
Molecular Interactions between Gasotransmitters in Patients with Obstructive Sleep Apnea

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Study Participants
2.2. Polysomnography (PSG)
2.3. Sample Collection
2.4. H2S Analysis
2.5. Nitric Oxide Analysis
2.6. Heme Oxygenase-1 (HO-1) Analysis
2.7. Biostatistical Analysis
3. Results
3.1. Participant Characteristics
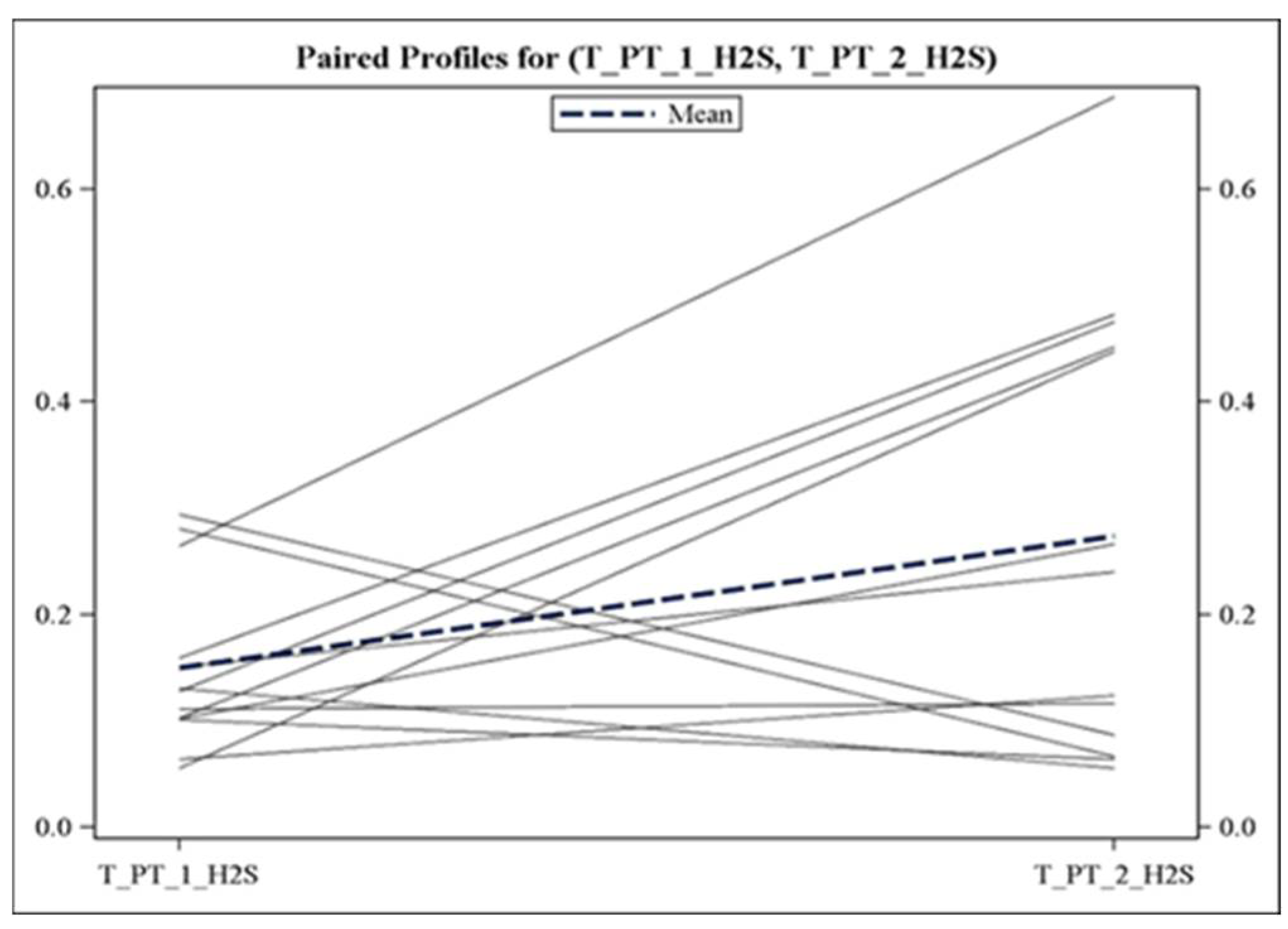
3.2. Alterations in Plasma Gasotransmitter Levels following Obstructive Events
3.3. Effect of Obstructive Events on Heme Oxygenase-1 (HO-1) Expression
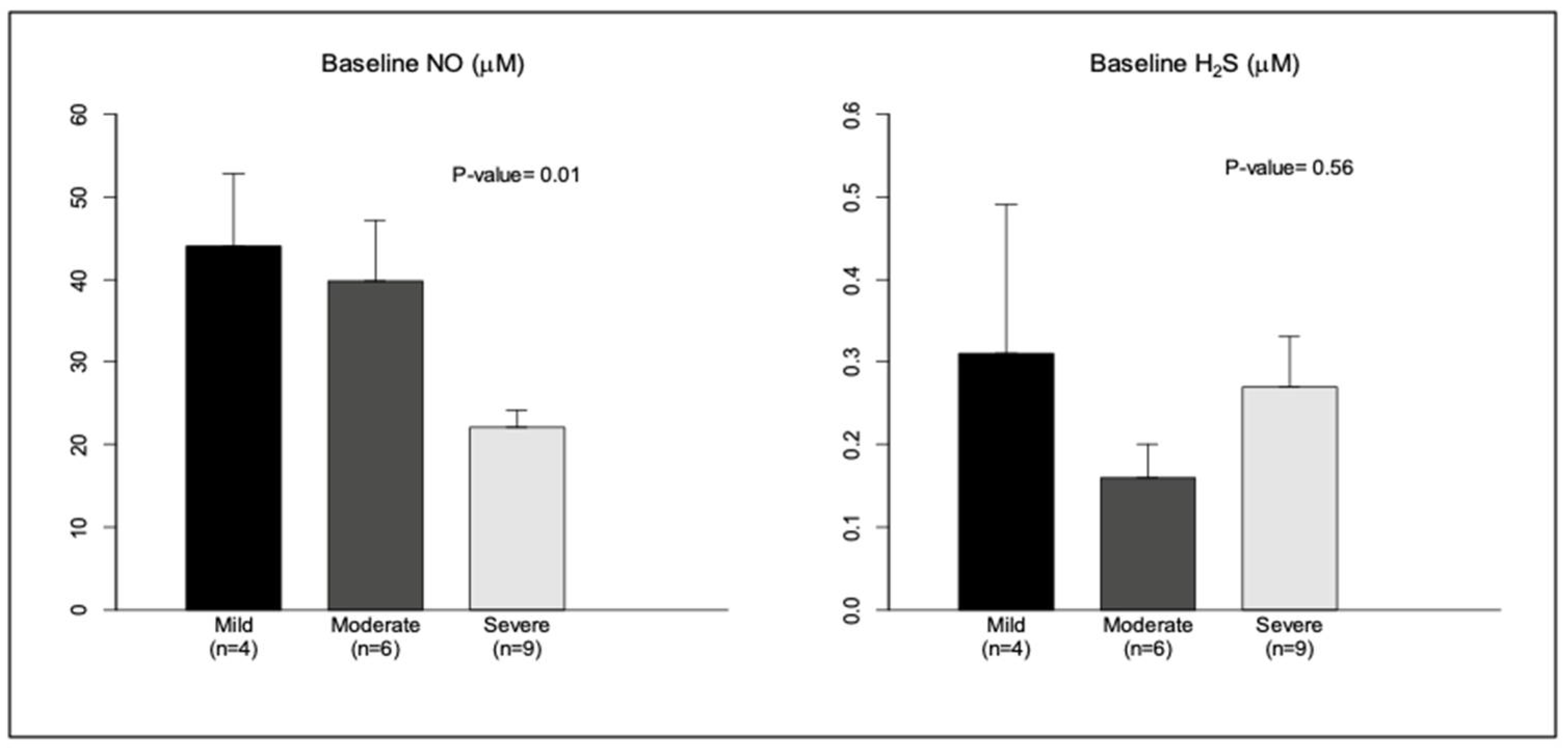
3.4. Effect of Severity of OSA on Baseline Levels of NO, H2S, and HO-1
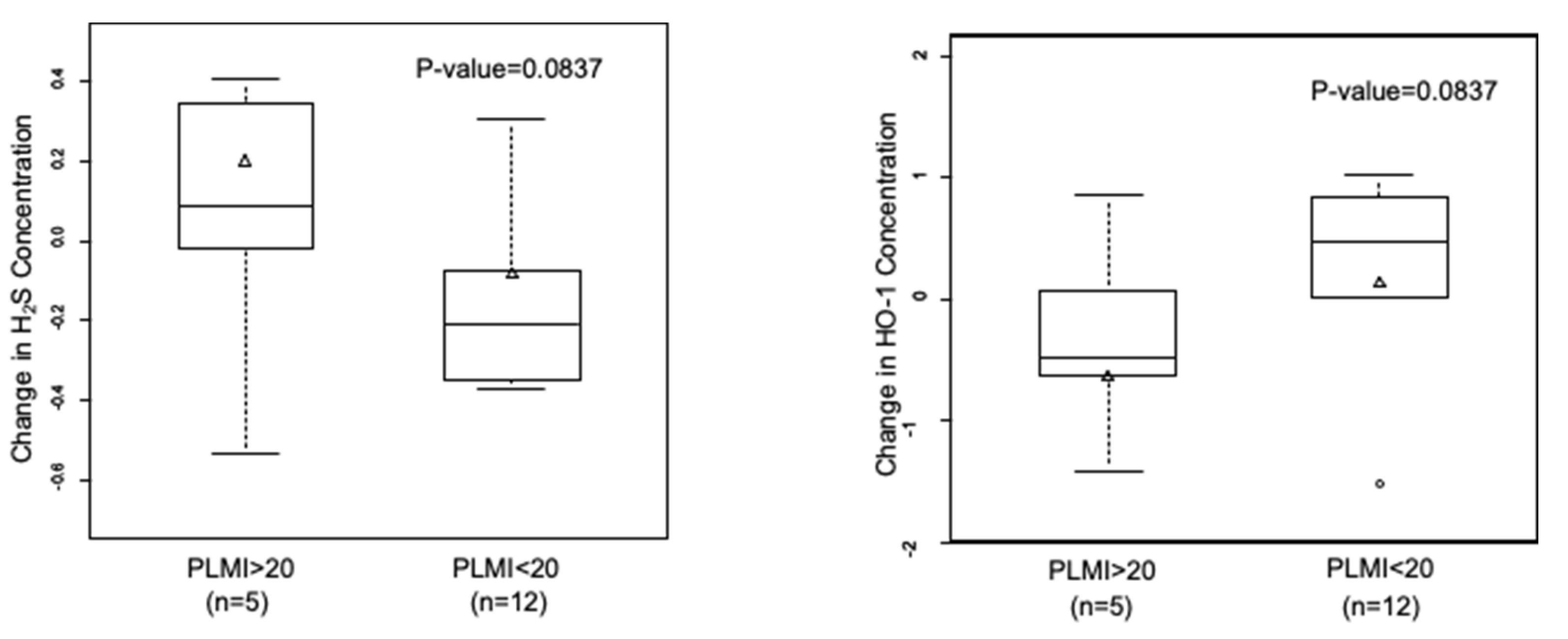
3.5. A Trend towards an Increase in H2S and Decrease in HO-1 with Frequent Periodic Limb Movements
4. Discussion
5. Limitations
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Competing Financial Interests
Abbreviations
| OSA | Obstructive sleep apnea |
| CVD | Cardiovascular disease |
| NO | Nitric oxide |
| H2S | Hydrogen sulfide |
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| e-NOS | Endothelial nitric oxide synthase |
| CPAP | Continuous positive airway pressure |
| PSG | Polysomnography |
| AHI | Apnea-hypopnea index |
| AASM | American Academy of Sleep Medicine |
| HPLC | High-performance liquid chromatography |
| CHF | Congestive heart failure |
| CAD | Coronary artery disease |
| Afib | Atrial fibrillation |
| REM | Rapid eye movement |
| PLMs | Periodic limb movements during sleep |
| PLMI | Periodic limb movement index |
| ROS | Reactive oxygen species |
References
- Senaratna, C.V.; Perret, J.L.; Lodge, C.J.; Lowe, A.J.; Campbell, B.E.; Matheson, M.C.; Hamilton, G.S.; Dharmage, S.C. Prevalence of obstructive sleep apnea in the general population: A systematic review. Sleep Med. Rev. 2017, 34, 70–81. [Google Scholar] [CrossRef] [PubMed]
