
BUB1 Inhibition Induces Ferroptosis in Triple-Negative Breast Cancer Cell Lines


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines and Culture
2.3. Drug Treatment and Radiation
2.4. Cell Viability
2.5. Quantitative PCR
2.6. CRISPR-CAS9 RNP Transfection
2.7. Lipid Peroxidation Assay (TBARS)
3. Results
3.1. BUB1 Inhibition Increases Cell Death Induced by Ferroptosis Activator RSL3
3.2. Ferroptosis Inhibitor Ferrostatin-1 Reverses BUB1 Inhibition-Induced Cell Death
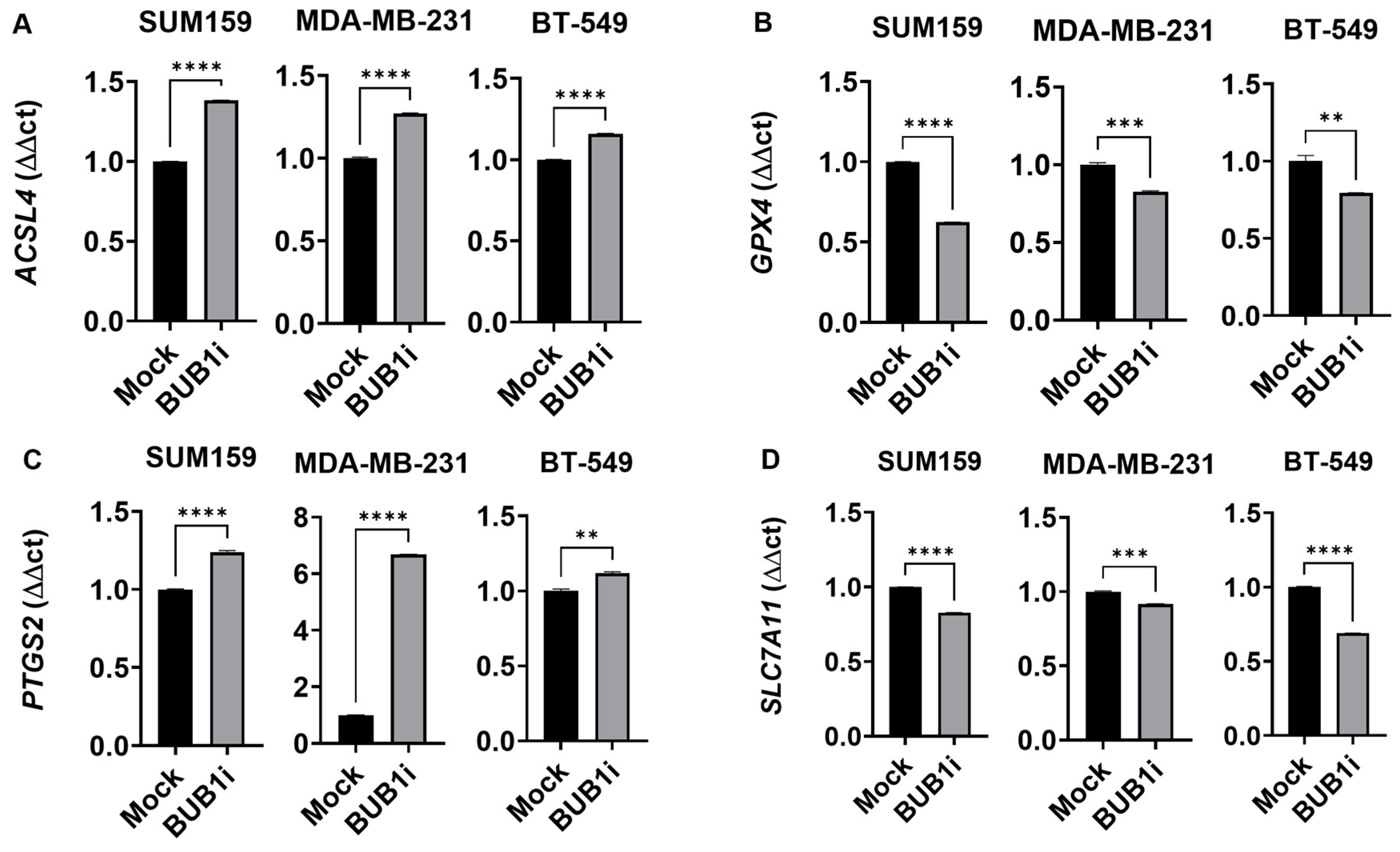
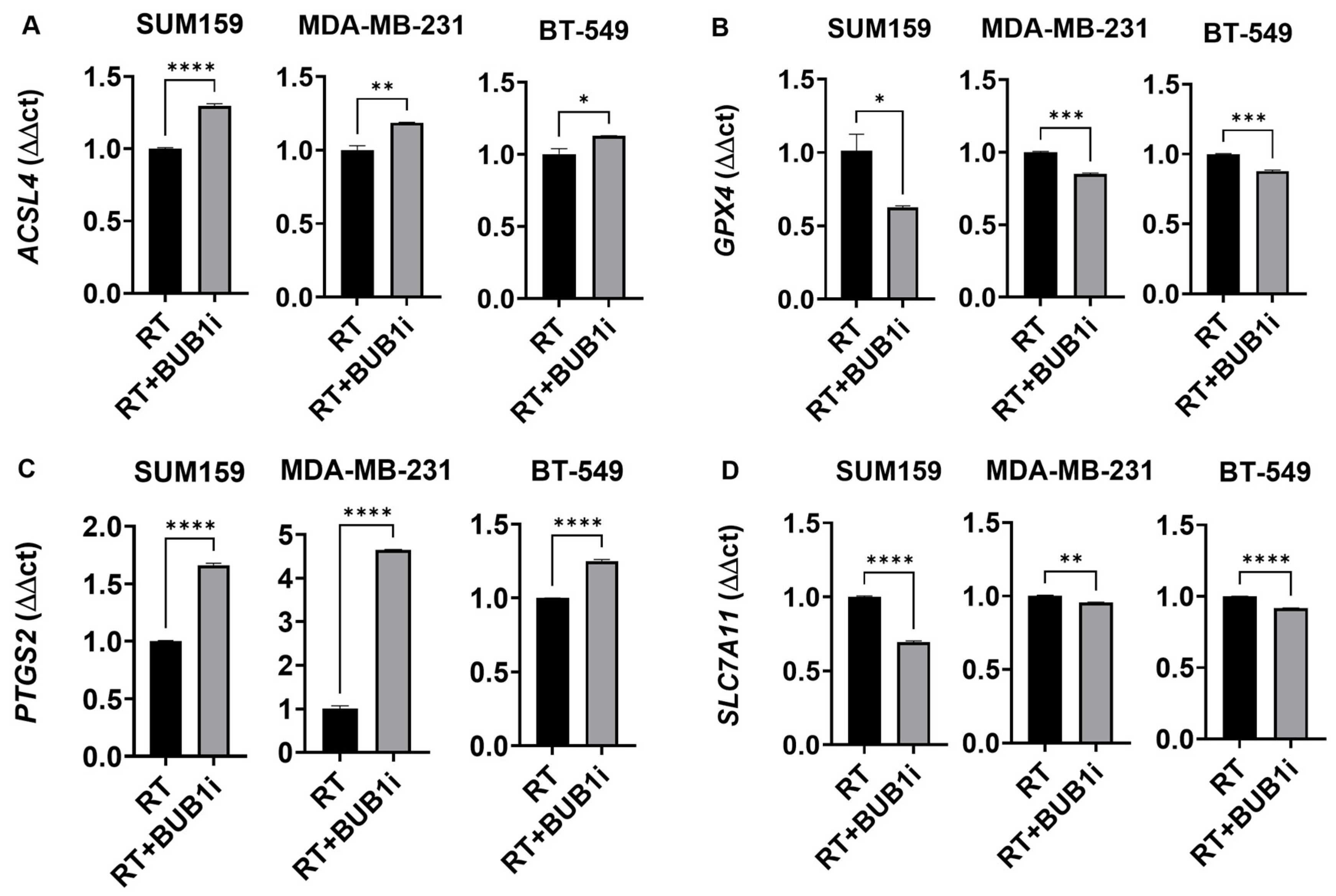
3.3. BUB1 Inhibition Alters Expression of Key Ferroptosis Genes
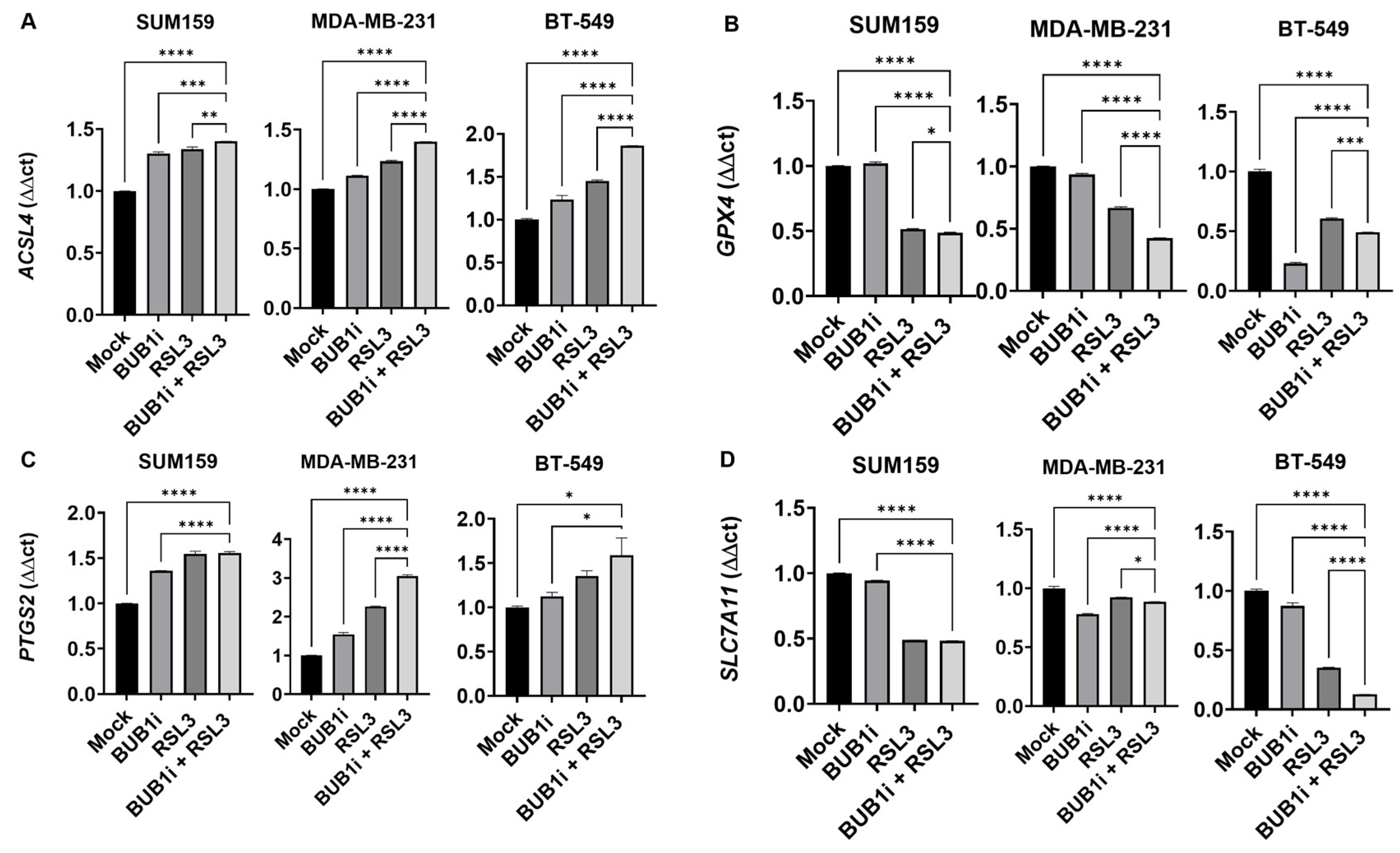
3.4. BUB1i and RSL3 Regulate Ferroptosis Markers in TNBC Cell Lines
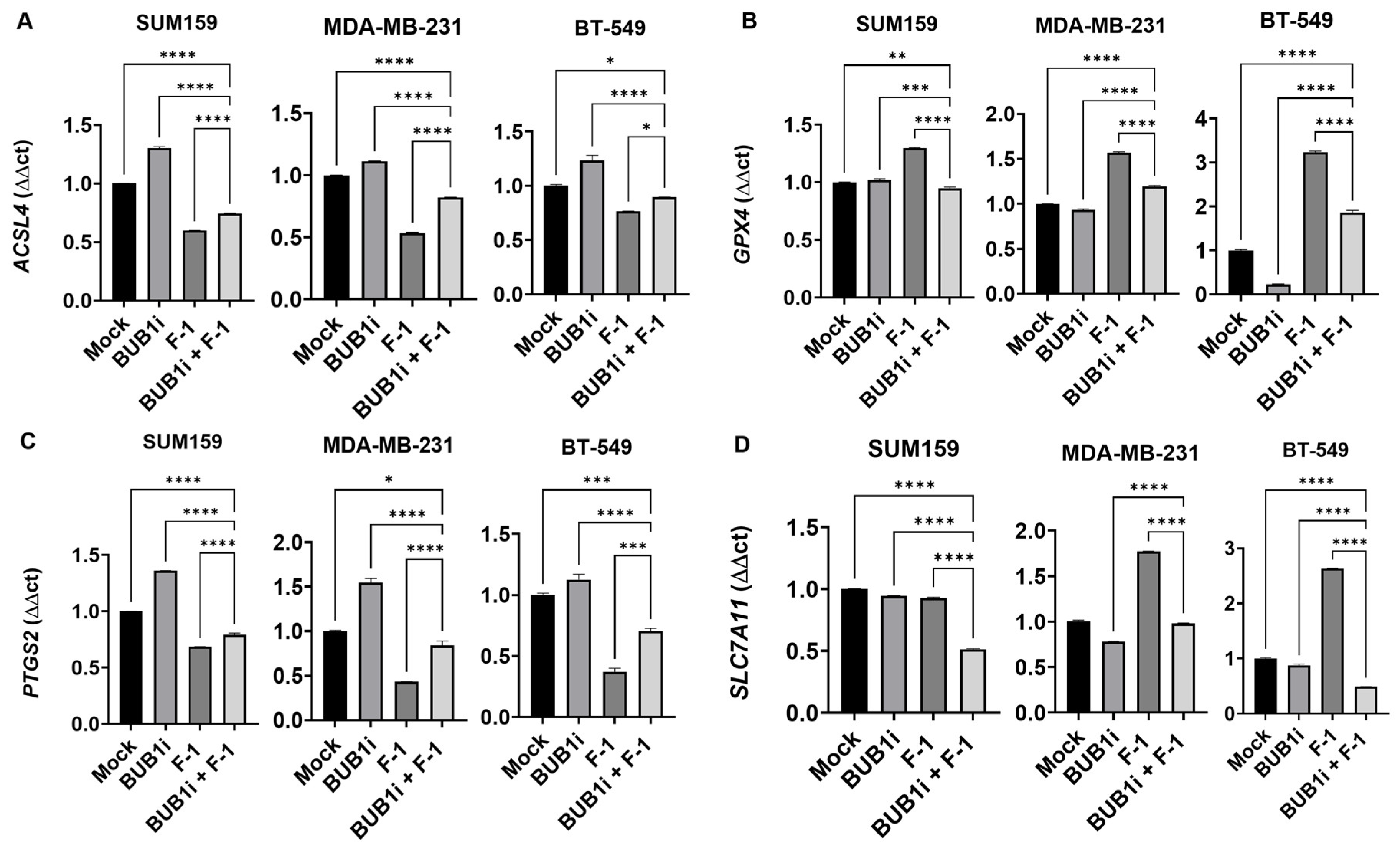
3.5. Ferrostatin-1 Reverses BUB1i-Induced Ferroptosis in TNBC Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Merkher, Y.; Chen, L.; Liu, N.; Leonov, S.; Chen, Y. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022, 15, 121. [Google Scholar] [CrossRef] [PubMed]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Y.; Song, B.; Yi, M.; Yan, Y.; Mei, Q.; Wu, K. Recent advances in targeted strategies for triple-negative breast cancer. J. Hematol. Oncol. 2023, 16, 100. [Google Scholar] [CrossRef]
- Zagami, P.; Carey, L.A. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer 2022, 8, 95. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Ghorayeb, T.; Saliba, F.; Allam, S.; Bou Zerdan, M.; Yaghi, M.; Bilani, N.; Jaafar, R.; Nahleh, Z. Triple Negative Breast Cancer: Updates on Classification and Treatment in 2021. Cancers 2022, 14, 1253. [Google Scholar] [CrossRef]
