
Partial Proliferating Cell Nuclear Antigen Functional Knockout Impairs Cisplatin Resistance and Clonogenic Potential in Lung Adenocarcinoma Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Partial PCNA Loss of Function Establishment Using CRISPR/Cas9 System
- PCNA _Forward: 5′ TGGCGGGAAAATCAAGGGTT 3′
- PCNA _Reverse: 5′ TATCGTGCCTGTGTACAGCA 3′
2.3. PCNA Overexpression Construct
2.4. Cell Viability and Cisplatin Resistance Assay
2.5. Colony Formation Assay
2.6. Western Blot
2.7. Statistical Analysis
3. Results
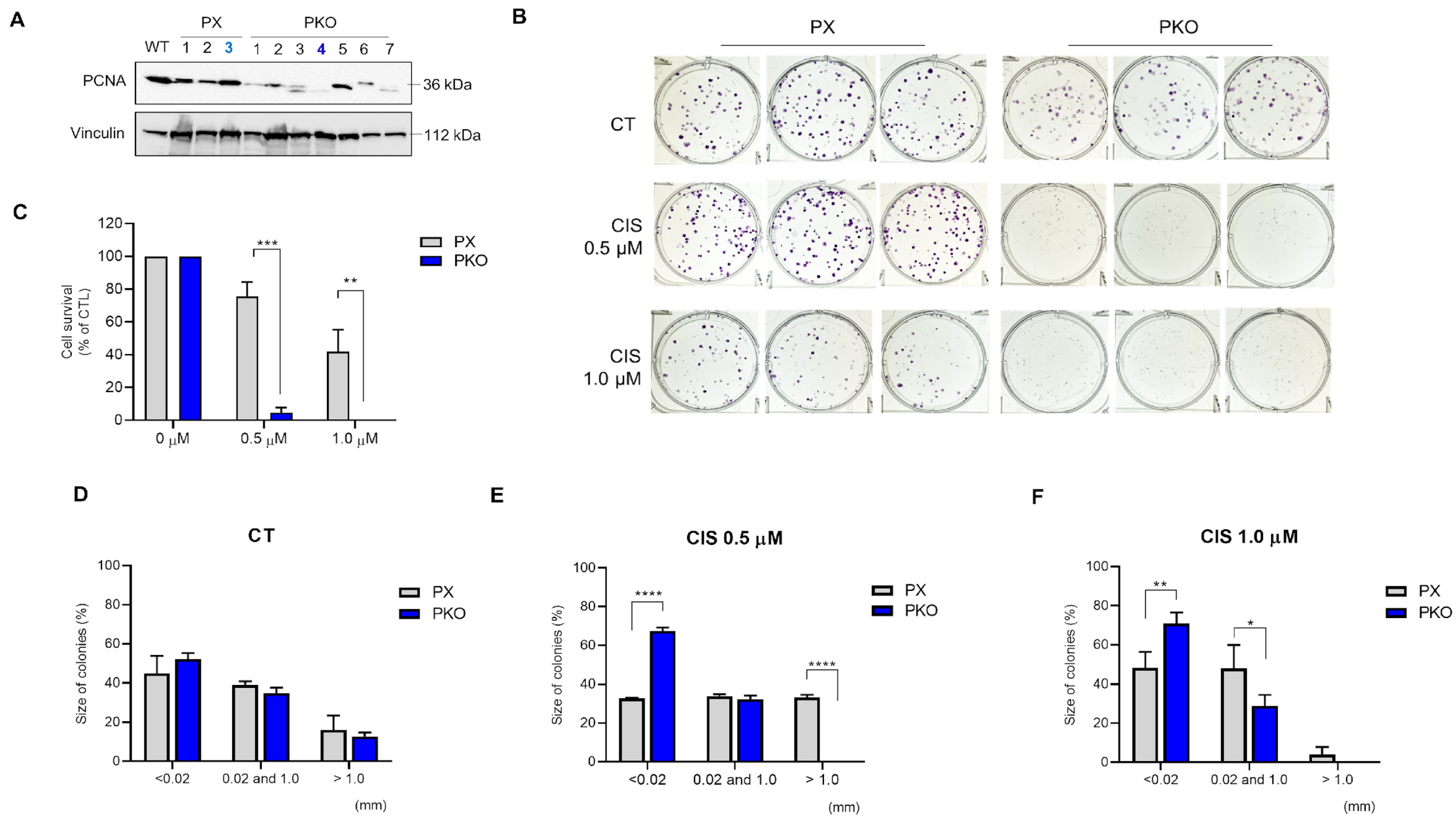
3.1. Generation of A549 PCNA Partial Loss Cells
3.2. Partial Loss of PCNA in PKO Cells Impaired Clonogenic Potential
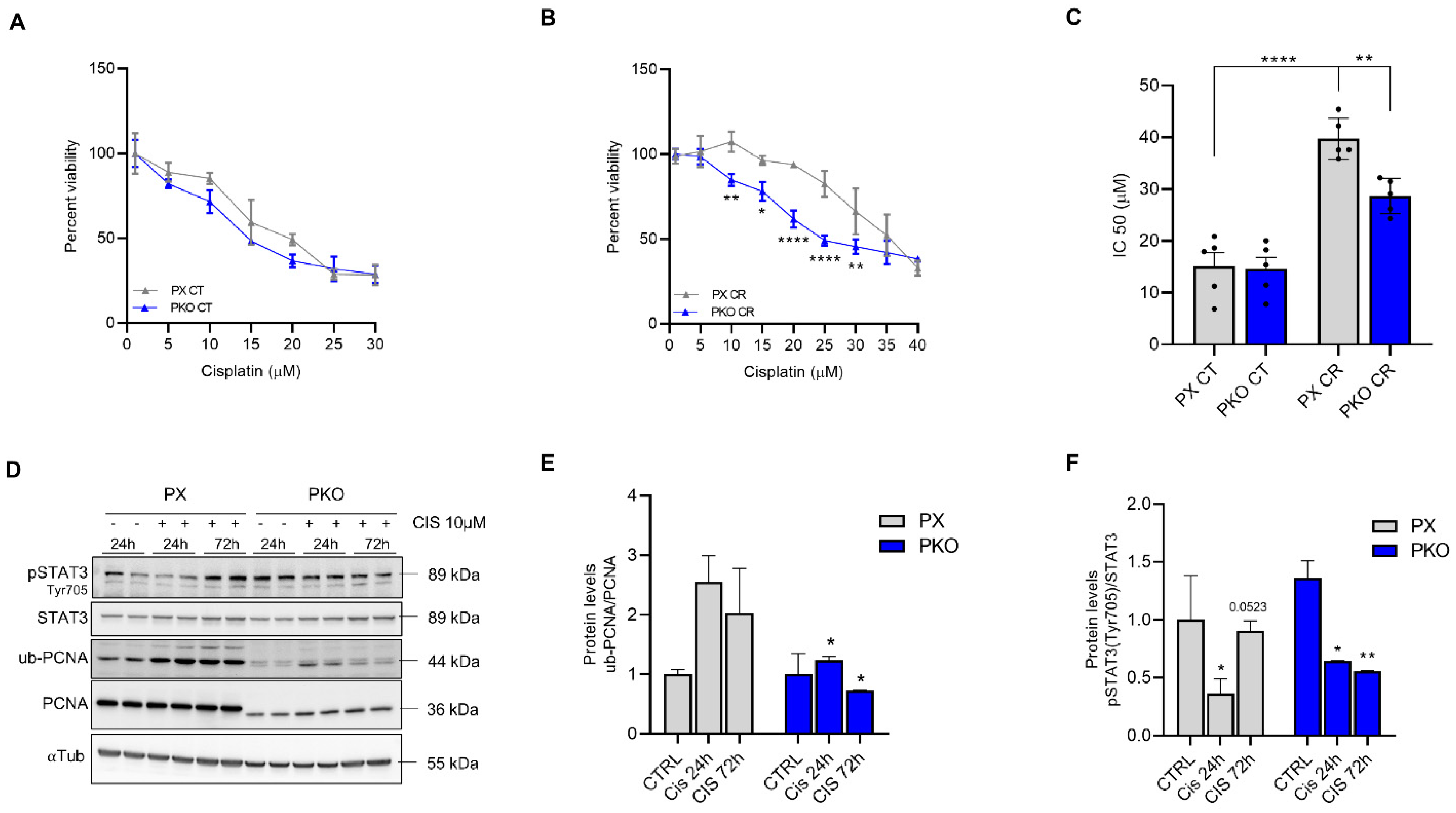
3.3. Cisplatin Is More Effective Against PKO Cells
3.4. STAT3 Is Related to PCNA Expression and Ubiquitylation in Cisplatin Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kakushadze, Z.; Raghubanshi, R.; Yu, W. Estimating Cost Savings from Early Cancer Diagnosis. Data 2017, 2, 30. [Google Scholar] [CrossRef]
- Steven, A.; Fisher, S.A.; Robinson, B.W. Immunotherapy for lung cancer. Respirology 2016, 21, 821–833. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.P.; Tortelli, T.C.; Mancini, M.C.S.; Pavan, I.C.B.; Silva, L.G.S.; Severino, M.B.; Granato, D.C.; Pestana, N.F.; Ponte, L.G.S.; Peruca, G.F.; et al. STAT3 contributes to cisplatin resistance, modulating EMT markers, and the mTOR signaling in lung adenocarcinoma. Neoplasia 2021, 23, 1048–1058. [Google Scholar] [CrossRef]
- Maga, G.; Hubscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef]
- Groeger, A.M.; Caputi, M.; Esposito, V.; Baldi, A.; Rossiello, R.; Santini, D.; Mancini, A.; E Kaiser, H.; Baldi, F. Expression of p21 in non small cell lung cancer relationship with PCNA. Anticancer Res. 2000, 20, 3301–3305. [Google Scholar] [PubMed]
