
Chronic Inhibition of Nitric Oxide Synthases Impairs Spatiotemporal Learning and Memory to a Similar Extent in C57BL/6 and hAPP23+/− Mice

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Set-Up
2.2. Spatiotemporal Learning and Memory
2.3. Blood Pressure Measurements
2.4. Echocardiography and PWV Measurements
2.5. Rodent Oscillatory Tension Set-Up for Arterial Compliance (ROTSAC)
2.6. Histology
2.7. Statistical Analysis
3. Results
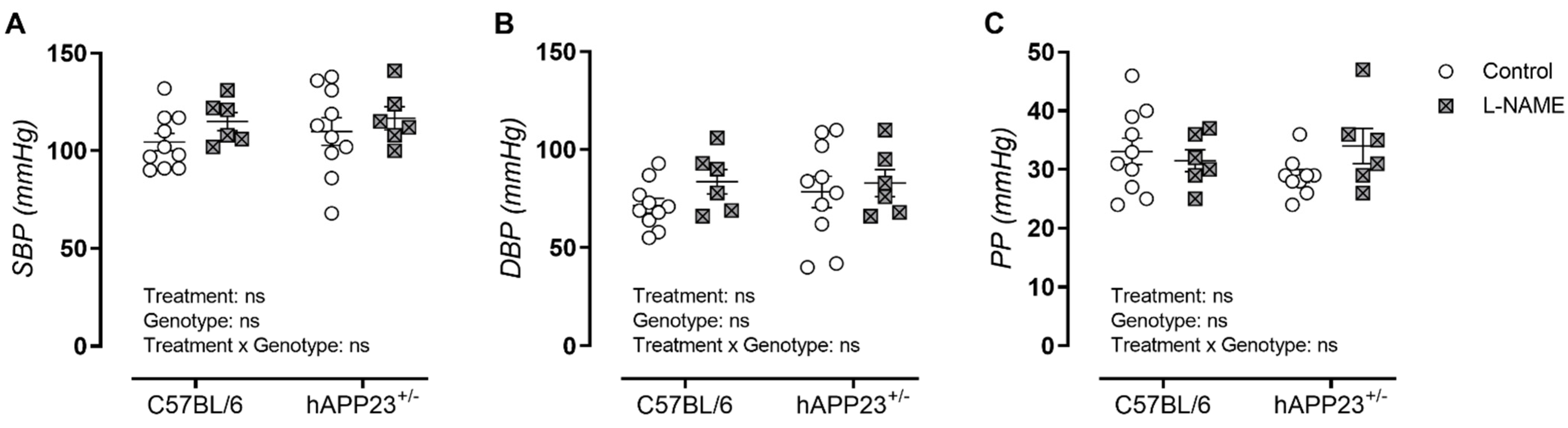
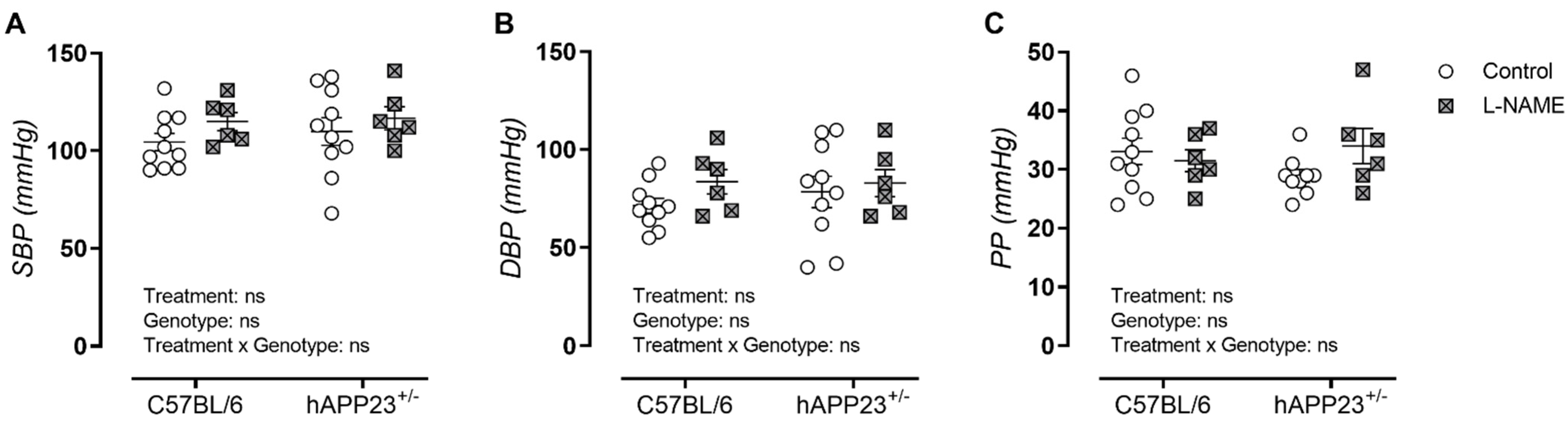
3.1. L-NAME Treatment Does Not Affect Peripheral Blood Pressure
3.2. L-NAME Treatment Affects Systolic Cardiac Function in hAPP23+/− and C57BL/6 Mice
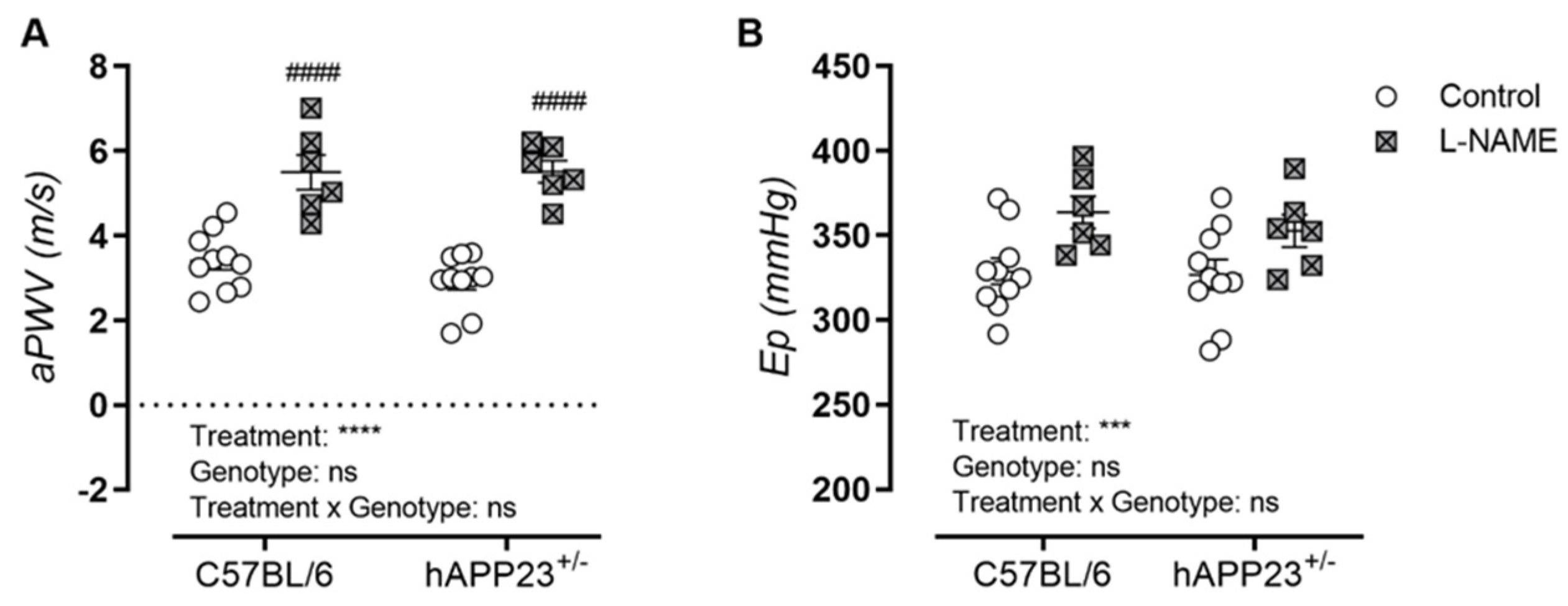
3.3. L-NAME Treatment Affects AS In Vivo and Ex Vivo in hAPP23+/− and C57BL/6 Mice
3.4. L-NAME Treatment Worsens Spatiotemporal Learning and Memory Equally in hAPP23+/− and C57BL/6 Mice
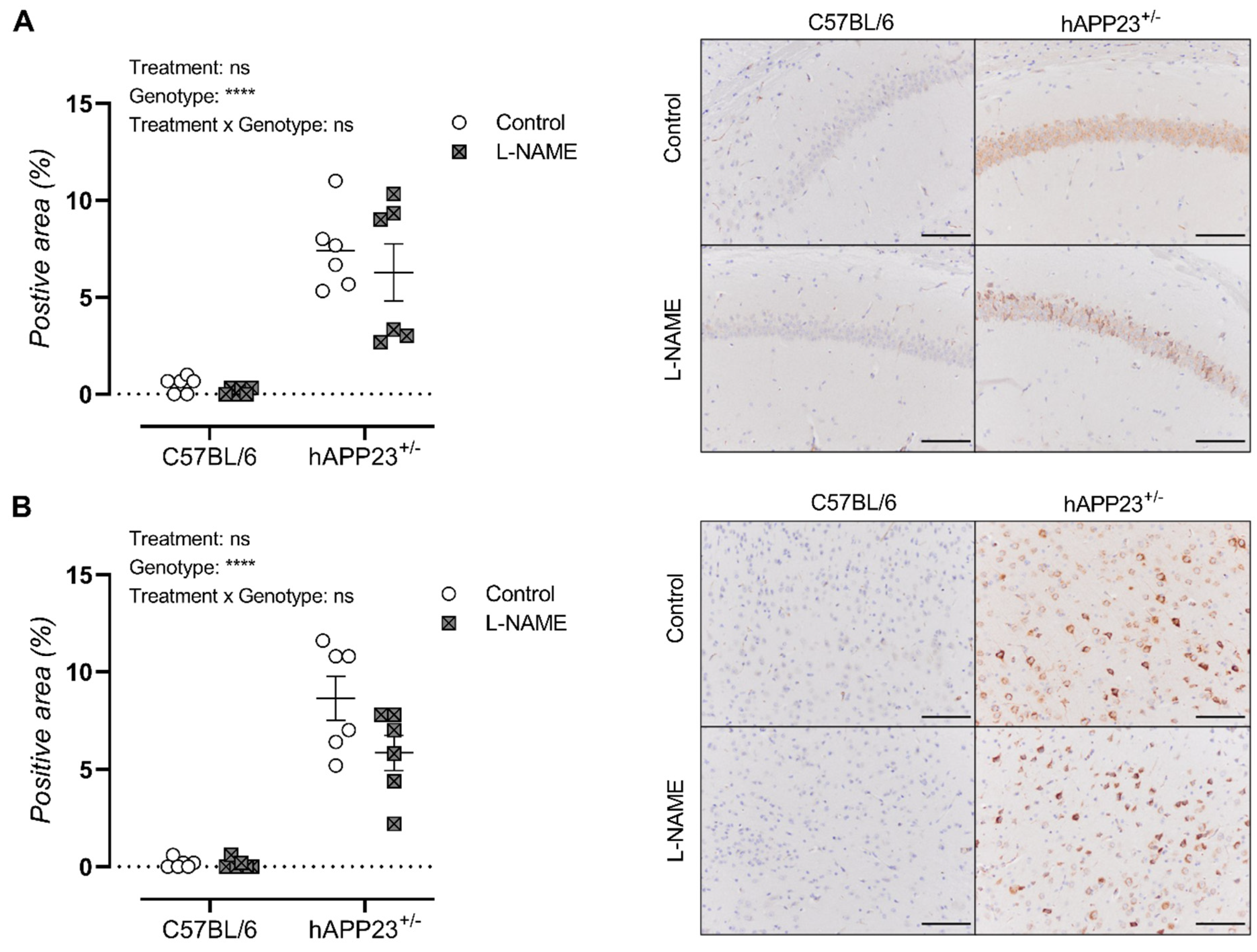
3.5. L-NAME Treatment Does Not Affect Cerebral Amyloid Load in hAPP23+/− Animals
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iulita, M.F.; Noriega de la Colina, A.; Girouard, H. Arterial stiffness, cognitive impairment and dementia: Confounding factor or real risk? J. Neurochem. 2018, 144, 527–548. [Google Scholar] [CrossRef] [PubMed]
- van Sloten, T.T.; Protogerou, A.D.; Henry, R.M.; Schram, M.T.; Launer, L.J.; Stehouwer, C.D. Association between arterial stiffness, cerebral small vessel disease and cognitive impairment: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2015, 53, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, J.O.; Martinet, W.; Van Dam, D.; De Meyer, G.R. Inflammation, Nitro-Oxidative Stress, Impaired Autophagy, and Insulin Resistance as a Mechanistic Convergence between Arterial Stiffness and Alzheimer’s Disease. Front. Mol. Biosci. 