- Twelves, C.; Jove, M.; Gombos, A.; Awada, A. Cytotoxic chemotherapy: Still the mainstay of clinical practice for all subtypes metastatic breast cancer. Crit. Rev. Oncol./Hematol. 2016, 100, 74–87. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Speers, C.; Nyati, S. Present and Future of Immunotherapy for Triple-Negative Breast Cancer. Cancers 2024, 16, 3250. [Google Scholar] [CrossRef]
- Alaluf, E.; Shalamov, M.M.; Sonnenblick, A. Update on current and new potential immunotherapies in breast cancer, from bench to bedside. Front. Immunol. 2024, 15, 1287824. [Google Scholar] [CrossRef]
- Rossi, V.; Turati, A.; Rosato, A.; Carpanese, D. Sacituzumab govitecan in triple-negative breast cancer: From bench to bedside, and back. Front. Immunol. 2024, 15, 1447280. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Wu, H.; Yu, Z. Advancements and challenges in triple-negative breast cancer: A comprehensive review of therapeutic and diagnostic strategies. Front. Oncol. 2024, 14, 1405491. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, Y.; Jiang, Y.; Bu, J.; Gu, X. Targeting ferroptosis, the achilles’ heel of breast cancer: A review. Front. Pharmacol. 2022, 13, 1036140. [Google Scholar] [CrossRef]
- Qi, X.; Wan, Z.; Jiang, B.; Ouyang, Y.; Feng, W.; Zhu, H.; Tan, Y.; He, R.; Xie, L.; Li, Y. Inducing ferroptosis has the potential to overcome therapy resistance in breast cancer. Front. Immunol. 2022, 13, 1038225. [Google Scholar] [CrossRef]