- May, A.M.; Mehra, R. Obstructive sleep apnea: Role of intermittent hypoxia and inflammation. Semin. Respir. Crit. Care Med. 2014, 35, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Peppard, P.E.; Young, T.; Palta, M.; Skatrud, J. Prospective study of the association between sleep-disordered breathing and hypertension. N. Engl. J. Med. 2000, 342, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Yaggi, H.K.; Concato, J.; Kernan, W.N.; Lichtman, J.H.; Brass, L.M.; Mohsenin, V. Obstructive sleep apnea as a risk factor for stroke and death. N. Engl. J. Med. 2005, 353, 2034–2041. [Google Scholar] [CrossRef]
- Peker, Y.; Kraiczi, H.; Hedner, J.; Loth, S.; Johansson, A.; Bende, M. An independent association between obstructive sleep apnoea and coronary artery disease. Eur. Respir. J. 1999, 14, 179–184. [Google Scholar] [CrossRef]
- Drager, L.F.; Polotsky, V.Y.; Lorenzi-Filho, G. Obstructive sleep apnea: An emerging risk factor for atherosclerosis. Chest 2011, 140, 534–542. [Google Scholar] [CrossRef]
- Hartmann, G.; Tschop, M.; Fischer, R.; Bidlingmaier, C.; Riepl, R.; Tschop, K.; Hautmann, H.; Endres, S.; Toepfer, M. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine 2000, 12, 246–252. [Google Scholar] [CrossRef]
- Narkiewicz, K.; Montano, N.; Cogliati, C.; van de Borne, P.J.; Dyken, M.E.; Somers, V.K. Altered cardiovascular variability in obstructive sleep apnea. Circulation 1998, 98, 1071–1077. [Google Scholar] [CrossRef]
- Dyugovskaya, L.; Lavie, P.; Lavie, L. Lymphocyte activation as a possible measure of atherosclerotic risk in patients with sleep apnea. Ann. N. Y. Acad. Sci. 2005, 1051, 340–350. [Google Scholar] [CrossRef]
- World Health Organization. Cardiovascular Diseases (CVDs). 2017. Available online: http://www.who.int/cardiovascular_diseases/en/ (accessed on 25 July 2022).
- Shamsuzzaman, A.S.; Gersh, B.J.; Somers, V.K. Obstructive sleep apnea: Implications for cardiac and vascular disease. JAMA 2003, 290, 1906–1914. [Google Scholar] [CrossRef]
