
Clinical Implementation of MicroRNAs in Cancer Immunology


Abstract
1. Introduction
2. Biogenesis and Function of miRNAs
3. Signaling Pathways
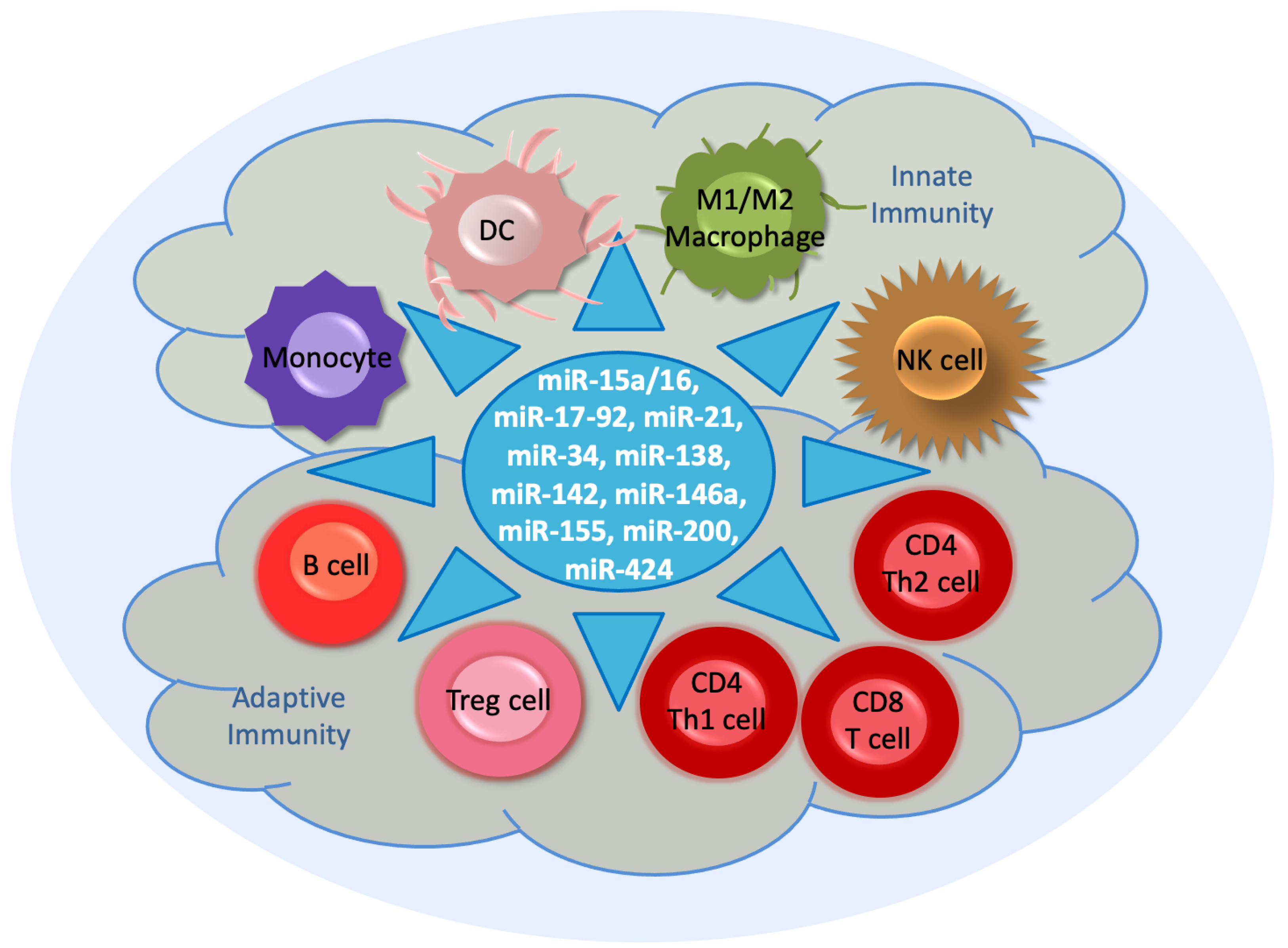
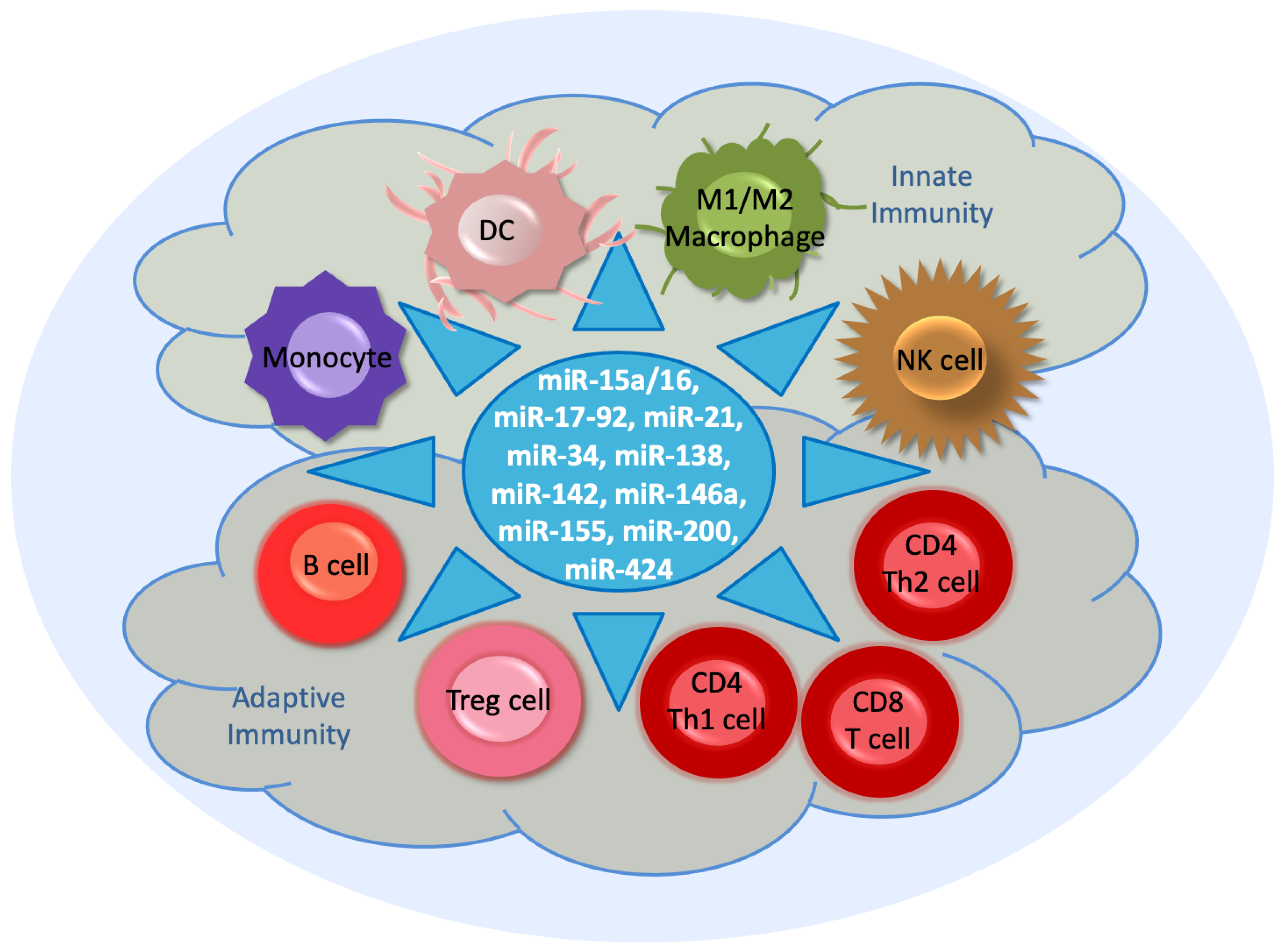
4. Immunoregulation by miRNAs
4.1. MiR-15a/16
4.2. MiR-17-92 Cluster
4.3. MiR-21
4.4. MiR-34
4.5. MiR-138
4.6. MiR-142
4.7. MiR-146a
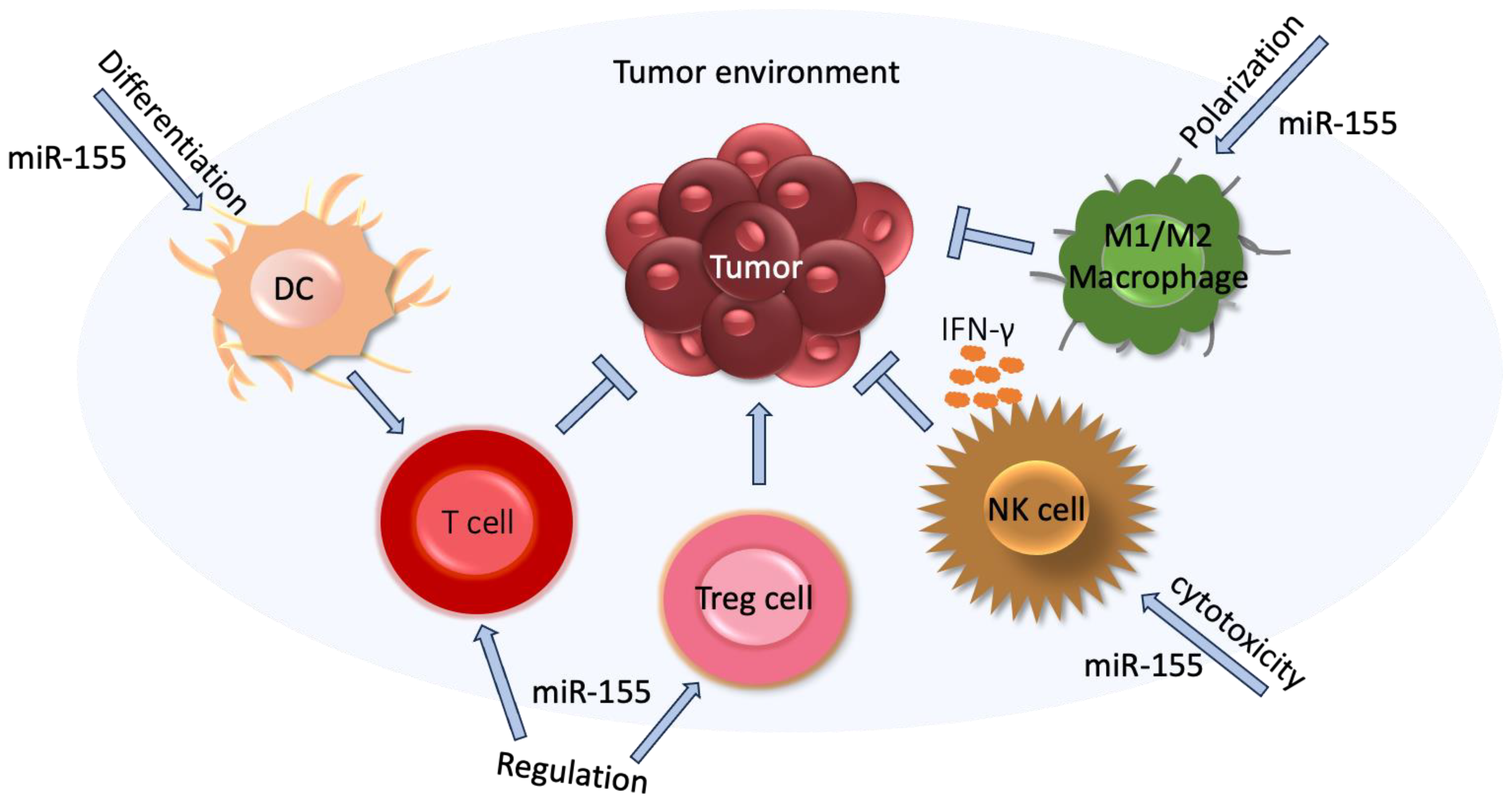
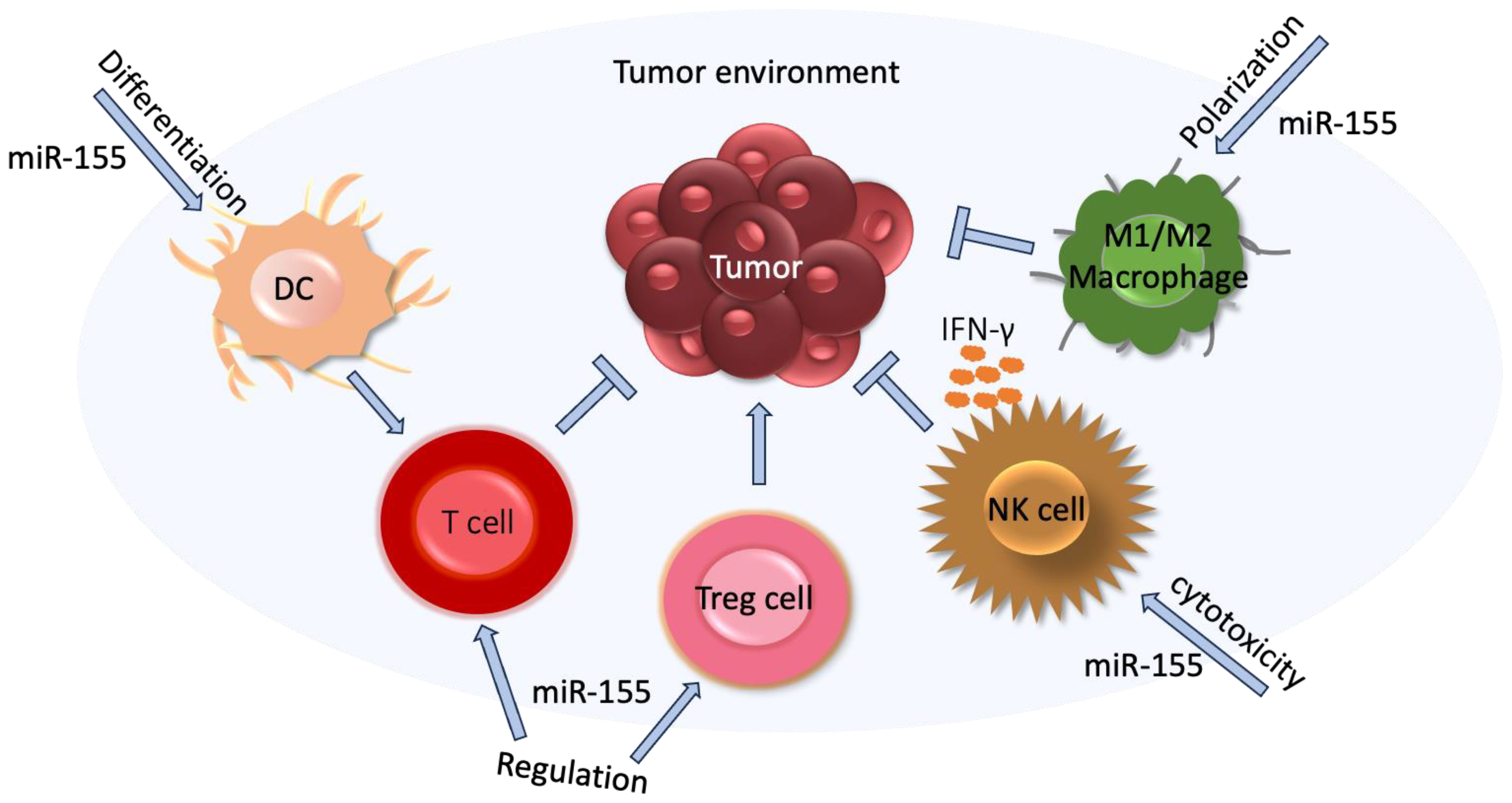
4.8. MiR-155
4.9. MiR-200 Family
4.10. MiR-424
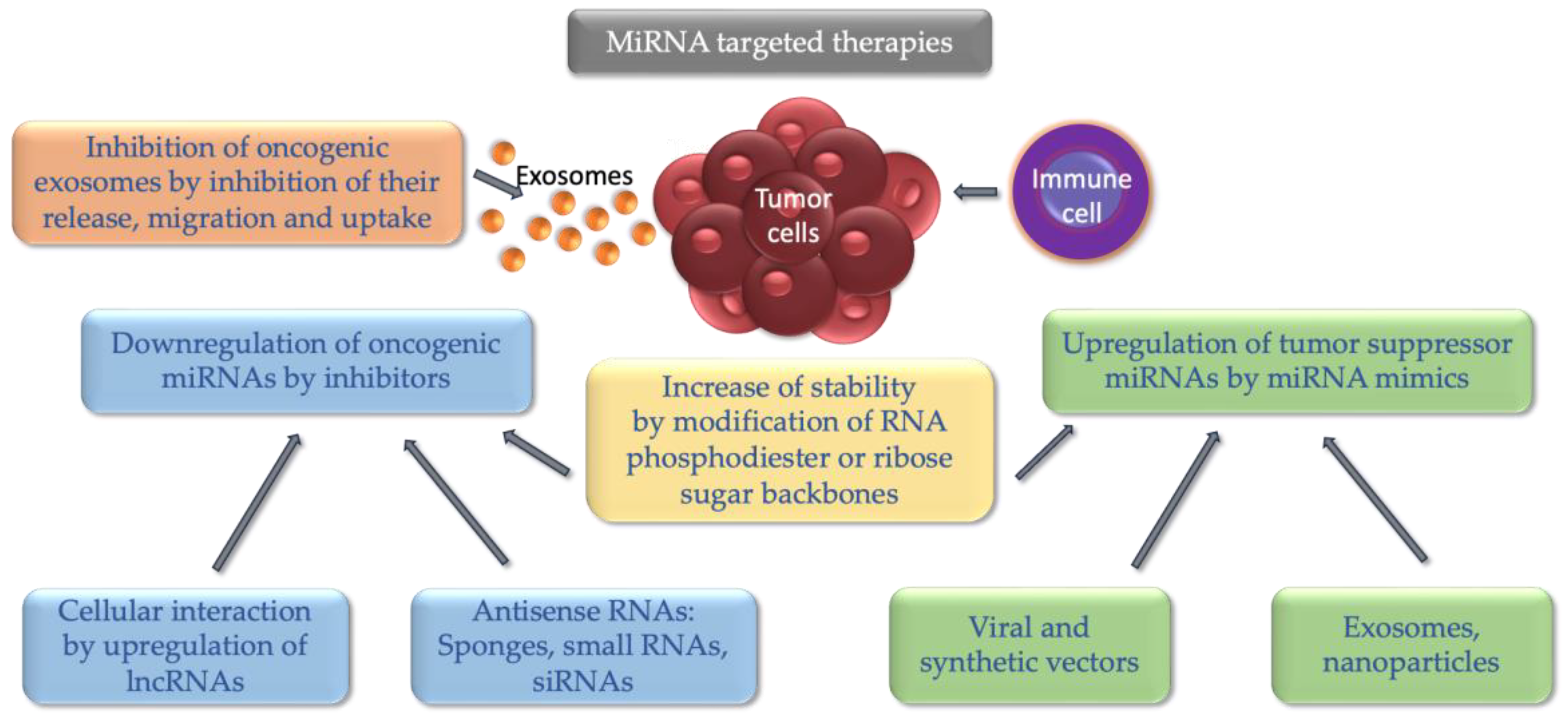
5. MiRNAs in Immunotherapies
5.1. MiR-15a/16
5.2. MiR-17-92 Cluster
5.3. MiR-21
5.4. MiR-34
5.5. MiR-138
5.6. MiR-142
5.7. MiR-146a
5.8. MiR-155
5.9. MiR-200 Family
5.10. MiR-424
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. S2), S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- van Vlerken-Ysla, L.; Tyurina, Y.Y.; Kagan, V.E.; Gabrilovich, D.I. Functional states of myeloid cells in cancer. Cancer Cell 2023, 41, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Rudensky, A.Y. Regulatory T Cells in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 459–477. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1979) 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, G.; Cava, C.; Castiglioni, I. Micrornas: New biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics 2015, 5, 1122–1143. [Google Scholar] [CrossRef] [PubMed]
- Kipkeeva, F.; Muzaffarova, T.; Korotaeva, A.; Mansorunov, D.; Apanovich, P.; Nikulin, M.; Malikhova, O.; Stilidi, I.; Karpukhin, A. The Features of Immune Checkpoint Gene Regulation by microRNA in Cancer. Int. J. Mol. Sci. 2022, 23, 9324. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Bortolin-Cavaille, M.L.; Dance, M.; Weber, M.; Cavaille, J. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res. 2009, 37, 3464–3473. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel Genes Coding for Small Expressed RNAs. Science (1979) 2001, 294, 853–858. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Wu, J.; Yang, J.; Cho, W.C.; Zheng, Y. Argonaute proteins: Structural features, functions and emerging roles. J. Adv. Res. 2020, 24, 317–324. [Google Scholar] [CrossRef]
- Slezak-Prochazka, I.; Durmus, S.; Kroesen, B.J.; van den Berg, A. MicroRNAs, macrocontrol: Regulation of miRNA processing. RNA 2010, 16, 1087–1095. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P. MicroRNA Shuttle from Cell-To-Cell by Exosomes and Its Impact in Cancer. Noncoding RNA 2019, 5, 28. [Google Scholar] [CrossRef]