- Xu, C.; Chen, Y.; Yu, Q.; Song, J.; Jin, Y.; Gao, X. Compounds targeting ferroptosis in breast cancer: Progress and their therapeutic potential. Front. Pharmacol. 2023, 14, 1243286. [Google Scholar] [CrossRef]
- Ge, A.; He, Q.; Zhao, D.; Li, Y.; Chen, J.; Deng, Y.; Xiang, W.; Fan, H.; Wu, S.; Li, Y.; et al. Mechanism of ferroptosis in breast cancer and research progress of natural compounds regulating ferroptosis. J. Cell Mol. Med. 2024, 28, e18044. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-l.; Huang, Z.-j.; Lin, Z.-t.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Yan, H.-f.; Zou, T.; Tuo, Q.-z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Tomitsuka, Y.; Imaeda, H.; Ito, H.; Asou, I.; Ohbayashi, M.; Ishikawa, F.; Kuwata, H.; Hara, S. Gene deletion of long-chain acyl-CoA synthetase 4 attenuates xenobiotic chemical-induced lung injury via the suppression of lipid peroxidation. Redox Biol. 2023, 66, 102850. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Del Rey, M.Q.; Kapner, K.S.; Zhang, H.; Gikandi, A.; Malcolm, C.; Poupault, C.; Kuljanin, M.; John, K.M.; Biancur, D.E. NCOA4-mediated ferritinophagy is a pancreatic cancer dependency via maintenance of iron bioavailability for iron–sulfur cluster proteins. Cancer Discov. 2022, 12, 2180–2197. [Google Scholar] [CrossRef]
- Yao, F.; Cui, X.; Zhang, Y.; Bei, Z.; Wang, H.; Zhao, D.; Wang, H.; Yang, Y. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol. Lett. 2021, 22, 657. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e3417. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.-L. Targeting Iron-Sulfur Clusters in Cancer: Opportunities and Challenges for Ferroptosis-Based Therapy. Cancers 2023, 15, 2694. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell 2024, 42, 513–534. [Google Scholar] [CrossRef]