- Gupta, R.M.; Libby, P.; Barton, M. Linking regulation of nitric oxide to endothelin-1: The Yin and Yang of vascular tone in the atherosclerotic plaque. Atherosclerosis 2020, 292, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Maines, M.D. Nitric oxide induces heme oxygenase-1 via mitogen-activated protein kinases ERK and p38. Cell. Mol. Biol. (Noisy-le-Grand) 2000, 46, 609–617. [Google Scholar] [PubMed]
- Bain, A.R.; Weil, B.R.; Diehl, K.J.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Insufficient sleep is associated with impaired nitric oxide-mediated endothelium-dependent vasodilation. Atherosclerosis 2017, 265, 41–46. [Google Scholar] [CrossRef]
- Huang, P.L. Endothelial nitric oxide synthase and endothelial dysfunction. Curr. Hypertens. Rep. 2003, 5, 473–480. [Google Scholar] [CrossRef]
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 2010, 11, 1504–1516. [Google Scholar] [CrossRef]
- Ishikawa, K.; Sugawara, D.; Wang, X.; Suzuki, K.; Itabe, H.; Maruyama, Y.; Lusis, A.J. Heme oxygenase-1 inhibits atherosclerotic lesion formation in ldl-receptor knockout mice. Circ. Res. 2001, 88, 506–512. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef]
- Ahmad, M.; Turkseven, S.; Mingone, C.J.; Gupte, S.A.; Wolin, M.S.; Abraham, N.G. Heme oxygenase-1 gene expression increases vascular relaxation and decreases inducible nitric oxide synthase in diabetic rats. Cell. Mol. Biol. (Noisy-le-Grand) 2005, 51, 371–376. [Google Scholar]
- Peng, Y.J.; Zhang, X.; Gridina, A.; Chupikova, I.; McCormick, D.L.; Thomas, R.J.; Scammell, T.E.; Kim, G.; Vasavda, C.; Nanduri, J.; et al. Complementary roles of gasotransmitters CO and H2S in sleep apnea. Proc. Natl. Acad. Sci. USA 2017, 114, 1413–1418. [Google Scholar] [CrossRef]
- Fortuna, A.M.; Miralda, R.; Calaf, N.; Gonzalez, M.; Casan, P.; Mayos, M. Airway and alveolar nitric oxide measurements in obstructive sleep apnea syndrome. Respir. Med. 2011, 105, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Nagai, Y.; Umemura, K.; Kimura, Y. Physiological roles of hydrogen sulfide: Synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid. Redox Signal. 2005, 7, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef]