- Schwarzenbach, H. CNAPS and General Medicine: Nucleic Acids in Early Diagnosis, Prognosis and Treatment Monitoring. An Introduction; Gahan, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 5, pp. 143–163. [Google Scholar]
- Berindan-Neagoe, I.; Monroig, P.d.C.; Pasculli, B.; Calin, G.A. MicroRNAome genome: A treasure for cancer diagnosis and therapy. CA Cancer J. Clin. 2014, 64, 311–336. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P.B. Interplay between LncRNAs and microRNAs in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 8095. [Google Scholar] [CrossRef]
- Müller, V.; Oliveira-Ferrer, L.; Steinbach, B.; Pantel, K.; Schwarzenbach, H. Interplay of lncRNA H19/miR-675 and lncRNA NEAT1/miR-204 in breast cancer. Mol. Oncol. 2019, 13, 1137–1149. [Google Scholar] [CrossRef]
- Venkatesh, J.; Wasson, M.C.D.; Brown, J.M.; Fernando, W.; Marcato, P. LncRNA-miRNA axes in breast cancer: Novel points of interaction for strategic attack. Cancer Lett. 2021, 509, 81–88. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Schwarzenbach, H. Biological and Clinical Relevance of H19 in Colorectal Cancer Patients. EBioMedicine 2016, 13, 9–10. [Google Scholar] [CrossRef]
- Vennin, C.; Spruyt, N.; Dahmani, F.; Julien, S.; Bertucci, F.; Finetti, P.; Chassat, T.; Bourette, R.P.; Le Bourhis, X.; Adriaenssens, E. H19 non coding RNA-derived miR-675 enhances tumorigenesis and metastasis of breast cancer cells by downregulating c-Cbl and Cbl-b. Oncotarget 2015, 6, 29209–29223. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Bright, J.J.; Sriram, S. TGF-β Inhibits IL-12-Induced Activation of Jak-STAT Pathway in T Lymphocytes. J. Immunol. 1998, 161, 1772–1777. [Google Scholar] [CrossRef]
- Tasian, S.K.; Doral, M.Y.; Borowitz, M.J.; Wood, B.L.; Chen, I.M.; Harvey, R.C.; Gastier-Foster, J.M.; Willman, C.L.; Hunger, S.P.; Mullighan, C.G.; et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012, 120, 833–842. [Google Scholar] [CrossRef]
- Karin, M.; Baud, V. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar]
- Xing, Y.; Wang, Z.; Lu, Z.; Xia, J.; Xie, Z.; Jiao, M.; Liu, R.; Chu, Y. MicroRNAs: Immune modulators in cancer immunotherapy. Immunother. Adv. 2021, 1, ltab006. [Google Scholar] [CrossRef]
- Yi, M.; Xu, L.; Jiao, Y.; Luo, S.; Li, A.; Wu, K. The role of cancer-derived microRNAs in cancer immune escape. J. Hematol. Oncol. 2020, 13, 25. [Google Scholar] [CrossRef]
- Huang, E.; Liu, R.; Chu, Y. MiRNA-15a/16: As tumor suppressors and more. Future Oncol. 2015, 11, 2351–2363. [Google Scholar] [CrossRef]
- Musumeci, M.; Coppola, V.; Addario, A.; Patrizii, M.; Maugeri-Saccá, M.; Memeo, L.; Colarossi, C.; Francescangeli, F.; Biffoni, M.; Collura, D.; et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 2011, 30, 4231–4242. [Google Scholar] [CrossRef]
