Abstract
Due to the increased and excessive consumption of fossil fuels, sustainable alternative energy sources are badly needed to replace fossil fuels. The conversion of biomass into energy and value-added chemicals is one of the most promising potential pathways to solve this problem. Millions of tons of lignin, one of the major components of biomass, are produced annually as a byproduct of various industries, where it is treated as a low-value material. However, since it has an aromatic polymer nature, lignin is a proven source for different value-added products. Studies suggest that the selective cleavage of a specific bond of the complex lignin structure is one of the major challenges of converting lignin to a targeted product. In this study, eight different lignin depolymerization methods, both traditional and green, are reviewed. Acid and base catalytic depolymerization methods are straightforward, but due to their low selectivity and comparatively severe reaction conditions, they are expensive and not eco-friendly. Pyrolysis-based depolymerization comes with similar problems but has a higher conversion. In contrast, greener approaches, such as oxidative, microwave-assisted, super/sub-critical fluids (SCF), ionic liquid (IL), and deep eutectic solvent (DES)-based depolymerization techniques, have shown higher efficiency in terms of converting the lignin into phenolic compounds even under milder reaction conditions. SCF, IL, and DES-based approaches will likely become more popular in the future for their greener nature. Overall, depolymerization of lignin with greener technologies could make this process more economically viable and sustainable.
1. Introduction
Fossil fuels have been extensively used for different purposes, such as producing chemicals and fuels for transportation, which has caused their rapid depletion [1,2]. Presently, approximately 80% of chemicals are produced from fossil fuels [3]. Since such sources are limited, an alternative source to produce energy and chemicals is urgently needed. Today, lignocellulosic biomass is considered the renewable carbon source with the most potential, which could produce energy in the form of biofuel and value-added chemicals [4].
Lignocellulose must undergo various treatments to be broken into its major components, such as lignin, hemicellulose, cellulose, protein, and oil [5]. Previously, biofuels were produced industrially—mainly from the cellulose part of the biomass—and the lignin was considered a waste product. Approximately 70 million tons of lignin per year are produced industrially, and only a tiny portion (1–2%) is used [6,7,8]. Because it is polyaromatic, lignin is a proven source of many value-added chemicals [9].
Lignin is a complex cross-linked organic polymer composed of phenylpropanoid units linked by ether and carbon–carbon bonds [10]. Determining the structure of lignin is complicated because of its irregular monomeric sequences. Additionally, its convoluted structure makes it less reactive and causes resistance to depolymerizing it into the monomeric units [11]. Therefore, it is important to understand the chemical structure, interactions, crystallinity, and lignin sources before selecting a depolymerization technique.
Phenolic monomers are one of the major products of lignin depolymerization [12]. To obtain them, intramolecular linkages, such as the C–C and ether bonds, must be cleaved [13]. Studies also suggest that the intermediates produced from lignin could be repolymerized under milder or stronger conditions such as high (>80 °C) and atmospheric temperatures [14]. Therefore, developing and selecting an appropriate depolymerization method is required to improve production efficacy.
Different depolymerization techniques, such as acid-catalyzed, base-catalyzed, oxidative, pyrolysis, microwave-assisted, and sub- or supercritical methods, have been used [15,16,17]. Acid catalyst, base catalyst, and oxidation-based methods are traditional chemical methods that show good lignin conversion. However, these methods require high temperature and pressure, expensive instrumental setups, and toxic chemicals, making them costly and not eco-friendly [18]. Pyrolysis-based depolymerization has inherent disadvantages, such as lower selectivity and higher temperature requirements [19,20]. The microwave-assisted method is considered an efficient approach, which can significantly decrease reaction time and selectively increase the yield of the targeted products [21]. Overall, these methods must be optimized to make them more efficient. Due to some of the inherent disadvantages of the conventional depolymerization methods, developing highly effective and greener methods is inevitable. Sub- and supercritical depolymerization of lignin are two greener methods. This technology does not require any hazardous materials [22]. Recently, ionic liquid (IL)-based and deep eutectic solvent (DES)-based depolymerization approaches have shown great promise for sustainable and more selective lignin depolymerization [23,24,25,26]. Careful selection of greener solvents and the addition of microwave-based techniques could enhance the efficacy of these methods [23,24,25,26].
