1. Introduction
Acute kidney injury (AKI) is a condition characterized by prolonged hospitalization and high mortality rates. Despite extensive research, no pharmacologic therapy has been successfully and effectively proven to treat AKI. While several studies have identified molecules that either attenuate or exacerbate ischemia-induced renal damage (IRD), no molecules with a significant impact on the phenotype or potential as future therapeutic targets have been conclusively identified so far.
The large Maf transcription factors MafB and c-Maf are known to be expressed in macrophage-lineage hematopoietic cells. In our previous study, we analyzed their expression patterns in macrophage subtypes and tissue-resident macrophages, demonstrating that normal kidney macrophages do not express MafB or c-Maf [
1].
MafB and c-Maf are transcription factors that play crucial roles in macrophage development and function. MafB is essential for macrophage differentiation, regulating genes necessary for their specific functions and lineage commitment. It also modulates inflammatory responses by influencing cytokine production, thereby maintaining immune homeostasis and preventing excessive inflammation. Additionally, MafB restricts macrophage proliferation by repressing self-renewal enhancers, ensuring proper control of macrophage numbers [
2,
3]. On the other hand, c-Maf drives the polarization of macrophages toward an anti-inflammatory phenotype by upregulating genes associated with anti-inflammatory responses, such as IL-10. It serves as a critical transcriptional regulator of IL-10, a cytokine involved in suppressing inflammatory responses and promoting immune tolerance. Furthermore, c-Maf influences macrophage function in various disease contexts, including shaping macrophage responses during infections and inflammatory diseases. Additionally, the study suggests that a high MAFB/MAF expression ratio in lung macrophages could serve as an accurate diagnostic tool for COVID-19 progression. Reversing this ratio might impair exacerbated inflammatory and profibrotic responses, potentially serving as a strategy to limit the severity of COVID-19 [
4].
Together, MafB and c-Maf coordinate macrophage function by balancing pro-inflammatory and anti-inflammatory responses. They influence tissue-specific macrophage identities, contributing to functional heterogeneity during homeostasis, inflammation, and repair processes. Their expression and function are dynamically regulated depending on the context, such as infection, injury, or disease states [
5].
However, this study seeks to elucidate their expression patterns in pathological kidney macrophages associated with AKI. Specifically, we aim to investigate the roles of MafB and c-Maf in the macrophages of the pathological case of AKI-affected kidneys and explore their potential implications in disease attenuation and recovery.
1.1. The Studied Pathological Case of Macrophage: IRD (Ischemia Renal Disease)
Acute kidney injury (AKI) is a dangerous disease that can be caused by many factors.
1.1.1. AKI and IRD
Previous research has shown that kidney ischemia and acute renal failure often result from a combination of factors but can generally be categorized into three main causes: pre-renal azotemia, where renal tubular and glomerular functions are intact but clearance is impaired; post-renal azotemia, caused by obstruction in the urinary outflow tract; and intrinsic causes, which include acute glomerulonephritis (5%), interstitial nephritis (10%), and tubular necrosis (85%). Among intrinsic causes, ischemia accounts for 50% of tubular necrosis cases, making it a significant and particularly relevant factor for this study.
It is well established that renal ischemia-reperfusion is one of the main causes of acute kidney injury (AKI), a dangerous disease known for its high mortality rate [
6]. Several in vivo and in vitro experimental models have been developed to analyze the pathological mechanisms underlying AKI. The most commonly used approach to create a mouse model of bilateral renal ischemia-reperfusion is kidney clamping [
7].
1.1.2. Immune Cells and Macrophages Involved in IRD Healing
It is well established that diverse immune cell populations are implicated in IRD, namely natural killer T cells (NKT), which are involved in tubular regeneration after injury; dendritic cells, which release proinflammatory factors; and neutrophils [
8]. As we previously mentioned in this work, the most extensively studied cells are dendritic cells and macrophages [
9].
Previous studies have highlighted the significant roles of dendritic cells and macrophages in ischemia-reperfusion injury (IRI). Utilizing electron microscopy and immunohistochemistry, research by Okusa and Li confirmed that both cell types are localized in the interstitial extracellular compartment of the kidney [
9].
Hughes and colleagues further analyzed how macrophages and dendritic cells are primary contributors to the initiation of IRI, including the injury effect, by acting as key players in inflammatory pathways, both directly through soluble mediators and indirectly via other effector cells [
10]. Their study demonstrated that macrophages are involved in the onset of injury following renal ischemia, while dendritic cells participate in the early innate injurious response after renal ischemia
Macrophages have the ability to promote tissue repair by transitioning between subtypes in a process known as polarization, which regulates their roles during different phases of the healing response. Specifically, the transition from M1 macrophages to alternatively activated M2a, M2b, and M2c macrophages plays a crucial role in the inflammation, proliferation, and remodeling phases of healing.