- Liu, Y.H.; Lu, M.; Hu, L.F.; Wong, P.T.; Webb, G.D.; Bian, J.S. Hydrogen sulfide in the mammalian cardiovascular system. Antioxid. Redox Signal. 2012, 17, 141–185. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Kostetski, I.; Chu, S.H.; Siau, J.L.; Bhatia, M.; Moore, P.K. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 2006, 343, 303–310. [Google Scholar] [CrossRef] [PubMed]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef]
- Iber, C.; Ancoli-Israel, S.; Chesson, A.J.; Quan, S. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications; American Academy of Sleep Medicine: Darien, IL, USA, 2007. [Google Scholar]
- Sparatore, A.; Perrino, E.; Tazzari, V.; Giustarini, D.; Rossi, R.; Rossoni, G.; Erdmann, K.; Schroder, H.; Del Soldato, P. Pharmacological profile of a novel H2S-releasing aspirin. Free. Radic. Biol. Med. 2009, 46, 586–592. [Google Scholar] [CrossRef]
- Kartha, R.V.; Zhou, J.; Basso, L.; Schroder, H.; Orchard, P.J.; Cloyd, J. Mechanisms of Antioxidant Induction with High-Dose N-Acetylcysteine in Childhood Cerebral Adrenoleukodystrophy. CNS Drugs 2015, 29, 1041–1047. [Google Scholar] [CrossRef]
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent hypoxemia and OSA: Implications for comorbidities. Chest 2015, 147, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Kroll, M.H.; Christodoulides, N.; Peyton, K.J.; Schafer, A.I. Nitric oxide induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Circ. Res. 1997, 80, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Epstein, L.J.; Kristo, D.; Strollo, P.J., Jr.; Friedman, N.; Malhotra, A.; Patil, S.P.; Ramar, K.; Rogers, R.; Schwab, R.J.; Weaver, E.M.; et al. Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J. Clin. Sleep Med. JCSM Off. Publ. Am. Acad. Sleep Med. 2009, 5, 263–276. [Google Scholar]