- Diao, J.; Jia, Y.; Dai, E.; Liu, J.; Kang, R.; Tang, D.; Han, L.; Zhong, Y.; Meng, L. Ferroptotic therapy in cancer: Benefits, side effects, and risks. Mol. Cancer 2024, 23, 89. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, X.; Liu, C.; Ge, W.; Wang, Q.; Hao, X.; Wang, M.; Chen, Y.; Zhang, Q. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J. Hematol. Oncol. 2021, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, X.; Kong, L.; Pan, Z.; Chen, G. BUB1 Promotes Gemcitabine Resistance in Pancreatic Cancer Cells by Inhibiting Ferroptosis. Cancers 2024, 16, 1540. [Google Scholar] [CrossRef] [PubMed]
- Klebig, C.; Korinth, D.; Meraldi, P. Bub1 regulates chromosome segregation in a kinetochore-independent manner. J. Cell Biol. 2009, 185, 841–858. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Walker, E.; Nyati, S. BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy. Biomolecules 2024, 14, 625. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Chen, W.-M.; Hassan, O.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Green, M.D.; Davis, A.J.; Speers, C.; et al. BUB1 regulates non-homologous end joining pathway to mediate radioresistance in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2024, 43, 163. [Google Scholar] [CrossRef]
- Thoidingjam, S.; Sriramulu, S.; Hassan, O.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Gadgeel, S.; Nyati, S. BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer. Cancers 2024, 16, 3291. [Google Scholar] [CrossRef]
- Xu, L.; Liu, Y.e.; Chen, X.; Zhong, H.; Wang, Y. Ferroptosis in life: To be or not to be. Biomed. Pharmacother. 2023, 159, 114241. [Google Scholar] [CrossRef]
- Thu, K.L.; Soria-Bretones, I.; Mak, T.W.; Cescon, D.W. Targeting the cell cycle in breast cancer: Towards the next phase. Cell Cycle 2018, 17, 1871–1885. [Google Scholar] [CrossRef]
- Berry, C.E.; Kendig, C.B.; An, N.; Fazilat, A.Z.; Churukian, A.A.; Griffin, M.; Pan, P.M.; Longaker, M.T.; Dixon, S.J.; Wan, D.C. Role of ferroptosis in radiation-induced soft tissue injury. Cell Death Discov. 2024, 10, 313. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Li, J.; He, D.; Li, S.; Xiao, J.; Zhu, Z. Ferroptosis: The emerging player in remodeling triple-negative breast cancer. Front. Immunol 2023, 14, 1284057. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Li, J.; Song, Y.; Luo, C. ACSL4-Mediated Ferroptosis and Its Potential Role in Central Nervous System Diseases and Injuries. Int. J. Mol. Sci. 2023, 24, 21. [Google Scholar] [CrossRef] [PubMed]
- Guo, N. Identification of ACSL4 as a biomarker and contributor of ferroptosis in clear cell renal cell carcinoma. Transl. Cancer Res. 2022, 11, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in cell death, autophagy, and disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef]