In our work, we concentrated only on macrophages and studied their ability to express MafB and c-Maf in the diseased kidney. M2 macrophages were reported to be involved in AKI repair [
10]. Interestingly, the Harris group demonstrated that the IL-4/IL-13-induced macrophages and dendritic renal cell populations are responsible for IRD-induced AKI recovery in mice [
11]. More importantly, we demonstrated that IL-4/IL-13 M2-induced macrophages express MafB [
1]. Thus, a better understanding of the main molecules engaged in this disease and their relation to MafB would help in elucidating more details about its signaling pathway and mechanism in order to speed up the procedure of developing better treatment strategies to promote AKI recovery by reversing or preventing the effects of IRD.
1.1.3. MafB and c-Maf Transcription Factors’ Importance in Kidney
Many previous studies have investigated the expression and functions of MafB and c-Maf in the kidney. In this report, we aim to contribute to understanding their potential functions in double ischemic renal disease (dIRD). We previously showed that MafB is essential for foot process formation in podocytes [
12]. Another study demonstrated that mutations in MafB lead to multicentric carpotarsal osteolysis (MCTO), a bone-related disease associated with progressive renal failure [
13]. Furthermore, we analyzed diabetic MafB transgenic (TG) mice and found in previous work that MafB overexpression in kidney podocytes might ameliorate diabetic nephropathy via Nephrin, Gpx3, and Notch2 [
14]. A previous report has shown that MafB and c-Maf expression may not overlap in the kidney—c-Maf is expressed in tubules and MafB in glomeruli—and that c-Maf could be involved in the embryological development and/or cell differentiation of the kidneys and liver [
15].
In the current study, we found that
Mafbf/f::Tie2-Cre mice had worse renal function than
Mafbf/f mice, as the serum and blood urea nitrogen (BUN) showed very high rates after 48 h from IRD-induced AKI. In addition, MafB-deficient mice had a higher mortality rate. Furthermore,
Mafbf/f::Tie2-Cre mice presented enhanced renal tubular injury after ischemia. Mac1
+F4/80
low-inflamed monocytes are enhanced in
Mafbf/f::Tie2-Cre mice, which is a notable feature in the pathophysiology of AKI, contributing to the inflammatory milieu and subsequent renal damage during injury consistent with previous work of Li and Okusa figure 3 [
9].
We also found downregulation of KIM-1 in
Mafbf/f::Tie2-Cre. Our data also showed the downregulation of AIM. On the other hand, we demonstrated in a previous report that MafB directly regulates AIM [
16]. We demonstrated that MafB is upregulated in IL-4/IL-13-induced mouse bone marrow-derived macrophages (BMDM) [
1]. In addition, it was confirmed that the IL-4/IL-13-induced M2a renal macrophage/dendritic cell phenotype is crucial for recovery from acute kidney injury (AKI) [
11]. Therefore, our data show that the signaling pathway of AKI recovery is likely to recruit IL-4/IL-13, which upregulates MafB, and the latter will regulate AIM. Next, AIM binds to KIM-1 and enhances intraluminal debris removal from bilateral IRD-induced AKI after 48 h [
17].
2. Materials and Methods
2.1. Mice
To create
Mafb conditional knock-out (cKO) mice, the
Mafb gene was flanked with a loxP element with a neomycin resistance gene using homologous recombination in ES cells (
Mafbf/f). Following this, the mice were crossed with Tie2-Cre knock-in mice (
Mafbf/f::Tie2-Cre mice) [
18]. The same procedure was used to create
Mafbf/f::Lys-Cre mice.
Wild-type (WT) Mafbf/f, considered as a negative control in all of this paper starting from Figure 2, and Mafb cKO mice were euthanized at the appropriate time points with carbon dioxide gas. The mice were maintained under specific pathogen-free conditions at the laboratory animal resource center of the University of Tsukuba. All experiments were performed in compliance with relevant Japanese and institutional laws and guidelines and were approved by the University of Tsukuba animal ethics committee (authorization number 15e179).
2.2. Surgical Generation of Acute Kidney Injury (AKI) Mouse Model by Bilateral Ischemic Renal Disease or Double IRD (dIRD)
Immediately after the mouse was confirmed to be anesthetized with isoflurane (until the mouse did not move), the skin of the surgical area was cleaned with 70% ethanol. Two bilateral incisions were precisely located on the backside of the mouse at 1 cm from the vertebral column and 1 cm from the last dorsal rib; the incision size was 1 cm along the back. The skin and muscle of the right flank were removed to expose the right kidney. The kidney was gently taken out, and the fat was removed. The renal pedicle blood vessels were then clamped with microaneurysm clamps (S & T AG Fine science Tools, 00398-02, Heidelberg, Germany) for 60 min. The clamped kidney was then partially returned to the abdomen cavity. After making sure the color of the kidney darkened, the same surgical procedure was applied to the left kidney no more than 5–10 min later, and it was left for another 60 min (different positions of the kidney and distances to the rib are taken into consideration). During the ischemia surgery, the mouse’s body temperature was maintained by placing it on a 37 °C heat pad. After 60 min, the clamps were removed, and the kidneys were gently returned to their original position inside the abdomen cavity after checking that their color had returned to red (blood refilling). Then, the animal’s body was carefully closed. The animal was injected with warm saline peritoneally to recover possible blood loss and was given free access to food and water to recover, as mentioned in our recent paper and previously described [
19,
20].