- Sullivan, R.P.; Leong, J.W.; Schneider, S.E.; Keppel, C.R.; Germino, E.; French, A.R.; Fehniger, T.A. MicroRNA-Deficient Murine NK Cells Exhibit Impaired Development and Survival but Enhanced IFN-γ Production In Vitro and In Vivo. Blood 2011, 118, 357. [Google Scholar] [CrossRef]
- Tao, Z.; Xu, S.; Ruan, H.; Wang, T.; Song, W.; Qian, L.; Chen, K. MiR-195/-16 Family Enhances Radiotherapy via T Cell Activation in the Tumor Microenvironment by Blocking the PD-L1 Immune Checkpoint. Cell. Physiol. Biochem. 2018, 48, 801–814. [Google Scholar] [CrossRef]
- Jia, X.; Li, X.; Shen, Y.; Miao, J.; Liu, H.; Li, G.; Wang, Z. MiR-16 regulates mouse peritoneal macrophage polarization and affects T-cell activation. J. Cell. Mol. Med. 2016, 20, 1898–1907. [Google Scholar] [CrossRef]
- Kuo, G.; Wu, C.Y.; Yang, H.Y. MiR-17-92 cluster and immunity. J. Formos. Med. Assoc. 2019, 118, 2–6. [Google Scholar] [CrossRef]
- Sasaki, K.; Kohanbash, G.; Hoji, A.; Ueda, R.; McDonald, H.A.; Reinhart, T.A.; Martinson, J.; Lotze, M.T.; Marincola, F.M.; Wang, E.; et al. MiR-17-92 expression in differentiated T cells—Implications for cancer immunotherapy. J. Transl. Med. 2010, 8, 8–17. [Google Scholar] [CrossRef]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat. Cell Biol. 2007, 9, 775–785. [Google Scholar] [CrossRef]
- Jiang, S.; Li, C.; Olive, V.; Lykken, E.; Feng, F.; Sevilla, J.; Wan, Y.; He, L.; Li, Q.J. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood 2011, 118, 5487–5497. [Google Scholar] [CrossRef]
- Fayyad-Kazan, H.; Rouas, R.; Fayyad-Kazan, M.; Badran, R.; El Zein, N.; Lewalle, P.; Najar, M.; Hamade, E.; Jebbawi, F.; Merimi, M.; et al. MicroRNA profile of circulating CD4-positive regulatory T cells in human adults and impact of differentially expressed microRNAs on expression of two genes essential to their function. J. Biol. Chem. 2012, 287, 9910–9922. [Google Scholar] [CrossRef]
- Sahraei, M.; Chaube, B.; Liu, Y.; Sun, J.; Kaplan, A.; Price, N.L.; Ding, W.; Oyaghire, S.; García-Milan, R.; Mehta, S.; et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J. Clin. Investig. 2019, 129, 5518–5536. [Google Scholar] [CrossRef]
- Arghiani, N.; Matin, M.M. MiR-21: A Key Small Molecule with Great Effects in Combination Cancer Therapy. Nucleic Acid Ther. 2021, 31, 271–283. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, N.; Hao, F.; Song, Y.; Wang, Z.; Ni, Y.; Ding, L. The Regulatory Role of Non-coding RNAs on Programmed Cell Death Four in Inflammation and Cancer. Front. Oncol. 2019, 9, 919. [Google Scholar] [CrossRef]