- Nandi, I.; Ji, L.; Smith, H.W.; Avizonis, D.; Papavasiliou, V.; Lavoie, C.; Pacis, A.; Attalla, S.; Sanguin-Gendreau, V.; Muller, W.J. Targeting fatty acid oxidation enhances response to HER2-targeted therapy. Nat. Commun. 2024, 15, 6587. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Yan, H.; Talty, R.; Aladelokun, O.; Bosenberg, M.; Johnson, C.H. Ferroptosis in colorectal cancer: A future target? Br. J. Cancer 2023, 128, 1439–1451. [Google Scholar] [CrossRef]
- Chen, G.-Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.-X.; Jiang, X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020, 27, 242–254. [Google Scholar] [CrossRef]
- Song, Q.; Peng, S.; Che, F.; Zhu, X. Artesunate induces ferroptosis via modulation of p38 and ERK signaling pathway in glioblastoma cells. J. Pharmacol. Sci. 2022, 148, 300–306. [Google Scholar] [CrossRef]
- Li, Q.; Chen, K.; Zhang, T.; Jiang, D.; Chen, L.; Jiang, J.; Zhang, C.; Li, S. Understanding sorafenib-induced ferroptosis and resistance mechanisms: Implications for cancer therapy. Eur. J. Pharmacol. 2023, 955, 175913. [Google Scholar] [CrossRef]
- von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Osburg, J.; Storch-Hagenlocher, B.; Sertel, S.; Elsässer, M.; Remppis, B.A.; Edler, L.; Munzinger, J.; et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res. Treat. 2017, 164, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Aspitia, A.; Morton, R.F.; Hillman, D.W.; Lingle, W.L.; Rowland, K.M., Jr.; Wiesenfeld, M.; Flynn, P.J.; Fitch, T.R.; Perez, E.A. Phase II trial of sorafenib in patients with metastatic breast cancer previously exposed to anthracyclines or taxanes: North Central Cancer Treatment Group and Mayo Clinic Trial N0336. J. Clin. Oncol. 