2021, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, M.F.; Safar, M.E. Relationship between aortic stiffening and microvascular disease in brain and kidney: Cause and logic of therapy. Hypertension 2005, 46, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W. Arterial stiffness: Detection and consequences in cognitive impairment and dementia of the elderly. J. Alzheimer’s Dis. 2012, 32, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.W.; Byun, M.S.; Yi, D.; Lee, J.H.; Jeon, S.Y.; Lee, Y.; Kee, B.S.; Lee, D.Y.; Group, K.R. The Ankle–Brachial Index Is Associated with Cerebral β-Amyloid Deposition in Cognitively Normal Older Adults. J. Gerontol. Ser. A 2019, 74, 1141–1148. [Google Scholar] [CrossRef]
- Hughes, T.M.; Wagenknecht, L.E.; Craft, S.; Mintz, A.; Heiss, G.; Palta, P.; Wong, D.; Zhou, Y.; Knopman, D.; Mosley, T.H. Arterial stiffness and dementia pathology: Atherosclerosis Risk in Communities (ARIC)-PET Study. Neurology 2018, 90, e1248–e1256. [Google Scholar] [CrossRef]
- Pasha, E.P.; Rutjes, E.; Tomoto, T.; Tarumi, T.; Stowe, A.; Claassen, J.; Munro Cullum, C.; Zhu, D.C.; Zhang, R. Carotid Stiffness is Associated with Brain Amyloid-beta Burden in Amnestic Mild Cognitive Impairment. J. Alzheimers Dis. 2020, 74, 925–935. [Google Scholar] [CrossRef]
- Hughes, T.M.; Kuller, L.H.; Barinas-Mitchell, E.J.; McDade, E.M.; Klunk, W.E.; Cohen, A.D.; Mathis, C.A.; DeKosky, S.T.; Price, J.C.; Lopez, O.L. Arterial stiffness and β-amyloid progression in nondemented elderly adults. JAMA Neurol. 2014, 71, 562–568. [Google Scholar] [CrossRef]
- Hendrickx, J.O.; De Moudt, S.; Calus, E.; De Deyn, P.P.; Van Dam, D.; De Meyer, G.R. Long-Term Pharmacological Inhibition of the Activity of All NOS Isoforms Rather Than Genetic Knock-Out of Endothelial NOS Leads to Impaired Spatial Learning and Memory in C57BL/6 Mice. Biomedicines 2021, 9, 1905. [Google Scholar] [CrossRef]
- Chiba, T.; Sakuma, K.; Komatsu, T.; Cao, X.; Aimoto, M.; Nagasawa, Y.; Shimizu, K.; Takahashi, M.; Hori, Y.; Shirai, K. Physiological role of nitric oxide for regulation of arterial stiffness in anesthetized rabbits. J. Pharmacol. Sci. 2019, 139, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Dubey, H.; Gulati, K.; Ray, A. Alzheimer’s Disease: A Contextual Link with Nitric Oxide Synthase. Curr. Mol. Med. 2020, 20, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Esch, T.; Ptacek, R.; Kream, R.M. Dysregulation of Nitric Oxide Signaling in Microglia: Multiple Points of Functional Convergence in the Complex Pathophysiology of Alzheimer Disease. Med. Sci. Monit. 