- Müller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Våtsveen, T.K.; Rø, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting Proliferating Cell Nuclear Antigen and Its Protein Interactions Induces Apoptosis in Multiple Myeloma Cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef]
- Lingeman, R.G.; Hickey, R.J.; Malkas, L.H. Expression of a novel peptide derived from PCNA damages DNA and reverses cisplatin resistance. Cancer Chemother. Pharmacol. 2014, 74, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Pavan, I.C.B.; Yokoo, S.; Granato, D.C.; Meneguello, L.; Carnielli, C.M.; Tavares, M.R.; Amaral, C.L.D.; de Freitas, L.B.; Leme, A.F.P.; Luchessi, A.D.; et al. Different interactomes for p70-S6K1 and p54-S6K2 revealed by proteomic analysis. Proteomics 2016, 16, 2650–2666. [Google Scholar] [CrossRef]
- Decker, T.; Müller, M. Jak-Stat Signaling: From Basics to Disease; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Stoimenov, I.; Helleday, T. PCNA on the crossroad of cancer. Biochem. Soc. Trans. 2009, 37, 605–613. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.-L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Malkas, L.H.; Herbert, B.S.; Abdel-Aziz, W.; Dobrolecki, L.E.; Liu, Y.; Agarwal, B.; Hoelz, D.; Badve, S.; Schnaper, L.; Arnold, R.J.; et al. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc. Natl. Acad. Sci. USA 2006, 103, 19472–19477. [Google Scholar] [CrossRef]
- González-Magaña, A.; Blanco, F.J. Human PCNA Structure, Function and Interactions. Biomolecules 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Boehm, E.M.; Gildenberg, M.S.; Washington, M.T. The Many Roles of PCNA in Eukaryotic DNA Replication. Enzymes 2016, 39, 231–254. [Google Scholar]
