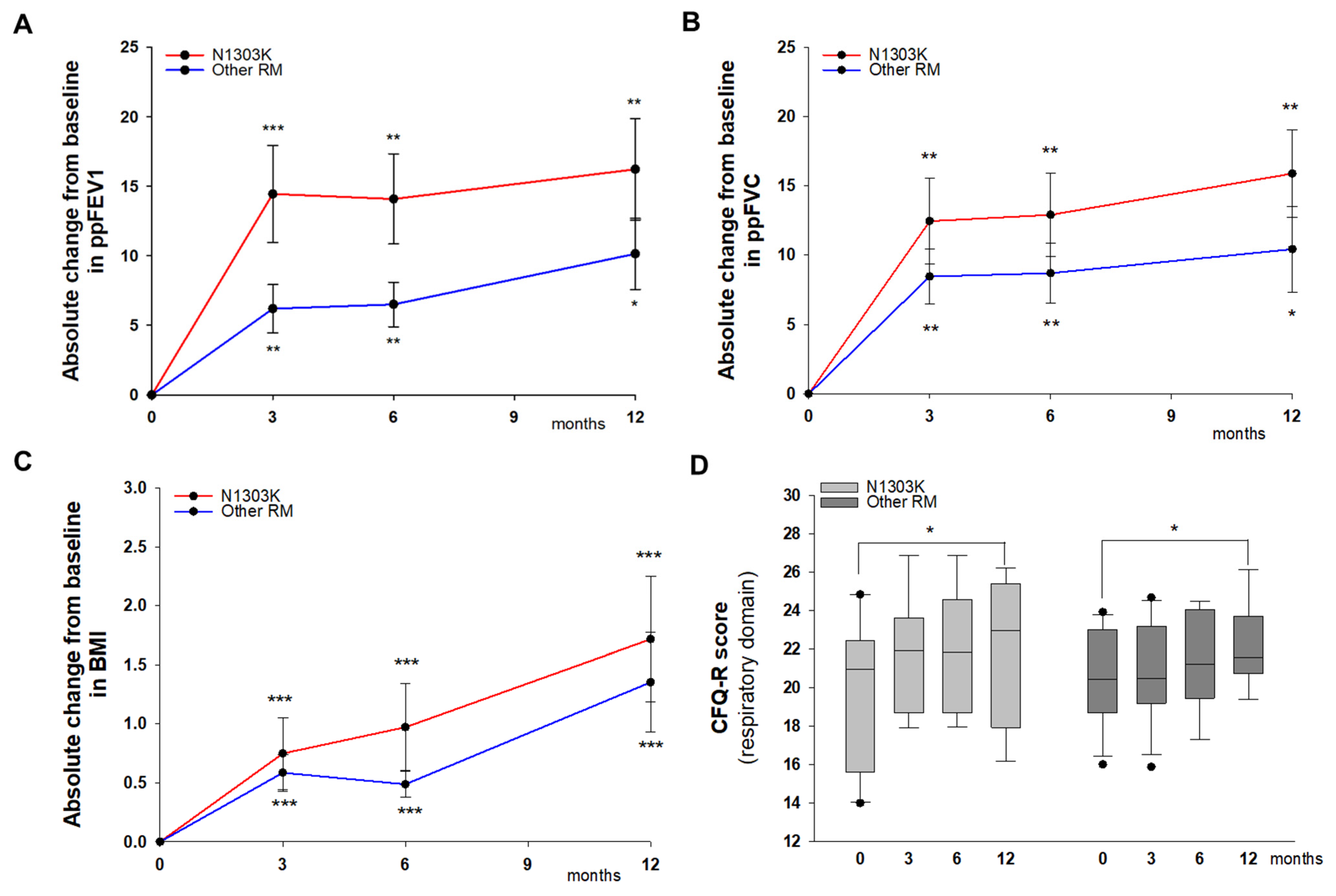
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mall, M.A.; Burgel, P.-R.; Castellani, C.; Davies, J.C.; Salathe, M.; Taylor-Cousar, J.L. Cystic fibrosis. Nat. Rev. Dis. Primers 2024, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Tupayachi Ortiz, M.G.; Baumlin, N.; Yoshida, M.; Salathe, M. Response to Elexacaftor/Tezacaftor/Ivacaftor in people with cystic fibrosis with the N1303K mutation: Case report and review of the literature. Heliyon 2024, 10, e26955. [Google Scholar] [CrossRef] [PubMed]
- Sutharsan, S.; Dillenhoefer, S.; Welsner, M.; Stehling, F.; Brinkmann, F.; Burkhart, M.; Ellemunter, H.; Dittrich, A.-M.; Smaczny, C.; Eickmeier, O.; et al. Impact of elexacaftor/tezacaftor/ivacaftor on lung function, nutritional status, pulmonary exacerbation frequency and sweat chloride in people with cystic fibrosis: Real-world evidence from the German CF Registry. Lancet Reg. Health-Eur. 2023, 32, 100690. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.; Sermet-Gaudelus, I.; Sheppard, D.N. N1303K: Leaving no stone unturned in the search for transformational therapeutics. J. Cyst. Fibros. 2018, 17, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Paul, G.; Lee, J.; Yarlagadda, S.; McCoy, K.; Naren, A.P. Elexacaftor/Tezacaftor/Ivacaftor Improved Clinical Outcomes in a Patient with N1303K-CFTR Based on In Vitro Experimental Evidence. Am. J. Respir. Crit. Care Med. 2021, 204, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.-R.; Sermet-Gaudelus, I.; Durieu, I.; Kanaan, R.; Macey, J.; Grenet, D.; Porzio, M.; Coolen-Allou, N.; Chiron, R.; Marguet, C.; et al. The French Compassionate Program of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur. Respir. J. 2023, 61, 2202437. [Google Scholar] [CrossRef] [PubMed]
- Solomon, G.M.; Linnemann, R.W.; Rich, R.; Streby, A.; Buehler, B.; Hunter, E.; Vijaykumar, K.; Hunt, W.R.; Brewington, J.J.; Rab, A.; et al. Evaluation of elexacaftor–tezacaftor–ivacaftor treatment in individuals with cystic fibrosis and CFTRN1303K in the USA: A prospective, multicentre, open-label, single-arm trial. Lancet Respir. Med. 2024, 12, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Destefano, S.; Gees, M.; Hwang, T.-C. Physiological and pharmacological characterization of the N1303K mutant CFTR. J. Cyst. Fibros. 2018, 17, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Balázs, A.; Ziegahn, N.; Rubil, T.; Vitzthum, C.; Piehler, L.; Drescher, M.; Seidel, K.; Rohrbach, A.; Röhmel, J.; et al. Personalized CFTR Modulator Therapy for G85E and N1303K Homozygous Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2023, 24, 12365. [Google Scholar] [CrossRef] [PubMed]