2020, 26, e927739. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Katusic, Z.S. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J. Cereb. Blood Flow. Metab. 2020, 40, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.; Li, H.; Chreifi, G.; Poulos, T.L.; Silverman, R.B. Optimization of Blood–Brain Barrier Permeability with Potent and Selective Human Neuronal Nitric Oxide Synthase Inhibitors Having a 2-Aminopyridine Scaffold. J. Med. Chem. 2019, 62, 2690–2707. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Doubal, F.N.; Valdes-Hernandez, M.; Wang, X.; Chappell, F.M.; Shuler, K.; Armitage, P.A.; Carpenter, T.C.; Dennis, M.S. Blood-brain barrier permeability and long-term clinical and imaging outcomes in cerebral small vessel disease. Stroke 2013, 44, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Katusic, Z.S. Loss of Endothelial Nitric Oxide Synthase Promotes p25 Generation and Tau Phosphorylation in a Murine Model of Alzheimer’s Disease. Circ. Res. 2016, 119, 1128–1134. [Google Scholar] [CrossRef]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.-H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Bürki, K.; Frey, P.; Paganetti, P.A. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Nagano, K.; Ishida, J.; Unno, M.; Matsukura, T.; Fukamizu, A. Apelin elevates blood pressure in ICR mice with L-NAME-induced endothelial dysfunction. Mol. Med. Rep. 2013, 7, 1371–1375. [Google Scholar] [CrossRef]
- Suda, O.; Tsutsui, M.; Morishita, T.; Tanimoto, A.; Horiuchi, M.; Tasaki, H.; Huang, P.L.; Sasaguri, Y.; Yanagihara, N.; Nakashima, Y. Long-term treatment with Nω-nitro-L-arginine methyl ester causes arteriosclerotic coronary lesions in endothelial nitric oxide synthase-deficient mice. Circulation 2002, 106, 1729–1735. [Google Scholar] [CrossRef]