- Cardano, M.; Tribioli, C.; Prosperi, E. Targeting Proliferating Cell Nuclear Antigen (PCNA) as an Effective Strategy to Inhibit Tumor Cell Proliferation. Curr. Cancer Drug Targets 2020, 20, 240–252. [Google Scholar] [CrossRef]
- Haerslev, T.; Jacobsen, G.K.; Zedeler, K. Correlation of growth fraction by Ki-67 and proliferating cell nuclear antigen (PCNA) immunohistochemistry with histopathological parameters and prognosis in primary breast carcinomas. Breast Cancer Res. Treat. 1996, 37, 101–113. [Google Scholar] [CrossRef]
- Lv, Q.; Zhang, J.; Yi, Y.; Huang, Y.; Wang, Y.; Wang, Y.; Zhang, W. Proliferating Cell Nuclear Antigen Has an Association with Prognosis and Risks Factors of Cancer Patients: A Systematic Review. Mol. Neurobiol. 2016, 53, 6209–6217. [Google Scholar] [CrossRef] [PubMed]
- Kordek, R.; Biernat, W.; Debiec-Rychter, M.; Alwasiak, J.; Liberski, P.P. Comparative Evaluation of p53-Protein Expression and the PCNA and Ki-67 Proliferating Cell Indices in Human Astrocytomas. Pathol. Res. Pract. 1996, 192, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Thakar, T.; Leung, W.; Nicolae, C.M.; Clements, K.E.; Shen, B.; Bielinsky, A.-K.; Moldovan, G.-L. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun. 2020, 11, 2147. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kong, W.; Liu, B.; Zhang, X. Proliferating cell nuclear antigen promotes cell proliferation and tumorigenesis by up-regulating STAT3 in non-small cell lung cancer. Biomed. Pharmacother. 2018, 104, 595–602. [Google Scholar] [CrossRef]