This review focuses on the chemical structure of lignin, the mechanisms of depolymerization, the studies conducted thus far using traditional methods, and the associated problems. The structural chemistry and mechanisms help us interpret the advantages and disadvantages of the current methods. While the greener methods and their approaches are scrutinized, the previously investigated chemistry helps to enhance the efficacy of greener methods. In this way, a solid understanding of the currently existing methods may achieve a highly efficient and sustainable lignin depolymerization method.
2. Lignin
In 1838, a French scientist named Anselme Payen first attempted to extract lignin from wood. He treated the wood with concentrated nitric acid and sodium hydroxide and obtained one soluble and another insoluble solid fraction [27]. The insoluble solid material was named “cellulose” because it mainly contains the sugar molecules of the plant cell wall [28,29]. He also observed that the soluble portion is richer in carbon than cellulose and named it “incrusting materials” [29]. In 1865, Payen’s “incrustation hypothesis” was supported by Schulze, who named the incrusting materials “lignin” [29]. The word “lignin” originates from the Latin word “lignum”, which means “wood”. Between 1897 and 1900, the Swedish chemist Johan Peter Klason studied the structure of lignin and proposed that it is a macromolecular compound consisting of coniferyl alcohol moieties connected through ether linkages [28].
2.1. Sources and Classification of Lignin
Plants and grasses are the primary sources of lignocellulose, which is composed of cellulose, hemicellulose, and lignin, along with a small quantity of extractives, such as proteins, salts, minerals, and wax [9]. This biomass contains 15–32% lignin, 25–40% hemicellulose, and 45–50% cellulose [30]. Table 1 gives the percentages of lignin in various biomasses.

Table 1.
Percentages of lignin in various lignocellulose.
Based on the extraction processes and modifications, lignin can be classified as (i) native or (ii) modified lignin. Native lignin is extracted without further modification, while lignin extracted through different modifications is called modified or technical lignin [36]. The primary sources of modified lignin are industrial byproducts. Depending on the extraction process used, modified lignin can be specifically identified as kraft lignin (KL), hydrolysis lignin (HL), organosolv lignin (OL), or pyrolytic lignin (PL) [37]. The molecular weight and composition of modified lignin vary based on the extraction method and source.
2.2. Structure of Lignin
Lignin, a class of organic polymer, has a complex, cross-linked, highly branched, amorphous, and irregular aromatic structure (Figure 1). It is a major structural component of the secondary cell wall of vascular plant cells, where it occurs alongside cellulose and hemicellulose [38,39,40]. In plant cells, lignin occupies the free space between the cellulose and the hemicellulose and cross-links them to form a rigid structure. This structure helps conduct water and nutrient materials in the plant stems.
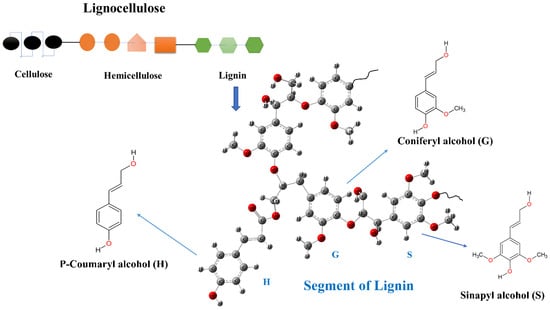
Figure 1.
Generalized structure of a segment of lignin.
Lignin is the second most abundant natural polymer after cellulose and makes up from 15% to 32% of the total biomass [30], depending on the age, environment, and sources of the biomass. It is a heterogeneous polymer without repetitive monomeric units, differentiating it from other polymers, such as cellulose and hemicellulose. Its helical structure is formed by the polymerization of three phenylpropanoid monomeric units: p-coumaryl alcohol (H), coniferyl alcohol (G), and sinapyl alcohol (S) (Figure 1) [36]. The phenolic monomers are interconnected by ether (-O-) and C–C linkages (Figure 2).