2.3. Histological Analysis
For histological analysis in the pathological macrophage study of dIRD, tissues from adult wild-type, sham, MafBf/f, MafBf/f::Lys-Cre, and MafBf/f::Tie2-Cre mice with and without dIRD were all sacrificed (if not dead like cKO) after 48 h. For frozen section preparation, tissues were fixed in 4% paraformaldehyde for 3 h at 4 °C and then incubated overnight in 30% sucrose (in PBS) at 4 °C. The next day, they were processed, and sections of 5 μm thickness were cut with a cryostat.
However, for the paraffin sections, the tissues were stored in Mildform overnight at 4 °C before slide processing. For Periodic acid-Schiff (PAS) and hematoxylin and eosin (HE) staining, kidney tissues were also fixed in Mildform overnight at 4 °C and embedded in paraffin. PAS and HE staining were performed in 5 µm sections of paraffin-embedded kidney blocks.
To analyze MafB, F4/80, and KIM-1 expression, frozen sections were subjected to a 1% SDS antigen retrieval method very shortly and then incubated with rabbit anti-MafB (BETHYL, Boston, MA, USA), rat polyclonal anti-mouse F4/80 (AbD Serotec, Neuried, Germany), and anti-mouse KIM-1 (R & D systems). On the other hand, for the immunohistochemical analysis of c-Maf, AIM, and Mac1, paraffin sections were subjected to antigen retrieval by autoclaving for 10 min at 120 °C and incubated with rabbit anti-c-Maf (BETHYL), anti-mouse AIM monoclonal antibody (Transgenic), and monoclonal anti-mouse CD11b (Mac1) (Immunotech, Ocala, FL, USA). Alexa Fluor 488 goat anti-rabbit IgG (Life Tech, Carlsbad, CA, USA) was used to detect MafB. Cy3-conjugated donkey anti-rat IgG (Jackson Immunology, Chester County, PA, USA) was used to detect the F4/80, KIM-1, c-Maf, AIM, and Mac1 antibodies.
Tissue section slides were mounted in 2 drops of PermaFluor (Thermo Fisher Scientific, Waltham, MA, USA) with Hoechst stain and dried for 2 h before microscopic observation. Slide observations and images were acquired using a BIOREVO BZ-9000 microscope (Keyence, Osaka, Japan). The scale bar in each panel represents 100 μm. To further support our data, the mean fluorescence of the KIM-1 expression level was analyzed. Using ImageJ software (v1.48), an outline was drawn around each fluorescent injured tubule area. The mean fluorescence of the injured tubules and the mean fluorescence background readings were all measured, and the total corrected cellular fluorescence (TCCF) was calculated using the following formula:
Five different areas from 3 independent different slides of the same mouse were used, and 3 mice from each group were tested.
2.4. Monitoring IRD Success by Measuring Creatinine and BUN Levels
At several steps, it is possible to monitor the progress of ischemic renal disease. At the start of the procedure, it was important to verify that the color of the kidney turned to dark purple a few minutes after clamping and then changed back to red after unclamping.
Next, kidney dysfunction, which shows the severity of the acute kidney injury (AKI) model, was detected by measuring the level of increase in blood urea nitrogen (BUN) and serum creatinine (CRE). The measurement was made using slides from a Dri-Chem 7000 machine (Fujifilm, Tokyo, Japan). Each parameter curve was drawn as a function of time (days after IRD).
2.5. Statistical Analysis
The data were calculated as the mean ± SD. Significant differences between means were evaluated by analysis of variance (Excel 2011), followed by t-tests for pairwise comparisons. Significant differences were defined as p ≤ 0.05.
3. Results
3.1. Bilateral/Double IRD-Induced MafB and c-Maf Expression In Vivo by Kidney Macrophages After 48 h
Scientists might argue that MafB and c-Maf do not have functions in kidney macrophages because it is known that MafB and c-Maf are not expressed in normal kidney macrophages, as we have confirmed in our previous research [
1], and it has also been reported that their expression may not overlap in the kidney considering that c-Maf expression is in tubules and MafB expression is in glomeruli [
15]. However, kidney disease unexpectedly triggered the expression of the two transcription factors in macrophages, which was highly surprising.
Immunohistochemistry data from kidney tissue after 48 h of bilateral/double ischemia-reperfusion injury (dIRI) showed the detection of MafB and c-Maf in WT kidney macrophages, respectively. Therefore, the data suggest that MafB and c-Maf might have functions in the IRD signaling pathway (
Figure 1A,B). M2 macrophages were reported to be involved in AKI repair [
10]. We speculated that M2 might be the source of MafB and c-Maf after injury and that these two transcription factors, or at least MafB, could have a crucial role in the disease. In previous research, we found that IL-4/IL-3 upregulates MafB [
1] and another group recently found that the IL-4/IL-13-induced macrophages are in charge of the AKI recovery [
11]. Taken together, we would like to concentrate on the effect of MafB in this pathology. Therefore, we decided in the coming part of this work to focus on examining the potential role of MafB in IRD and elucidate the effect of its absence on kidney dysfunction, taking advantage of our previously generated MafB cKO mice and aiming to contribute to understanding its role in the pathological macrophage recruited in IRD.