- Kleinfelder, K.; Lotti, V.; Eramo, A.; Amato, F.; Lo Cicero, S.; Castelli, G.; Spadaro, F.; Farinazzo, A.; Dell’Orco, D.; Preato, S.; et al. In silico analysis and theratyping of an ultra-rare CFTR genotype (W57G/A234D) in primary human rectal and nasal epithelial cells. iScience 2023, 12, 108180. [Google Scholar] [CrossRef]
- Kroes, S.; Bierlaagh, M.C.; Lefferts, J.W.; Boni, A.; Muilwijk, D.; Viscomi, C.; Keijzer-Nieuwenhuijze, N.D.A.; Cristiani, L.; Niemöller, P.J.; Verburg, T.F.; et al. Elexacaftor/tezacaftor/ivacaftor efficacy in intestinal organoids with rare CFTR variants in comparison to CFTR-F508del and CFTR-wild type controls. J. Cyst. Fibros. 2025, 24, 175–182. [Google Scholar] [CrossRef]
- Kleinfelder, K.; Villella, V.R.; Hristodor, A.M.; Laudanna, C.; Castaldo, G.; Amato, F.; Melotti, P.; Sorio, C. Theratyping of the Rare CFTR Genotype A559T in Rectal Organoids and Nasal Cells Reveals a Relevant Response to Elexacaftor (VX-445) and Tezacaftor (VX-661) Combination. Int. J. Mol. Sci. 2023, 24, 10358. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| ID | Age | Sex | CFTR mut# 1 | CFTR mut# 2 | PI/PS | Sweat Chloride Conc. (mmol/L) | ppFEV1 | BMI | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | ||||||
| 1 | 33 | M | N1303K | 2183AA>G | PI | 119 | 102 | 36 | 48 | 21.6 | 22.3 |
| 2 | 35 | M | N1303K | 711+5G>A | PI | 107 | 103 | 68 | 87 | 21.1 | 23.7 |
| 3 | 37 | M | N1303K | 1717-1G>A | PI | 107 | 106 | 44 | 71 | 24.2 | 26.9 |
| 4 | 13 | F | N1303K | 1717-1G>A | PI | 104 | 93 | 81 | 93 | 17.2 | 18.0 |
| 5 | 13 | F | N1303K | 2183AA>G | PI | 87 | 84 | 82 | 87 | 17.3 | 18.1 |
| 6 | 32 | M | N1303K | 2183AA>G | PI | 93 | 98 | 91 | 99 | 21.6 | 21.4 |
| 7 | 23 | F | N1303K | 2183AA>G | PI | 89 | 108 | 14 | 21 | 14.0 | 16.2 |
| 8 | 48 | M | N1303K | P205S * | PI | 78 | 41 | 37 | 38 | 21.1 | 24.6 |
| 9 | 21 | M | N1303K | G542X | PI | 109 | 102 | 48 | 84 | 21.3 | 25.9 |
| 10 | 58 | F | N1303K | R1162X | PI | 103 | 94 | 44 | 55 | 20.1 | 20.4 |
| 11 | 23 | F | N1303K | W1282X | PI | 113 | 151 | 28 | 50 | 24.8 | 24.9 |
| 12 | 51 | F | 3849+10KbC>T | R1162X | PS | 71 | 54 | 57 | 64 | 22.4 | 21.8 |
| 13 | 56 | M | 3849+10KbC>T | R1162X | PS | 71 | 35 | 57 | 58 | 20.1 | 20.3 |
| 14 | 58 | M | 3849+10KbC>T | G542X | PS | 92 | 13 | 43 | 45 | 23.9 | 24.5 |
| 15 | 45 | F | 2789+5G>A | R553X | PS | 97 | 81 | 39 | 45 | 23.9 | 24.5 |
| 16 | 31 | F | R347P * | W1282X | PI | 84 | 44 | 38 | 56 | 20.4 | 22.2 |
| 17 | 56 | F | G85E * | 1584+18672bpA>G | PS | 67 | 52 | 91 | 100 | 20.4 | 20.6 |