- Underwood, W.; Anthony, R. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition. Retrieved March 2013, 30. [Google Scholar]
- Kilkenny, C.; Browne, W.; Cuthill, I.; Emerson, M.; Altman, D.G. The ARRIVE guidelines. ReqartoCom 2010, 8, 1–2. [Google Scholar]
- Van Dam, D.; d’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, D.; Lenders, G.; De Deyn, P.P. Effect of Morris water maze diameter on visual-spatial learning in different mouse strains. Neurobiol. Learn. Mem. 2006, 85, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Whitesall, S.; Zhang, Y.; Beibel, M.; Alecy, L.D.; DiPetrillo, K. Validation of volume–pressure recording tail-cuff blood pressure measurements. Am. J. Hypertens. 2008, 21, 1288–1291. [Google Scholar] [CrossRef]
- Di Lascio, N.; Stea, F.; Kusmic, C.; Sicari, R.; Faita, F. Non-invasive assessment of pulse wave velocity in mice by means of ultrasound images. Atherosclerosis 2014, 237, 31–37. [Google Scholar] [CrossRef]
- Maugard, M.; Doux, C.; Bonvento, G. A new statistical method to analyze Morris Water Maze data using Dirichlet distribution. F1000Research 2019, 8, 1601. [Google Scholar] [CrossRef]
- Leloup, A.J.; Fransen, P.; Van Hove, C.E.; Demolder, M.; De Keulenaer, G.W.; Schrijvers, D.M. Applanation tonometry in mice: A novel noninvasive technique to assess pulse wave velocity and arterial stiffness. Hypertension 2014, 64, 195–200. [Google Scholar] [CrossRef]
- Isabelle, M.; Simonet, S.; Ragonnet, C.; Sansilvestri-Morel, P.; Clavreul, N.; Vayssettes-Courchay, C.; Verbeuren, T.J. Chronic reduction of nitric oxide level in adult spontaneously hypertensive rats induces aortic stiffness similar to old spontaneously hypertensive rats. J. Vasc. Res. 2012, 49, 309–318. [Google Scholar] [CrossRef]