- Wang, S.-C.; Nakajima, Y.; Yu, Y.-L.; Xia, W.; Chen, C.-T.; Yang, C.-C.; McIntush, E.W.; Li, L.-Y.; Hawke, D.H.; Kobayashi, R.; et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 2006, 8, 1359–1368. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morelli, A.P.; Quintero-Ruiz, N.; Mancini, M.C.S.; Pavan, I.C.B.; Flores, I.L.; Silva, L.G.S.; Severino, M.B.; Bezerra, R.M.N.; Simabuco, F.M. Partial Proliferating Cell Nuclear Antigen Functional Knockout Impairs Cisplatin Resistance and Clonogenic Potential in Lung Adenocarcinoma Cells. DNA 2025, 5, 7. https://doi.org/10.3390/dna5010007
Morelli AP, Quintero-Ruiz N, Mancini MCS, Pavan ICB, Flores IL, Silva LGS, Severino MB, Bezerra RMN, Simabuco FM. Partial Proliferating Cell Nuclear Antigen Functional Knockout Impairs Cisplatin Resistance and Clonogenic Potential in Lung Adenocarcinoma Cells. DNA. 2025; 5(1):7. https://doi.org/10.3390/dna5010007
Chicago/Turabian StyleMorelli, Ana Paula, Nathalia Quintero-Ruiz, Mariana Camargo Silva Mancini, Isadora Carolina Betim Pavan, Isabelle Lima Flores, Luiz Guilherme Salvino Silva, Matheus Brandemarte Severino, Rosangela Maria Neves Bezerra, and Fernando Moreira Simabuco. 2025. "Partial Proliferating Cell Nuclear Antigen Functional Knockout Impairs Cisplatin Resistance and Clonogenic Potential in Lung Adenocarcinoma Cells" DNA 5, no. 1: 7. https://doi.org/10.3390/dna5010007
APA StyleMorelli, A. P., Quintero-Ruiz, N., Mancini, M. C. S., Pavan, I. C. B., Flores, I. L., Silva, L. G. S., Severino, M. B., Bezerra, R. M. N., & Simabuco, F. M. (2025). Partial Proliferating Cell Nuclear Antigen Functional Knockout Impairs Cisplatin Resistance and Clonogenic Potential in Lung Adenocarcinoma Cells. DNA, 5(1), 7. https://doi.org/10.3390/dna5010007