- Kato, M.; Roberts-Thomson, P.; Phillips, B.G.; Haynes, W.G.; Winnicki, M.; Accurso, V.; Somers, V.K. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 2000, 102, 2607–2610. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkoski, M.; Soriano, R.N.; Francescato, H.D.; Batalhao, M.E.; Coimbra, T.M.; Carnio, E.C.; Branco, L.G. Hydrogen sulfide as a cryogenic mediator of hypoxia-induced anapyrexia. Neuroscience 2012, 201, 146–156. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L. Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid. Redox Signal. 2010, 12, 1219–1234. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L.; Bearden, S.E.; St Leger, J.; Nilson, E.; Gao, Y.; Madden, J.A. Hypoxic pulmonary vasodilation: A paradigm shift with a hydrogen sulfide mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R51–R60. [Google Scholar] [CrossRef]
- Ali, M.Y.; Ping, C.Y.; Mok, Y.Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P.K. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef]
- Peter, E.A.; Shen, X.; Shah, S.H.; Pardue, S.; Glawe, J.D.; Zhang, W.W.; Reddy, P.; Akkus, N.I.; Varma, J.; Kevil, C.G. Plasma free H2S levels are elevated in patients with cardiovascular disease. J. Am. Heart Assoc. 2013, 2, e000387. [Google Scholar] [CrossRef]
- Dang-Thi-Mai, K.; Le-Dong, N.N.; Le-Thuong, V.; Tran-Van, N.; Duong-Quy, S. Exhaled Nitric Oxide as a Surrogate Marker for Obstructive Sleep Apnea Severity Grading: An In-Hospital Population Study. Nat. Sci. Sleep 2021, 13, 763–773. [Google Scholar] [CrossRef]
- Al-Alawi, A.; Mulgrew, A.; Tench, E.; Ryan, C.F. Prevalence, risk factors and impact on daytime sleepiness and hypertension of periodic leg movements with arousals in patients with obstructive sleep apnea. J. Clin. Sleep Med. 2006, 2, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Variable | Cases n = 19 Mean ± SE or Number | Control n = 6 Mean ± SE or Number | p-Value |
|---|---|---|---|