- Chang, C.C.; Zhang, Q.Y.; Liu, Z.; Clynes, R.A.; Suciu-Foca, N.; Vlad, G. Downregulation of Inflammatory MicroRNAs by Ig-like Transcript 3 Is Essential for the Differentiation of Human CD8+ T Suppressor Cells. J. Immunol. 2012, 188, 3042–3052. [Google Scholar] [CrossRef]
- Iurca, I.; Tirpe, A.; Zimta, A.A.; Moldovan, C.; Gulei, D.; Slabý, O.; Condorelli, G.; Berindan-Neagoe, I. Macrophages Interaction and MicroRNA Interplay in the Modulation of Cancer Development and Metastasis. Front. Immunol. 2020, 11, 870. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, X.; Li, M.; Zhao, J.; Wang, X.; Zhu, H. ADAR1 improved Treg cell function through the miR-21b/Foxp3 axis and inhibits the progression of acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Int. Immunopharmacol. 2023, 115, 109620. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2022, 211, 157–182. [Google Scholar] [CrossRef]
- Wu, X.; Cheng, Y.S.L.; Matthen, M.; Yoon, A.; Schwartz, G.K.; Bala, S.; Taylor, A.M.; Momen-Heravi, F. Down-regulation of the tumor suppressor miR-34a contributes to head and neck cancer by up-regulating the MET oncogene and modulating tumor immune evasion. J. Exp. Clin. Cancer Res. 2021, 40, 70. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef]
- Xie, M.; Wang, J.; Gong, W.; Xu, H.; Pan, X.; Chen, Y.; Ru, S.; Wang, H.; Chen, X.; Zhao, Y.; et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J. Autoimmun. 2019, 102, 96–113. [Google Scholar] [CrossRef]
- Sha, H.H.; Wang, D.D.; Chen, D.; Liu, S.W.; Wang, Z.; Yan, D.L.; Dong, S.C.; Feng, J.F. MiR-138: A promising therapeutic target for cancer. Tumor Biol. 2017, 39, 1010428317697575. [Google Scholar] [CrossRef]
- Wei, J.; Nduom, E.K.; Kong, L.Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.; Huang, N.; Qia, W.; Zhou, S.; et al. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro Oncol. 2016, 18, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Li, P.; Song, P.; Li, Y.; Zhou, S.; Su, Q.; Li, X.; Yu, Y.; Li, P.; Feng, M.; et al. MicroRNA-138-5p Suppresses Non-small Cell Lung Cancer Cells by Targeting PD-L1/PD-1 to Regulate Tumor Microenvironment. Front. Cell. Dev. Biol. 2020, 8, 540. [Google Scholar]
- Rasoolnezhad, M.; Safaralizadeh, R.; Hosseinpourfeizi, M.A.; Banan-Khojasteh, S.M.; Baradaran, B. MiRNA-138–5p: A strong tumor suppressor targeting PD-L-1 inhibits proliferation and motility of breast cancer cells and induces apoptosis. Eur. J. Pharmacol. 2021, 896, 173933. [Google Scholar] [CrossRef]