3.2. Kidney Dysfunction Monitoring After 48 h of Bilateral Ischemic Renal Disease (IRD)
Ischemic renal injury (IRI) is widely recognized as one of the most common causes of acute kidney injury (AKI) in humans, often leading to severe outcomes, including death. To study this condition, we developed an AKI mouse model by inducing ischemic renal injury. This was achieved by clamping both kidneys for 60 min, followed by spontaneous reperfusion, while maintaining a controlled mouse body temperature of 37 °C throughout the surgical procedures.
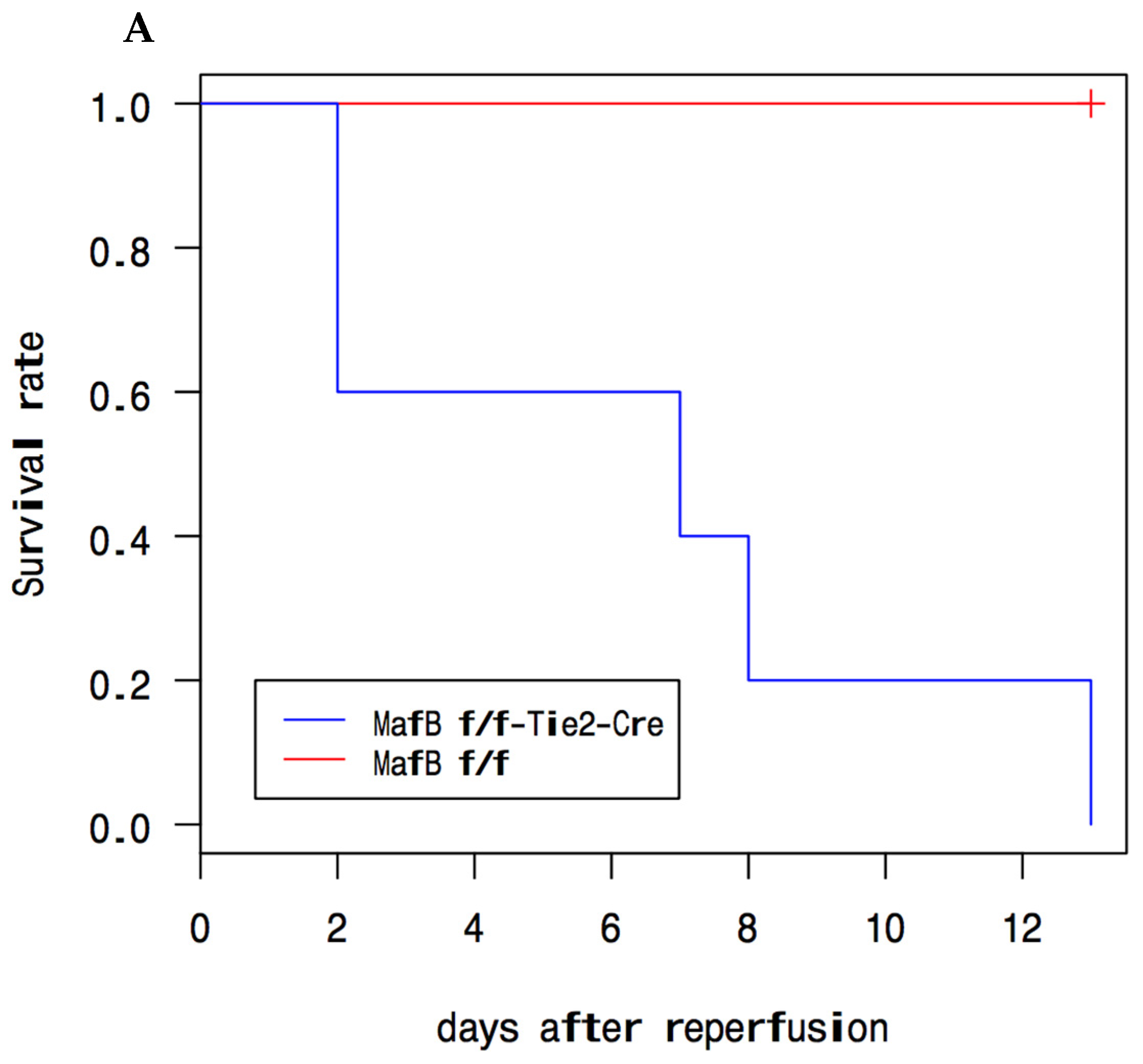
Serum blood urea nitrogen (BUN) and creatinine (CRE) levels are the main markers of kidney dysfunction. These two parameters were identified to monitor the health situation of the kidneys. Blood was collected from mice on a daily basis, and the parameters were determined accordingly. The
MafBf/f BUN and CRE levels presented an important peak around day one, then a decrease that showed up in almost all our pilot mouse experiments and main experiment mouse groups (
Figure 2A,B), which was in concordance with other previous reports [
17,
20]. The general health status of the mice at day 1 (N = 3 and N = 5 groups) was very weak and diseased. Interestingly, in previous reports, the mice survival rate was not affected until later times, such as for AIM-deficient (AIM−) mice: more than 40% died by day 3 post-ischemia-reperfusion injury (IRI), and the mortality rate increased to 67% by day 7 [
17], in contrast to our study where
Mafbf/f::Tie2-Cre mice died earlier. In particular,
Mafbf/f::Tie2-Cre mice showed extreme symptoms of weakness and were severely emaciated before dying at day 2 after reperfusion (
Figure 2A). In detail, all the MafB cKO mice in the main experiment (N = 3) died after 2 days from reperfusion and a majority of MafB cKO mice in the pilot experiment (N = 5) were as follows: two died on day 2, one died on day 7, one died on day 8, and one died on day 13. On the other hand, the
Mafbf/f::Tie2-Cre BUN and CRE levels exhibited more intense peaks at day1 than those of their wild-type littermates (
Figure 2B) in comparison with
Mafbf/f::Lys-Cre.