| Age (Years) | 36.2 (4.8) | 51 (2.7) | 0.0127 |
| Gender, Male | 11 (57.9) | 1 (16.7) | 0.0780 |
| BMI (Kg/m2) | 38.5 (1.3) | 36.8 (2.3) | 0.53 |
| Height (Meters) | 1.73 (0.02) | 1.64 (0.04) | 0.06 |
| Hypertension | 10 | 2 | 0.4095 |
| Diabetes Mellitus | 5 | 1 | 0.6295 |
| Congestive Heart Failure | 1 | 1 | 0.3694 |
| Coronary Artery Disease | 3 | 2 | 0.3490 |
| Atrial Fibrillation | 1 | 0 | 0.5663 |
| Hyperlipidemia | 7 | 2 | 0.8760 |
| Current or Former Smoker | 11 | 1 | 0.0780 |
| Variable | OSA (n = 19) | Control (n = 6) | p-Value |
|---|---|---|---|
| NO (measured as nitrites and nitrites in μM) | 32.28 (16.16) | 38.30 (34.00) | 0.9749 |
| H2S (in μM) | 0.25(0.21) | 0.30 (0.25) | 0.6153 |
| H2S/NO ratio | 9.88 (9.33) | 13.40 (16.80) | 0.9749 |
| HO-1 (ng/mL) | 2.92 (1.38) | 2.96 (1.89) | 0.9749 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pusalavidyasagar, S.; Hovde, L.B.; Lee, J.; Zhang, L.; Abbasi, A.; Kartha, R.V. Molecular Interactions between Gasotransmitters in Patients with Obstructive Sleep Apnea. Int. J. Transl. Med. 2022, 2, 408-418. https://doi.org/10.3390/ijtm2030032
Pusalavidyasagar S, Hovde LB, Lee J, Zhang L, Abbasi A, Kartha RV. Molecular Interactions between Gasotransmitters in Patients with Obstructive Sleep Apnea. International Journal of Translational Medicine. 2022; 2(3):408-418. https://doi.org/10.3390/ijtm2030032
Chicago/Turabian StylePusalavidyasagar, Snigdha, Laurie B. Hovde, Jessie Lee, Lei Zhang, Adnan Abbasi, and Reena V. Kartha. 2022. "Molecular Interactions between Gasotransmitters in Patients with Obstructive Sleep Apnea" International Journal of Translational Medicine 2, no. 3: 408-418. https://doi.org/10.3390/ijtm2030032
APA StylePusalavidyasagar, S., Hovde, L. B., Lee, J., Zhang, L., Abbasi, A., & Kartha, R. V. (2022). Molecular Interactions between Gasotransmitters in Patients with Obstructive Sleep Apnea. International Journal of Translational Medicine, 2(3), 408-418. https://doi.org/10.3390/ijtm2030032