- Xun, J.; Du, L.; Gao, R.; Shen, L.; Wang, D.; Kang, L.; Chen, C.; Zhang, Z.; Zhang, Y.; Yue, S.; et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-Associated macrophages through inhibition of KDM6B. Theranostics 2021, 11, 6847–6859. [Google Scholar] [CrossRef]
- Olson, W.J.; Derudder, E. The miR-142 miRNAs: Shaping the naïve immune system. Immunol. Lett. 2023, 261, 37–46. [Google Scholar] [CrossRef]
- Wan, J.; Ling, X.; Peng, B.; Ding, G. MiR-142-5p regulates CD4+ T cells in human non-small cell lung cancer through PD-L1 expression via the PTEN pathway. Oncol. Rep. 2018, 40, 272–282. [Google Scholar] [CrossRef]
- Jia, L.; Xi, Q.; Wang, H.; Zhang, Z.; Liu, H.; Cheng, Y.; Guo, X.; Zhang, J.; Zhang, Q.; Zhang, L.; et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem. Biophys. Res. Commun. 2017, 488, 425–431. [Google Scholar] [CrossRef]
- Sonda, N.; Simonato, F.; Peranzoni, E.; Calì, B.; Bortoluzzi, S.; Bisognin, A.; Wang, E.; Marincola, F.M.; Naldini, L.; Gentner, B.; et al. MiR-142-3p Prevents Macrophage Differentiation during Cancer-Induced Myelopoiesis. Immunity 2013, 38, 1236–1249. [Google Scholar] [CrossRef]
- Xu, S.; Wei, J.; Wang, F.; Kong, L.Y.; Ling, X.Y.; Nduom, E.; Gabrusiewicz, K.; Doucette, T.; Yang, Y.; Yaghi, N.K.; et al. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J. Natl. Cancer Inst. 2014, 106, dju162. [Google Scholar] [CrossRef]
- Iacona, J.R.; Lutz, C.S. miR-146a-5p: Expression, regulation, and functions in cancer. Wiley Interdiscip. Rev. RNA 2019, 10, e1533. [Google Scholar] [CrossRef]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in Controlling Treg Cell-Mediated Regulation of Th1 Responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.M.; Callen, D.F. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1236–1243. [Google Scholar] [CrossRef]
- Vigorito, E.; Kohlhaas, S.; Lu, D.; Leyland, R. miR-155: An ancient regulator of the immune system. Immunol. Rev. 2013, 253, 146–157. [Google Scholar] [CrossRef]
- Schmeier, S.; MacPherson, C.R.; Essack, M.; Kaur, M.; Schaefer, U.; Suzuki, H.; Hayashizaki, Y.; Bajic, V.B. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genom. 2009, 10, 595. [Google Scholar] [CrossRef]
- Trotta, R.; Chen, L.; Ciarlariello, D.; Josyula, S.; Mao, C.; Costinean, S.; Yu, L.; Butchar, J.B.; Tridandapani, S.; Croce, C.M.; et al. miR-155 regulates IFN-γ production in natural killer cells. Blood 2012, 119, 3478–3485. [Google Scholar] [CrossRef]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science (1979) 2007, 316, 608–611. [Google Scholar] [CrossRef]