After peaking on day 1, the BUN levels in MafBf/f::Tie2-Cre mice began to decrease, whereas CRE levels remained steady until the mice succumbed on day 2. The CRE and BUN levels showed a significant difference in the absence of MafB. These findings, coupled with the death of the mice on day 2, suggest that MafB cKO mice, particularly the MafBf/f::Tie2-Cre model, are more severely affected by ischemic renal disease.
In summary, the absence of MafB resulted in the inability of mice to survive ischemic renal disease beyond 2 days, highlighting the critical role of MafB in this condition. This promising result has inspired us to further advance our research to better understand the role of MafB in ischemic renal disease
3.3. Histological Analysis by PAS Staining Showed Impaired Recovery from AKI in the Absence of MafB
We next analyzed the influence of MafB on the removal of intraluminal debris. We also looked at its effect on the existence of the brush border in the epithelium and its potential to contribute to injured tubules severity, disease worsening, and early signs of recovery 48 h after IRD-induced AKI (possible brush border reestablishment in the epithelium and its contribution to injured tubules recovery).
Data from periodic acid-Schiff (PAS) staining of the kidney corticomedullary region showed severe tubular dilatation and damage with possible cast formation in both
Mafbf/f and
Mafbf/f::Lys-Cre mice, but a more severe phenotype was clearly distinguishable in the cKO mice (
Figure 3A,E).
This general phenotype was accompanied by an accumulation of intraluminal debris in Mafbf/f::Lys-Cre mice that was significantly higher than in their Mafbf/f littermates. This observation represented potential evidence that intraluminal debris removal was abrogated in the absence of the MafB transcription factor.
Furthermore, we noticed a significant (p = 0.04) loss of brush border in the absence of MafB that was clearly marked by an average of total brush borders number of 34.33 ± 0.04 in Mafbf/f::Lys-Cre mice versus a higher average of approximately 121.89 ± 0.04 total brush borders detected in MafBf/f mice (n = 3, p = 0.04). This finding indicated that the brush borders were more plentiful in Mafbf/f and were absent without MafB, which probably means that after two days from dIRD, the absence of MafB caused the removal of the brush borders or the kidney failed to reestablish its brush border without MafB. This represents potential evidence of the contribution of MafB to disease attenuation or to the recovery mechanism. Comparison of the tubules in Mafbf/f::Lys-Cre mice and their Mafbf/f littermates showed that without MafB, dIRD causes more lesions of the kidney cortex and corticomedullary zones, clear damage to the general kidney anatomical structure, very enlarged proximal tubules, accumulation of many dead cells and blood, and many holes in the center of the tubules, confirming the loss of the brush border structure and very deep damage to the diseased kidney.
We believe this explains why the absence of MafB caused a significant worsening of the disease phenotype and likely delayed the recovery process, ultimately leading to the death of the mice within 48 h of IRD. This is illustrated by the H&E staining shown in
Figure 3E. To further support our findings, we conducted a detailed analysis of proximal tubule injury by examining the expression levels of kidney injury molecule-1 (KIM-1) at the protein level, as discussed in the following paragraph.
3.4. Proximal Tubule Injury Analysis by HE Staining and Examination of KIM-1 Expression Showed That MafB Contributes to Recovery from AKI
Previous research found that kidney injury molecule-1 (KIM-1) protein and mRNA are at low levels in the normal kidney but increase very significantly in the post-ischemic kidney [
21]. This protein is reported to be expressed in the repair stage after ischemic renal disease and is highly expressed after 5 days. Its urinary level is a biomarker of acute kidney injury.
In our study, 48 h following the ischemia reperfusion,
Mafbf/f::Lys-Cre mice died and control mice were sacrificed, and then proximal tubule injury was studied with histological techniques by measuring kidney injury molecule-1 (KIM-1) protein expression and examining the injured tubules with HE staining (
Figure 3E).