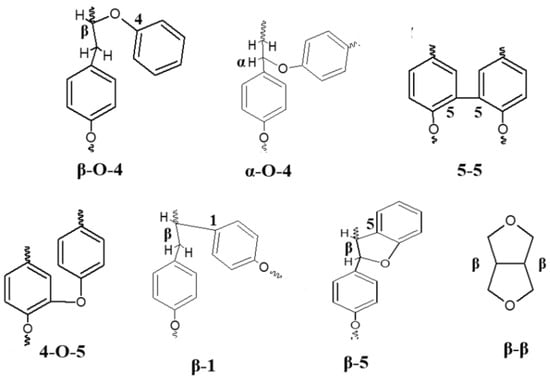
Figure 2.
The most common carbon–carbon and ether interlinkages in lignin structure; adapted from [23].
2.3. Monomers of Lignin
The H, G, and S moieties are randomly connected through ether (-O-) and C–C linkages (Figure 1 and Figure 2) [41]. Their monomeric units differ from each other by methoxy groups, where the H moiety does not have any methoxy groups, but the G and S moieties have one and two methoxy groups, respectively (Figure 1). These three monomers are connected through free radical reactions and form the highly interconnected three-dimensional complex ligand structure. Lignin in hardwood species mainly contains G and S moieties, whereas lignin in softwood species mostly contains G units [9]. All three moieties are usually present in grass lignin [42].
2.4. Major Hindrances to Depolymerizing Lignin
Since it is abundant and polyaromatic, lignin could be an excellent resource for the renewable energy industry. However, the industrial-scale transformation of lignin into value-added chemicals requires the successful selective breakdown of its structure. Studies have shown that the depolymerization of KL is difficult because of its higher percentage of refractory carbon–carbon linkages [43,44]. In a typical lignin structure, the three different moieties are interconnected to each other through the ether and C–C bonds, such as the β-O-4, 4-O-5, 5-5, β-5, β-1, and β-β bonds (Figure 2), which make the structure resistant to decomposition [43,45].
Several depolymerization approaches, including chemical, physical, thermochemical, and enzymatic methods, have been developed to decompose lignin into aromatic compounds and biomaterials [46]. All methods have advantages and disadvantages, and none are perfectly successful in terms of cost-effectiveness, re-condensation, hazards, product yield, or greenness. This review discusses their comparative strengths, weaknesses, and possible improvements based on green chemistry principles and recent advances.
3. Depolymerization of Lignin
Among the ether and C–C linkages, ether bonds are more frequent, constituting 56% of the total network [47]. Among them, 50–60% of the bonds are β-aryl ether (β-O-4) bonds, depending on the source of the lignin [48]. Therefore, cleaving β-O-4 bonds is considered one of the critical steps of lignin depolymerization [49].
Thermal analysis is a well-proven degradation technique widely used to determine the physical and chemical properties of lignin as a function of temperature [50]. This method has shortcomings, such as providing only partial information regarding the polymeric structure, complicating the data analysis. Catalytic degradation methods can selectively break down the ether linkages of lignin and produce various monomeric products. In the following subsection, we compare the chemistry behind traditional and greener methods’ efficiency, advantages, and disadvantages.
3.1. Acid-Catalyzed Lignin Depolymerization
Depolymerization of lignin based on acid catalysis is a well-known method that has been used since the mid-twentieth century. This method decomposes the β-O-4 linkages of lignin using acid [15]. Different acids, such as hydrochloric, sulfuric, formic, and peracetic, have been used as catalysts to break down the ether linkages [51]. During hydrolysis, the acids provide H3O+ to break the ether bonds, producing phenolic compounds. An acid catalyst mechanism for the cleavage of β-O-4 is shown in Figure 3.
These kinds of depolymerizations are usually conducted at high temperatures and pressures. A depolymerization study is typically performed using ethanol and acid by varying the solvent-acid ratio (e.g., hydrochloric acid/ethanol and formic acid/ethylene glycol) to separate the products into water-soluble and water-insoluble lignin. Mahmood et al. showed that a temperature range of 78–200 °C is insufficient to break the lignin structure into monomeric units [52].

Figure 3.
Cleavage of β-O-4 linkages by acid catalysts, adapted from [53].