- Kohlhaas, S.; Garden, O.A.; Scudamore, C.; Turner, M.; Okkenhaug, K.; Vigorito, E. Cutting Edge: The Foxp3 Target miR-155 Contributes to the Development of Regulatory T Cells. J. Immunol. 2009, 182, 2578–2582. [Google Scholar] [CrossRef]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M.; et al. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593–602. [Google Scholar] [CrossRef]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R.; et al. MicroRNA-155 is required for effector cd8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef]
- Lu, L.F.; Thai, T.H.; Calado, D.P.; Chaudhry, A.; Kubo, M.; Tanaka, K.; Loeb, G.B.; Lee, H.; Yoshimura, A.; Rajewsky, K.; et al. Foxp3-Dependent MicroRNA155 Confers Competitive Fitness to Regulatory T Cells by Targeting SOCS1 Protein. Immunity 2009, 30, 80–91. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Etzrodt, M.; De Palma, M.; Pittet, M.J. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 2013, 34, 350–359. [Google Scholar] [CrossRef]
- Zhang, H.F.; Xu, L.Y.; Li, E.M. A Family of Pleiotropically Acting MicroRNAs in Cancer Progression, miR-200: Potential Cancer Therapeutic Targets. Curr. Pharm. Des. 2014, 20, 1896–1903. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Chen, L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Lin, W.; Diao, L.; Wang, J.; Roybal, J.; et al. Regulation of tumor cell PD-L1 expression by microRNA-200 and control of lung cancer metastasis. J. Clin. Oncol. 2014, 32 (Suppl. S15), 5s. [Google Scholar] [CrossRef]
- Han, S.; Gonzalo, D.H.; Feely, M.; Delitto, D.; Behrns, K.E.; Beveridge, M.; Zhang, D.Y.; Thomas, R.; Trevino, J.G.; Schmittgen, T.D.; et al. The pancreatic tumor microenvironment drives changes in miRNA expression that promote cytokine production and inhibit migration by the tumor associated stroma. Oncotarget 2017, 8, 54054–54067. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Askari, A.; Hussen, B.M.; Taheri, M.; Akbari Dilmaghani, N. Role of miR-424 in the carcinogenesis. Clin. Transl. Oncol. 2024, 26, 16–38. [Google Scholar] [CrossRef]
- Xu, S.; Tao, Z.; Hai, B.; Liang, H.; Shi, Y.; Wang, T.; Song, W.; Chen, Y.; QuYang, J.; Chen, J.; et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat. Commun. 2016, 7, 11406. [Google Scholar] [CrossRef]
- Cortez, M.A.; Anfossi, S.; Ramapriyan, R.; Menon, H.; Atalar, S.C.; Aliru, M.; Welsh, J.; Calin, G.A. Role of miRNAs in immune responses and immunotherapy in cancer. Genes Chromosomes Cancer 2019, 58, 244–253. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Guo, W.; Wu, Z.; Chen, J.; Guo, S.; You, W.; Wang, S.; Ma, J.; Wang, H.; Wang, X.; Wang, H.; et al. Nanoparticle delivery of miR-21-3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J. Immunother. Cancer 2022, 10, e004381. [Google Scholar] [CrossRef]