First, the tubules injured by ischemic renal disease are essentially marked by the loss of brush border, cell detachment, and nuclear condensation. After 48 h of IRD, we noticed by HE staining that a more deteriorated kidney anatomical structure and more enlarged and enflamed kidney tubules were detected in the absence of MafB. A great loss of brush border and cell detachment from the basement were seen also in
Mafbf/f::Lys-Cre mice (
Figure 3E). We next calculated injured tubules and found that
Mafbf/f::Lys-Cre mice contained over 4-fold more injured tubules, with an estimated average of 186.89 ± 0.03 injured tubules versus an estimated average of 43.56 ± 0.03 injured tubules in their
Mafbf/f littermates (n = 3 in each genotype). This significantly higher number of injured tubules is evidence that without MafB, the kidneys are more affected by IRD.
Second, the immunohistochemistry data showed that KIM-1, at a magnification of 10× (
Figure 3C), seemed to be less expressed in the injured proximal TEC (tubular epithelial cells) at the corticomedullary junction region of
Mafbf/f::Lys-Cre mice than their
Mafbf/f littermates after dIRD. To more comprehensively assess the KIM-1 fluorescence quantification, the KIM-1 fluorescence level of injured tubules was determined using ImageJ software (v1.48, NIH).
The mean fluorescence of the injured tubules and the mean fluorescence background readings were all measured, and the total corrected cellular fluorescence (TCCF) was calculated for each case.
Mafbf/f presented a TCCF value of 106 ± 0.17 versus a TCCF value of 45 ± 0.17 for
Mafbf/f::Lys-Cre. Low KIM-1 expression in the MafB cKO after IRD is another piece of evidence of the severity of the disease in these mice compared to
Mafbf/f. Taking into consideration the fact that AIM is a ligand for KIM-1 and that AIM-KIM-1 interaction is required to enhance the debris uptake process by epithelial cells [
17], our data also increase the likelihood of the defect or at least the delay of the recovery processes in these MafB cKO mice.
The data suggest that the disease severity and the possible delay of Mafbf/f::Lys-Cre recovery after 48 h of dIRD is probably caused by a drastic downregulation of AIM and KIM-1, which might be explained by the absence of MafB transcription factor.
Altogether, our data suggest the importance of the role of MafB to attenuate AKI and contribute to its recovery.
3.5. F4/80+ Macrophage Number Decreased in Mafbf/f::Lys-Cre Mice After 48 h of Bilateral Ischemic Renal Disease (IRD), While Mac1+ Cells Were Upregulated
We checked macrophage infiltration to the site of injury by immunohistochemistry of the macrophage marker F4/80. Its expression after 48 h of IRD showed that the macrophage number was significantly less in
Mafbf/f::Lys-Cre mice (average of total macrophage 84.89 ± 0.02) compared to their WT littermates (
Mafbf/f) (average of total macrophages 242.33 ± 0.02, n = 3 for each genotype, and
p = 0.02) (
Figure 3B). As reported previously by Duffield and colleagues, macrophages are in charge of repairing the injury mainly by inducing epithelial regeneration by cell–cell cross-talk. On the other hand, immunohistochemistry of Mac1 showed an opposite result: the number of macrophages expressing a positive signal was significantly higher in
Mafbf/f::Lys-Cre mice compared to their littermates
Mafbf/f.
Taking into consideration our previous report of Moriguchi’s work [
12] that confirmed that the absence of
MafB does not affect the development of the Mac1-positive macrophage population, our results showed that a factor related to ischemic renal disease was behind the increase in Mac1 in
Mafbf/f::Lys-Cre. We suggest that Mac1
+ cells might contribute to the injury 48 h after the onset of reperfusion. These data were in total concordance with the trafficking of monocytes in mice following this disease, which was summarized by the work of Li and Okusa [
9]. In this research, the authors showed that in the injured tissue, the macrophages derived from inflamed monocytes are characterized as being CD11b
+ F4/80
low. Considering that CD11b is the same as Mac1, our results make sense.
3.6. AIM Expression After 48 h of Bilateral Ischemia Renal Disease (IRD)
Confirming the idea that AIM in ischemia is expressed in intraluminal debris of kidney tubules at day 1, as reported by the Arai group [
17], our IHC data showed the same localization for
Mafbf/f (
Figure 3D). This clearly suggests that in the presence of MafB (
MafBf/f), translocation of AIM from the necrotic cell debris of the injured proximal tubules (within 48 h) to the urine occurs. Thus, the majority of the protein moves to the urine, enhancing phagocytic debris removal by epithelial cells and preparing the kidneys for the repair stage.
In addition, our data also showed that AIM was sometimes slightly induced in the interstitial zones of corticomedullary regions of
Mafbf/f mice. We hypothesize that infiltrated interstitial macrophages express AIM to recover ischemia. Infiltration by macrophages to repair IRD in mice was previously studied by Yano [
22], who confirmed the interstitial accumulation of leukocytes after renal ischemia/reperfusion on day 1 and their decrease by day 5. In contrast, AIM was not induced in
Mafbf/f::Lys-Cre mice. We hypothesize that AIM downregulation in the
Mafbf/f::Lys-Cre kidney after IRD suggests the involvement of MafB in processes related to the progress of the disease.