Recently, some research groups have investigated acid-catalyzed depolymerization at high temperatures and pressure using different proportions of acids [54,55]. One study suggested that a 10:77 (wt%) formic acid/ethanol mixture effectively depolymerized wheat straw lignin [54]. Another group reported that 10:81 (wt%) of formic acid/ethanol is the optimal ratio to depolymerize lignin from wheat straw [55]. Dilute sulfuric acid mixed with an ethanol/water solution was used for the depolymerization reaction, where the reaction achieved a high yield under 2 MPa and 250 °C for 1 h. Approximately 70 wt% of depolymerization was achieved when sulfuric acid was used as the catalyst in a 1:1 water/ethanol solvent system [51]. Conditions for the acid-catalyzed depolymerization of lignin and the major outcomes are listed in Table 2.
Table 2.
Acid-catalyzed depolymerization of lignin.
Although acid-catalyzed depolymerization is one of the most widely used methods, it has some disadvantages. Extreme reaction conditions, repolymerization, and toxic chemical use are unavoidable aspects of this method. Repolymerization mainly occurs between the reactive sites of the phenols and the α-carbon of phenol propanol [66,67]. This technique requires a comparatively longer reaction time, higher temperature, and higher pressure. It also produces environmentally corrosive waste materials, which significantly increase the reaction costs due to handling and disposal requirements.
3.2. Base-Catalyzed Depolymerization of Lignin
Base-catalyzed depolymerization (BCD) is another commonly used method to extract phenolic monomers from lignin. This method is performed at a higher temperature, where different bases, such as NaOH, KOH, and Ca(OH)2, are used as catalysts [68,69]. This depolymerization technique is considered one of the most promising methods due to its excellent catalytic performance [54,70]. It is usually conducted at >300 °C and high pressure. The most abundant linkage in lignin is the β-O-4 bond; cleavage of this bond begins at 270 °C. During the reaction, cations from the base assist in forming cation adducts, which catalyze a six-membered transition on the β-O-4 bond resulting in the formation of phenolic monomers [71]. A base catalyst mechanism is shown in Figure 4.
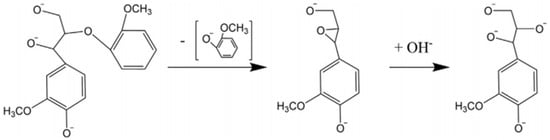
Figure 4.
Base catalyst mechanism for the cleavage of β-O-4 linkages, adapted from [49].
BCD is also well known for its controlled hydrolytic cleavage of the ether bonds of lignin [72]. A study has shown that weak bases, such as Ca(OH)2 and LiOH, can produce smaller amounts of depolymerized product with lower depolymerization rates than strong bases, such as KOH and NaOH [69]. Although the strong bases showed efficient depolymerization of lignin, bases such as LiOH, KOH, and CsOH were inefficient because of char deposition [70]. BCD could be used to achieve high yields for selective monomeric products. Therefore, selectivity and yield depend on the temperature, pressure, reaction time, base concentration, and solvent type [66,73].
An alkaline-based catalyst is a homogeneous catalyst that causes complex post-separation. MgO is an inexpensive solid base catalyst alternative to the homogeneous catalyst. It is also considered robust, with excellent catalytic activity [74]. The reaction conditions and promising outcomes using BCD of lignin are summarized in Table 3.
Table 3.
Base-catalyzed depolymerization of lignin.
Table 3 shows that most of the base-catalyzed transformations of lignin are performed at ≥300 °C and >10 MPa, where monomeric products vary with the reaction conditions. Beauchet et al. depolymerized KL at 270–315 °C and 130 bar at two different NaOH concentrations [72]. Their results demonstrated that NaOH efficiently acted as a catalyst to increase the production of aromatic monomers. The monomeric production increased significantly with increased temperature, where the total yield of 8.4 wt% was reached at 315 °C. Pyrocatechol phenolic monomers were abundant (up to 25.8%) at 315 °C with high selectivity.
Besides temperature, pressure is another critical factor in obtaining a better yield. Lavoie et al. studied softwood lignin depolymerization using 5 wt% NaOH at temperatures of 300–330 °C and pressures of 9–13 MPa. Their chromatographic study identified 26 compounds; the major products were vanillin, guaiacol, and catechol [70]. A similar study was performed by Roberts et al. at elevated pressure (25 MPa), and the major products, syringol, hydroxyacetophenone, and catechol, differed [71]. These studies revealed that the nature of phenolic monomers depends significantly on the reaction pressure.