- Xi, J.; Huang, Q.; Wang, L.; Ma, X.; Deng, Q.; Kumar, M.; Zhou, Z.; Li, L.; Zeng, Z.; Young, K.H.; et al. MiR-21 depletion in macrophages promotes tumoricidal polarization and enhances PD-1 immunotherapy. Oncogene 2018, 37, 3151–3165. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Chen, Y.; Yang, M.; Zhang, H.; Wang, W. Concomitant inhibition of B7-H3 and PD-L1 expression by a novel and synthetic microRNA delivers potent antitumor activities in colorectal tumor models. Investig. New Drugs 2021, 39, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Khani, A.T.; Sharifzad, F.; Mardpour, S.; Hassan, Z.M.; Ebrahimi, M. Tumor extracellular vesicles loaded with exogenous Let-7i and miR-142 can modulate both immune response and tumor microenvironment to initiate a powerful anti-tumor response. Cancer Lett. 2021, 501, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Achberger, S.; Aldrich, W.; Elson, P.; Garcia, J.; Dreicer, R. Differential immunologic and microRNA effects of 2 dosing regimens of recombinant human granulocyte/macrophage colony stimulating factor. J. Immunother. 2012, 35, 587–594. [Google Scholar] [CrossRef]
- Witten, L.; Slack, F.J. MiR-155 as a novel clinical target for hematological malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef]
- Byun, Y.; Choi, Y.C.; Jeong, Y.; Lee, G.; Yoon, S.; Jeong, Y.; Yoon, J.; Baek, K. MiR-200c downregulates HIF-1α and inhibits migration of lung cancer cells. Cell Mol. Biol. Lett. 2019, 24, 28. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, S.; Wu, H.; Zhang, L.; Dai, X.; Hu, J.; Xue, J.; Liu, T.; Liang, Y.; Wu, G. MiR-200c increases the radiosensitivity of non-small-cell lung cancer cell line A549 by targeting VEGF-VEGFR2 pathway. PLoS ONE 2013, 8, e78344. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Phung, C.D.; Tran, T.H.; Pham, T.T.; Pham, L.M.; Nguyen, T.T.; Jeong, J.H.; Choi, H.G.; Ku, S.K.; Yong, C.S.; et al. Manipulating immune system using nanoparticles for an effective cancer treatment: Combination of targeted therapy and checkpoint blockage miRNA. J. Control. Release 2021, 329, 524–537. [Google Scholar] [CrossRef]
- Buck, A.K.; Serfling, S.E.; Lindner, T.; Hänscheid, H.; Schirbel, A.; Hahner, S.; Fassnacht, M.; Einsele, H.; Werner, R.A. CXCR4-targeted theranostics in oncology. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4133–4144. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From garbage bins to promising therapeutic targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef]