- Park, S.U.; Jung, W.S.; Moon, S.K.; Ko, C.N.; Cho, K.H.; Kim, Y.S.; Bae, H.S. Chunghyul-Dan (Qingxie-dan) improves arterial stiffness in patients with increased baPWV. Am. J. Chin. Med. 2006, 34, 553–563. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Lewis, M.R.; Van Nostrand, W.E.; Davis, J.; Previti, M.L.; Gharkholonarehe, N.; Vitek, M.P.; Colton, C.A. Progression of amyloid pathology to Alzheimer’s disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J. Neurosci. 2008, 28, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Van Skike, C.E.; Hussong, S.A.; Banh, A.; Galvan, V. Nitric Oxide Synthase Dysfunction Underlies Cerebrovascular Deficits in a Mouse Model of Tauopathy. Innov. Aging 2019, 3 (Suppl. S1), S91. [Google Scholar] [CrossRef]
- Gasulla, J.; Calvo, D.J. Enhancement of tonic and phasic GABAergic currents following nitric oxide synthase inhibition in hippocampal CA1 pyramidal neurons. Neurosci. Lett. 2015, 590, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Tricoire, L.; Tania, V. Neuronal nitric oxide synthase expressing neurons: A journey from birth to neuronal circuits. Front. Neural Circuits 2012, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Perrenoud, Q.; Rossier, J.; Férézou, I.; Geoffroy, H.; Gallopin, T.; Vitalis, T.; Rancillac, A. Activation of cortical 5-HT3 receptor-expressing interneurons induces NO mediated vasodilatations and NPY mediated vasoconstrictions. Front. Neural. Circuits 2012, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Gholipour, T.; Ghasemi, M.; Riazi, K.; Ghaffarpour, M.; Dehpour, A.R. Seizure susceptibility alteration through 5-HT3 receptor: Modulation by nitric oxide. Seizure 2010, 19, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, K.; Jayakumar, R.; Venkatachalam, K. Increased neuronal nitric oxide synthase (nNOS) activity triggers picrotoxin-induced seizures in rats and evidence for participation of nNOS mechanism in the action of antiepileptic drugs. Brain Res. 2003, 979, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Pall, M.L. Nitric oxide synthase partial uncoupling as a key switching mechanism for the NO/ONOO–cycle. Med. Hypotheses 2007, 69, 821–825. [Google Scholar] [CrossRef]