2009, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, Z. Recent progress in ferroptosis: Inducers and inhibitors. Cell Death Discov. 2022, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACSL4 |
| Forward: 5′-ATAAAGCAGAGTACCCTGAAG |
| Reverse: 5′-CAAGTTTTCTGGGTTAGATCC |
| GPX4 |
| Forward: 5′-GAAGTAAACTACACTCAGCTC |
| Reverse: 5′-CTCTTTGATCTCTTCGTTACTC |
| PTGS2 |
| Forward: 5′-AAGCAGGCTAATACTGATAGG |
| Reverse: 5′-TGTTGAAAAGTAGTTCTGGG |
| SLC7A11 |
| Forward: 5′-GGTTATTCTATGTTGCGTCTC |
| Reverse: 5′-AATAACAGCTGGTAGAGGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sriramulu, S.; Thoidingjam, S.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Nyati, S. BUB1 Inhibition Induces Ferroptosis in Triple-Negative Breast Cancer Cell Lines. DNA 2025, 5, 16. https://doi.org/10.3390/dna5010016
Sriramulu S, Thoidingjam S, Brown SL, Siddiqui F, Movsas B, Nyati S. BUB1 Inhibition Induces Ferroptosis in Triple-Negative Breast Cancer Cell Lines. DNA. 2025; 5(1):16. https://doi.org/10.3390/dna5010016
Chicago/Turabian StyleSriramulu, Sushmitha, Shivani Thoidingjam, Stephen L. Brown, Farzan Siddiqui, Benjamin Movsas, and Shyam Nyati. 2025. "BUB1 Inhibition Induces Ferroptosis in Triple-Negative Breast Cancer Cell Lines" DNA 5, no. 1: 16. https://doi.org/10.3390/dna5010016
APA StyleSriramulu, S., Thoidingjam, S., Brown, S. L., Siddiqui, F., Movsas, B., & Nyati, S. (2025). BUB1 Inhibition Induces Ferroptosis in Triple-Negative Breast Cancer Cell Lines. DNA, 5(1), 16. https://doi.org/10.3390/dna5010016