- Tenchov, R.; Sasso, J.M.; Wang, X.; Liaw, W.S.; Chen, C.A.; Zhou, Q.A. Exosomes Nature’s Lipid Nanoparticles, a Rising Star in Drug Delivery and Diagnostics. ACS Nano 2022, 16, 17802–17846. [Google Scholar] [CrossRef]
- Xie, J.; Shen, Q.; Huang, K.; Zheng, T.; Cheng, L.; Zhang, Z.; Yu, Y.; Liao, G.; Wang, X.; Li, C. Oriented Assembly of Cell-Mimicking Nanoparticles via a Molecular Affinity Strategy for Targeted Drug Delivery. ACS Nano 2019, 13, 5268–5277. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A Novel Nanoparticle Drug Delivery System: The Anti-inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| miRNAs | Targets |
|---|---|
| miR-10a | MDSCs |
| miR-18a | PD-L1 |
| miR-20a | MHC-I chain-related molecules A/B, T cells |
| miR-24 | T cells |
| miR-27a | Fibroblasts |
| miR-29a | MDSCs, MHC-I chain-related molecules B |
| miR-93 | MHC-I chain-related molecules A, PD-L1 |
| miR-106a | MHC-I chain-related molecules A/B, T cells |
| miR-106b | PD-L1 |
| miR-107 | MHC-I chain-related molecules B, MDSCs |
| miR-125b | MHC-I chain-related molecules A, macrophages |
| miR-140 | PD-L1 |
| miR-148a | PD-L1 |
| miR-153 | MHC-I chain-related molecules A |
| miR-181d | Macrophages |
| miR-193a | PD-L1 |
| miR-195 | MHC-I chain-related molecules B, PD-L1 |
| miR-197 | PD-L1 |
| miR-222 | Macrophages |
| miR-302c | MHC-I chain-related molecules A/B |
| miR-340 | PD-L1 |
| miR-373 | MHC-I chain-related molecules A/B |
| miR-375 | PD-L1 |
| miR-383 | PD-L1 |
| miR-497 | PD-L1 |
| miR-519a | MHC-I chain-related molecules A |
| miR-520c | MHC-I chain-related molecules A/B |
| miR-690 | T cells |
| miR-873 | PD-L1 |
| miR-891a | T cells |
| miR-940 | Macrophages |
| miR-1246 | Macrophages |
| miR-1247 | Fibroblasts |
| miR-1908 | T cells |
| miR-3127 | PD-L1 |
| miR-3609 | PD-L1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarzenbach, H. Clinical Implementation of MicroRNAs in Cancer Immunology. Int. J. Transl. Med. 2024, 4, 53-71. https://doi.org/10.3390/ijtm4010003
Schwarzenbach H. Clinical Implementation of MicroRNAs in Cancer Immunology. International Journal of Translational Medicine. 2024; 4(1):53-71. https://doi.org/10.3390/ijtm4010003
Chicago/Turabian StyleSchwarzenbach, Heidi. 2024. "Clinical Implementation of MicroRNAs in Cancer Immunology" International Journal of Translational Medicine 4, no. 1: 53-71. https://doi.org/10.3390/ijtm4010003
APA StyleSchwarzenbach, H. (2024). Clinical Implementation of MicroRNAs in Cancer Immunology. International Journal of Translational Medicine, 4(1), 53-71. https://doi.org/10.3390/ijtm4010003