Solvents also play an important role in the BCD of lignin. Therefore, solvent selection is crucial for these reactions. A depolymerization reaction with a low base catalyst concentration in water shows very little depolymerization, sometimes yielding almost no depolymerized products [83]. This indicates that an aqueous environment is not the correct solvent for this reaction. As a result, most researchers have used organic solvents, such as ethanol, isopropanol, tetrahydrofuran, and polyethylene glycerol, for BCD of lignin [84,85]. Long et al. have shown that tetrahydrofuran (THF) works well as a solvent for the depolymerization of lignin [86]. Their study revealed that THF significantly promoted the catalytic activity of MgO. Hence, the production of phenolic monomers at 250 °C for 15 min increased by 13.2% compared to conventional solvents.
3.3. Oxidative Depolymerization of Lignin
Studies have shown that acid- or base-catalyzed depolymerization is a promising method to produce phenolic monomers by cleaving the ether linkages of lignin. However, these methods require relatively extreme reaction conditions, making the methods expensive, environmentally unfriendly, and difficult to handle.
Oxidative depolymerization is a promising method for converting lignin into monomers at milder conditions than the acid or base catalyst methods [87]. A wide range of oxidants is used for the depolymerization reaction, such as permanganate, nitrobenzene, copper oxide, hydrogen peroxide, chlorine, and hypochlorite [17,88,89]. The oxidative degradation approach selectively breaks down the ether linkages by preserving the aromatic character [90]. Strong oxidants can break the linkages among the aromatic moieties of the lignin with good efficiency at milder conditions than catalysis methods [8]. Therefore, successful oxidative depolymerization depends on the proper selection of the oxidant. Reaction conditions and the major outcomes of oxidative depolymerization of lignin are listed in Table 4.
Table 4.
Oxidative depolymerization of lignin.
Hydrogen peroxide, an important oxidant, has been used to depolymerize lignin for the past few decades. It is an eco-friendly and economic catalyst that donates oxygen during lignin depolymerization [98]. Studies have demonstrated that a minimum of 0.1% hydrogen peroxide can depolymerize the alkali lignin into monomers [99]. Other studies have stated that H2O2 can selectively break down the β-O-4 and β-1 linkages at low temperatures [100]. Although H2O2 shows a higher degradation rate than other methods, it produces a lower monophenolic compound yield. H2O2 in the presence of a metallic catalyst CuO (as Cu2+) can cleave more ether bonds and side chains. Furthermore, in the presence of Fe2(SO4)3 (as Fe3+), the oxidation ability of H2O2 is significantly increased, which could increase the production of monophenols. In addition, the performance of hydrogen peroxide-based depolymerization can be significantly increased by acid [100,101].
Oxidative depolymerization can be performed without using metallic or other toxic materials. Wet air and molecular oxygen are considered less hazardous and more benign oxidants than metallic or traditional oxidants. Results have shown that wet air oxidation of lignin can substantially increase the production of phenolic monomers at low temperatures and pressures [97]. Molecular oxygen depolymerization efficiently converts lignin into aromatic compounds in moderate reaction environments [93].
3.4. Depolymerization of Lignin by Pyrolysis
Pyrolysis is another method frequently used to depolymerize lignin into its monomeric products. This method uses heat to convert the non-volatile compounds to a volatile mixture in the absence of oxygen. The pyrolytic mixture contains different aromatic monomer types, which indicates that the pyrolysis method could effectively convert lignin into biomaterials [102]. It is a very fast method, taking only a few milliseconds to reach the pyrolytic temperature, compared to other thermal methods, for example, a thermogravimetric analyzer coupled with a Fourier Transform Infrared spectrometer (TG-FTIR) [103]. The efficiency of the depolymerization and the molecular weight of the monomeric products can be modified by tuning the reaction conditions, such as the reaction time [104], catalysts [105], and the sources of lignin [106]. The pyrolytic depolymerization of lignin starts with the cleavage of weak linkages at lower temperatures (<450 °C), followed by further breaking stronger linkages at higher temperatures (>450 °C). At higher temperatures, larger molecules decompose at selective sites to form smaller molecules, which are then separated by gas chromatography and detected by mass spectrometry [107]. This method can be performed differently, but most pyrolysis methods are divided into two stages [108]. The reaction conditions and promising outcomes of pyrolysis depolymerization of lignin are summarized in Table 5.