In the previous work of the Miyazaki group [
23], they found that AIM increases in the blood with obesity, and MafB cKO mice are known to be obese according to our previous research [
18]. Taken together, AIM is expected to increase in the blood of MafB cKO mice. In ischemia, AIM comes from serum in a major part and from macrophages in a minor part, as Arai’s research explained [
17].
At least at this level, we hypothesize that this represents evidence that the absence of the AIM in cKO might be due to ischemia and the absence of MafB leading. This exposes the kidneys of Mafbf/f::Lys-Cre mice to a longer injury stage by blocking the epithelial cells’ phagocytic debris removal and therefore to a lethal and severe pathological situation that may explain their death within 48 h from dIRD.
In summary, our data showed that the major part of AIM was expressed in the injured tubules, and the minor part came from slight induction in interstitial macrophages in
Mafbf/f. However, AIM was not detected in
Mafbf/f::Lys-Cre mice (
Figure 3D). In the latter case, we hypothesize that AIM is suppressed by the absence of its regulator (MafB), preventing AIM from binding to KIM-1, which abrogates intraluminal debris removal and causes the severity and worsening of the disease and may be behind the delay of the recovery signs in
Mafbf/f::Lys-Cre kidneys, and that might be the reason why they die sooner. This implies that MafB protects the kidney from or attenuates the IRD. However, it may be too early to confirm our hypothesis. More studies are required to improve understanding of the mechanism and how MafB is part of, or influences, the IRD signaling pathway.
4. Discussion
Different large Maf transcription factors are usually expressed in different cell types within the same tissue, such as the pancreas (α cells express MafB, while β cells express MafA), lens (lens epithelial cells express MafB, while lens fiber cells express c-Maf), and inner root sheath (Henle’s layers express MafB, while Huxley’s layers express c-Maf). Thus, the different expression patterns of MafB and c-Maf in macrophage subtypes may be similar in such tissues and provide insights into the heterogeneity of macrophages in vivo.
Pathological cases might affect the expression of transcription factors in certain tissues, which in many cases implies that they actually have a role in the disease-signaling pathway. Hence, it becomes a necessity to check if MafB and c-Maf are always not expressed in pathological tissue-resident macrophages, for instance, the kidney, as they are not expressed in normal adult mouse kidney macrophages [
1].
Our data have unexpectedly revealed that these two transcription factors are expressed in kidney macrophages following IRD-induced AKI. This finding suggests that MafB and c-Maf can also be expressed in macrophages with wound-healing functions, both in vivo and in vitro, as supported by our data and findings from other research groups. Additionally, this points to their potential involvement in the AKI signaling pathway.
To further investigate the role of MafB in AKI, we utilized our previously generated MafB conditional knock-out mouse model. However, our study does not clarify whether the expression of MafB and c-Maf originates from tissue-resident macrophages or infiltrating ones. Further experiments are necessary to address this question and to expand our understanding of their roles in AKI.
In fact, when evaluating the direct function of MafB deficiency after 1 h of bilateral IRD-induced AKI, we found that
Mafbf/f::Tie2-Cre mice had worse renal function than
Mafbf/f mice, as the serum and blood urea nitrogen (BUN) showed very high rates after 48 h of IRD-induced AKI. In addition, MafB-deficient mice had a higher mortality rate, and most of them died within 48 h after reperfusion due to severe ischemia, which means that MafB deficiency worsens AKI-induced mortality in mice. However, in other examples of IRD studies, mice survive for two weeks, such as the case of AIM
−/− in Arai’s group study [
17]. This proves that the disease model we generated is more severe. Furthermore,
Mafbf/f::Tie2-Cre mice presented enhanced renal tubular injury after ischemia. Periodic acid-Schiff (PAS) staining of
Mafbf/f::Tie2-Cre injured kidneys showed a remarkable loss of brush border and higher accumulation of intraluminal debris, and hematoxylin and eosin (HE) showed more injured proximal tubules than their
Mafbf/f littermates.
Several markers have been reported for the IRD model of AKI disease. For example, proinflammatory cytokines such as IL-18 present a pathogenic role in kidney IRI in addition to its downstream molecules [
22]. In this case, the absence of this protein helps in recovery from IRD. In the case of our study, the opposite seems to be true since the presence of MafB helps to accelerate the recovery or at least contributes to attenuating the disease. However, it was similar to the AIM protein case. The proteins’ roles in certain diseases depend on their level of expression, timing, and signaling pathway. Further analysis of the relation between MafB and c-Maf and some of the IRD markers, such as IL-18 and more proinflammatory cytokines, need to be done to help understand more about the disease progress and repair.
Our immunohistochemistry assays analyzing the influence of MafB deficiency on the macrophages after IRD-induced AKI suggest that Mac1
+ F4/80
low inflamed monocytes are enhanced in
Mafbf/f::Tie2-Cre mice, which is consistent with the previous work injury phenotype [
9]. However, we did not have a chance yet to identify the specific macrophage subtype expressing them in this paper or whether they were secreted by infiltrated or resident macrophages. It is known that M2a and M2b play crucial role in AKI wound healing [
11], so we suspect them to be the types responsible for these two transcription factors’ expression especially that we found Il4/IL13- induced M2a is expressing MafB [
1]. So far, some studies have looked at the subtypes of macrophages involved in IRD. For example, CD169
+ monocytes and macrophages have been reported to prevent excessive inflammation in IRD [
24]. Further analysis to identify which macrophages are expressing MafB and c-Maf needs to be performed to provide more understanding of the disease mechanism and signaling pathway. This will contribute to a better understanding of their functions in this disease.