- Barford, P.; Blair, J.; Eggar, C.; Hamon, C.; Morar, C.; Whitburn, S. Tetrahydrobiopterin metabolism in the temporal lobe of patients dying with senile dementia of Alzheimer type. J. Neurol. Neurosurg. Psychiatry 1984, 47, 736–738. [Google Scholar] [CrossRef]
- Thorns, V.; Hansen, L.; Masliah, E. nNOS expressing neurons in the entorhinal cortex and hippocampus are affected in patients with Alzheimer’s disease. Exp. Neurol. 1998, 150, 14–20. [Google Scholar] [CrossRef]
- Montaser, A.B.; Jarvinen, J.; Löffler, S.; Huttunen, J.; Auriola, S.; Lehtonen, M.; Jalkanen, A.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Enables the Efficient Brain Delivery of Small-Sized Prodrug across the Blood–Brain Barrier and into Human and Mouse Brain Parenchymal Cells. ACS Chem. Neurosci. 2020, 11, 4301–4315. [Google Scholar] [CrossRef] [PubMed]
- Majzúnová, M.; Pakanová, Z.; Kvasnička, P.; Bališ, P.; Čačányiová, S.; Dovinová, I. Age-dependent redox status in the brain stem of NO-deficient hypertensive rats. J. Biomed. Sci. 2017, 24, 72. [Google Scholar] [CrossRef] [PubMed]
- Reif, D.W.; McCreedy, S.A. N-nitro-L-arginine and N-monomethyl-L-arginine exhibit a different pattern of inactivation toward the three nitric oxide synthases. Arch. Biochem. Biophys. 1995, 320, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, S.; Leopold, E.; Schmidt, K.; Brunner, F.; Mayer, B. Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): Requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br. J. Pharmacol. 1996, 118, 1433–1440. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6 | hAPP23+/− | ||||||
|---|---|---|---|---|---|---|---|
| Control (n = 10) | L-NAME (n = 6) | Control (n = 9) | L-NAME (n = 6) | ptreatment | pgenotype | ptreatment×genotype | |
| Heart weight (mg) | 144 ± 4 | 155 ± 7 | 154 ± 8 | 138 ± 4 | ns | ns | * |
| Heart weight (%) | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 | ns | ns | ns |
| IVS,d (mm) | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | ns | ns | ns |
| LVID,d (mm) | 3.4 ± 0.1 | 3.0 ± 0.2 | 3.4 ± 0.1 | 3.2 ± 0.2 | 0.06 | ns | ns |
| LVPW,d (mm) | 1.1 ± 0.1 | 1.3 ± 0.1 | 0.9 ± 0.1 | 1.0 ± 0.1 | 0.08 | ** | ns |