| 18 | 28 | F | G85E * | 621+1G>T | PI | 100 | 68 | 34 | 36 | 18.3 | 19.1 |
| 19 | 42 | F | W57G | A234D * | PS | 104 | 14 | 62 | 65 | 19.5 | 21.0 |
| 20 | 51 | F | 2183AA>G | 711+3A>G | PS | 53 | 56 | 70 | 77 | 19.5 | 21.0 |
| 21 | 29 | F | 2183AA>G | 711+5G>A | PS | 96 | 73 | 28 | 36 | 16.8 | 17.3 |
| 22 | 52 | M | 2789+5G>A | 711+5G>A | PS | 125 | 92 | - | - | 23.2 | 24.4 |
| 23 | 33 | M | A559T | A559T | PI | 107 | 92 | 46 | 52 | 20.0 | 22.6 |
| ID | Treatment Duration (Months) | Adverse Effects (PEx Excluded) |
|---|---|---|
| 1 | 12 | skin erythema |
| 2 | 12 | None |
| 3 | 12 | None |
| 4 | 12 | None |
| 5 | 12 | None |
| 6 | 12 | None |
| 7 | 12 | None |
| 8 | 11 | None |
| 9 | 12 | None |
| 10 | 12 | abdominal swelling, short-term memory deficit, skin erythema |
| 11 | 12 | None |
| 12 | 12 | None |
| 13 | 12 | None |
| 14 | 12 | skin erythema |
| 15 | 12 | skin erythema, mild hypertension |
| 16 | 12 | short-term memory deficit, transient anomia |
| 17 | 11 | None |
| 18 | 12 | None |
| 19 | 12 | None |
| 20 | 8 | None |
| 21 | 12 | None |
| 22 | 12 | mild hypertension |
| 23 | 12 | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucca, F.; Volpi, S.; Ros, M.; Fabrizzi, B.; Meneghelli, I.; Bordicchia, M.; Buniotto, F.; Lancini, A.; Brignole, C.; Pauro, F.; et al. Elexacaftor/Tezacaftor/Ivacaftor Efficacy in a Cohort of Italian Patients with CFTR Rare Mutations. Int. J. Transl. Med. 2025, 5, 11. https://doi.org/10.3390/ijtm5010011
Lucca F, Volpi S, Ros M, Fabrizzi B, Meneghelli I, Bordicchia M, Buniotto F, Lancini A, Brignole C, Pauro F, et al. Elexacaftor/Tezacaftor/Ivacaftor Efficacy in a Cohort of Italian Patients with CFTR Rare Mutations. International Journal of Translational Medicine. 2025; 5(1):11. https://doi.org/10.3390/ijtm5010011
Chicago/Turabian StyleLucca, Francesca, Sonia Volpi, Mirco Ros, Benedetta Fabrizzi, Ilaria Meneghelli, Marica Bordicchia, Francesca Buniotto, Alessia Lancini, Cecilia Brignole, Francesca Pauro, and et al. 2025. "Elexacaftor/Tezacaftor/Ivacaftor Efficacy in a Cohort of Italian Patients with CFTR Rare Mutations" International Journal of Translational Medicine 5, no. 1: 11. https://doi.org/10.3390/ijtm5010011
APA StyleLucca, F., Volpi, S., Ros, M., Fabrizzi, B., Meneghelli, I., Bordicchia, M., Buniotto, F., Lancini, A., Brignole, C., Pauro, F., Bezzerri, V., & Cipolli, M. (2025). Elexacaftor/Tezacaftor/Ivacaftor Efficacy in a Cohort of Italian Patients with CFTR Rare Mutations. International Journal of Translational Medicine, 5(1), 11. https://doi.org/10.3390/ijtm5010011