Furthermore, we confirmed our analysis of the injured tubules’ histological status by assessing the level of protein expression of the kidney injury molecule (KIM-1). Our data showed downregulation of KIM-1 in Mafbf/f::Tie2-Cre mice compared to their Mafbf/f control littermates, which supports again the hypothesis of the worsening of ischemia severity and possibly the delay of kidney recovery in MafB-deficient mice.
However, our data do not seem consistent with previous reports of IRD-induced AKI in AIM
−/− mice, which demonstrated by immunohistochemistry that KIM-1 protein was induced to comparable levels in proximal TEC at the corticomedullary zone of both wild-type and AIM
−/− mice after ischemia renal injury and remained constant until day 7 when a higher level of the protein expression was observed in the AIM
−/− mice [
17].
More interestingly, we have shown in our previous work that MafB directly regulates the AIM gene [
16]. This theoretically implies that when MafB is downregulated (cKO mice), AIM is also downregulated or suppressed. Therefore, we expect that in the absence of MafB, there will be suppression or at least a decrease in AIM. Taken together, this evidence would obviously lead to the observation of a similar effect on MafB cKO and AIM
−/− after dIRD, which means we expected a comparable level of expression of KIM-1 in proximal tubular epithelial cells (TECs) of the corticomedullary area between the wild-type and
Mafbf/f::Lys-Cre mice, similarly to AIM
−/−. However, our data showed that KIM-1 was less induced in the injured proximal TEC at the corticomedullary junction region of
Mafbf/f::Lys-Cre mice after dIRD.
Therefore, this observation may suggest that in the absence of MafB, IRD becomes more severe than in the absence of AIM, or it may support the hypothesis that AIM influences KIM-1 expression and, therefore, downregulation of AIM leads to downregulation of KIM-1; however, more experiments would be necessary to confirm this idea. Therefore, we hypothesize that the absence of MafB, a regulator of AIM, led to the downregulation of AIM and KIM-1, thus preventing them from binding together. This suggests an explanation of the abrogation of debris uptake and the delay of the recovery of the kidney injury in the absence of MafB. However, this hypothesis requires more experiments for confirmation in the future, possibly by rescuing the absence of MafB by injection of wild-type macrophages (expressing MafB) into the cKO mouse after dIRD and studying its effect.
This evidence suggests that MafB promotes the recovery of IRD-induced AKI after 48 h. we next wanted to understand the signaling pathway behind the mechanism. Therefore, we checked AIM protein expression. Our data showed downregulation of AIM, which was reported to bind with KIM-1 in order to remove the intraluminal debris [
17]. On the other hand, our group demonstrated in a previous report that MafB directly regulates AIM. Taking these findings together, we think that the severe IRD-induced AKI phenotype of
Mafbf/f::Tie2-Cre after 48 h was due to the abrogated intraluminal debris removal caused by the absence of the MafB.
Our study has shown that MafB is upregulated in IL-4/IL-13-induced mouse bone marrow-derived macrophages (BMDM) [
1]. Another group has confirmed that IL-4/IL-13-induced M2a renal macrophages/dendritic cells phenotype is crucial for recovery from acute kidney injury (AKI) [
11]. Our data show that the signaling pathway of AKI recovery is likely to recruit IL-4/IL-13 combined action that upregulates MafB, and the latter one will regulate AIM. Next, AIM binds to KIM-1 and enhances intraluminal debris removal from bilateral IRD-induced AKI after 48 h. Further analysis to further support the suggested signaling pathway needs to be carried out to increase the understanding of the mechanism behind AKI.
In summary, our study showed that the expression patterns of MafB and c-Maf vary from non-pathological to pathological macrophages, even within the same tissue-resident macrophages. This observation is supported by a change in the localization of the two transcription factors in the two cases. Our data show that MafB can switch its function in kidney macrophages from a homeostasis keeper in non-pathological-condition macrophages to a disease recovery contributor in pathological-condition macrophages.
Even though previous reports did not show an obvious effect of MafB on ischemia renal disease so far, our study shows that MafB decreases the effect of ischemia renal disease because the significantly high loss of brush borders observed in the Mafbf/f::Lys-Cre mice compared to their littermate MafBf/f and absence of MafB led to absence/decrease of macrophages and, therefore, failure of the ischemia recovery, which is why we clearly see loss of brush border (significantly less brush border comparing to MafBf/f). With this research and previous recent findings, we suggest that using gene targeting techniques or fate mapping analysis with several cell populations such as M2a and transcription factors such as MafB will take AKI disease to near eradication. However, further analysis needs to be conducted to elucidate the exact mechanism of the signaling pathway.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}