| LV mass (mg) | 125 ± 7 | 134 ± 12 | 127 ± 7 | 122 ± 11 | ns | ns | ns |
| LV mass/BW (10−3) | 4.4 ± 0.3 | 4.9 ± 0.4 | 4.5 ± 0.2 | 4.7 ± 0.4 | ns | ns | ns |
| LV volume,d (µL) | 48 ± 4 | 37 ± 4 | 49 ± 4 | 43 ± 5 | 0.06 | ns | ns |
| Stroke volume (µL) | 39 ± 3 | 31 ± 4 | 36 ± 2 | 34 ± 3 | ns | ns | ns |
| EF (%) | 75 ± 4 | 86 ± 3 | 75 ± 3 | 80 ± 3 | 0.05 | ns | ns |
| FS (%) | 45 ± 4 | 54 ± 4 | 43 ± 2 | 48 ± 3 | 0.06 | ns | ns |
| E/A (none) | 1.6 ± 0.1 | 1.5 ± 0.2 | 1.4 ± 0.1 | 1.3 ± 0.1 | ns | ns | ns |
| E/E’ (none) | 28 ± 5 | 29 ± 7 | 28 ± 3 | 24 ± 4 | ns | ns | ns |
| IVRT (ms) | 15 ± 1 | 14 ± 1 | 20 ± 1 | 22 ± 2 | ns | **** | ns |
| Deceleration (ms) | 19 ± 2 | 16 ± 2 | 18 ± 2 | 15 ± 1 | ns | ns | ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrickx, J.O.; Calus, E.; De Deyn, P.P.; Van Dam, D.; De Meyer, G.R.Y. Chronic Inhibition of Nitric Oxide Synthases Impairs Spatiotemporal Learning and Memory to a Similar Extent in C57BL/6 and hAPP23+/− Mice. Int. J. Transl. Med. 2023, 3, 516-526. https://doi.org/10.3390/ijtm3040036
Hendrickx JO, Calus E, De Deyn PP, Van Dam D, De Meyer GRY. Chronic Inhibition of Nitric Oxide Synthases Impairs Spatiotemporal Learning and Memory to a Similar Extent in C57BL/6 and hAPP23+/− Mice. International Journal of Translational Medicine. 2023; 3(4):516-526. https://doi.org/10.3390/ijtm3040036
Chicago/Turabian StyleHendrickx, Jhana O., Elke Calus, Peter Paul De Deyn, Debby Van Dam, and Guido R. Y. De Meyer. 2023. "Chronic Inhibition of Nitric Oxide Synthases Impairs Spatiotemporal Learning and Memory to a Similar Extent in C57BL/6 and hAPP23+/− Mice" International Journal of Translational Medicine 3, no. 4: 516-526. https://doi.org/10.3390/ijtm3040036
APA StyleHendrickx, J. O., Calus, E., De Deyn, P. P., Van Dam, D., & De Meyer, G. R. Y. (2023). Chronic Inhibition of Nitric Oxide Synthases Impairs Spatiotemporal Learning and Memory to a Similar Extent in C57BL/6 and hAPP23+/− Mice. International Journal of Translational Medicine, 3(4), 516-526. https://doi.org/10.3390/ijtm3040036