Table 5.
Depolymerization of lignin by pyrolysis.
The first pyrolysis stage occurs in the temperature range of 150–400 °C. Only basic side chains are broken during this stage, and other linkages, such as aromatic methylated groups and condensed linkages, remain stable [106]. Therefore, softwood lignin produces a larger amount of residue than hardwood lignin because it contains more condensed and phenolic linkages; the major products in this stage are vanillin, coniferyl alcohol, isoeugenol, and other unsaturated compounds [19,117].
The second pyrolysis stage occurs in the temperature range between 400 °C and 800 °C. Most condensed methoxy groups and phenolic ether linkages are cleaved during this stage and the hydroxyl or methylated groups bond to the aromatic groups [118]. Guaiacol, o-vanillin, and o-quinone are produced in this stage [119]. The cleavage of benzene rings produces non-condensable gases at 550 °C [120].
Pyrolysis depolymerization has been performed with different combinations of catalysts and solvents to obtain better performance. Since pyrolysis is performed in the absence of oxygen, catalysts and solvents may improve the performance of depolymerization by assisting in demethylation and preventing repolymerization and condensation by providing an oxidant or hydrogen donor. Studies showed that zeolite (ZSM-5) metal is a common catalyst and has two roles (acidic sites and pores), which help to control the reaction and yield more stable and desired products [45]. Another study has proven that the zeolite (ZSM-5), along with a mixture of Si/Al, improved the pyrolysis efficiency and increased the production of aromatic monomers [113,117]. However, the phenolic character of lignin can also deactivate the zeolite [121]. Some other studies have stated that Cu and Ni are better catalysts than zeolite and can selectively cleave the linkages in lignin to generate particular aromatic products [122].
3.5. Microwave-Assisted Depolymerization of Lignin
Microwave-assisted technology is attractive because of its unique heating potential [118]. This is an alternative fast method for degrading samples compared to traditional heating technology. Microwave electromagnetic radiation is applied to degrade the lignin into the value-added products [123]. This method does not require physical contact between the heat source and the materials. It can create a huge amount of heat by rotating the polar molecules and causing ionic conduction [21,124]. Microwave-assisted technology is an economical and quick method for converting biomass and lignin into valuable products [125]. The reaction conditions and promising outcomes of microwave-assisted depolymerization of lignin are summarized in Table 6.
Table 6.
Depolymerization of lignin by microwave irradiation.
Recent studies have demonstrated that microwave-assisted technology has many advantages over traditional technology. It is fast, highly efficient, uniform, selective, and eco-friendly [136]. The heating system for this method is favorable for large-scale industries because of the instantaneous start and stop ability. Research has shown that polar materials preferentially absorb more energy, which helps penetrate the interior part of the raw materials, significantly reduces the processing time, and eventually increases the reaction rate [137]. This technology can add non-thermal effects to the reactions, which causes the reaction to proceed more quickly [138]. Godey et al. have shown that microwave-assisted reactions significantly increased the reaction rates compared to conventional methods [139].
Microwave-assisted technology can improve the reaction selectivity by cleaving the condensed bonds of lignin in the presence of a metal catalyst at lower temperatures. Zhu et al. used this method to depolymerize lignin into phenolic monomers through selective cleavage of the Cα-Cβ linkages [140], using ferric sulfate as the catalyst. Their study showed that the microwave method could selectively break down the Cα-Cβ bonds in phenolic and non-phenolic dimers, where the cleavage rate for the phenolic dimer was significantly higher than for non-phenolic dimers. Furthermore, the metal catalyst significantly decreased the activation energy of the depolymerization reaction and inhibited the formation of char [141].
Microwave-assisted technology could also play an important role in the liquefaction or solvolysis of lignin due to its rapid heating rate and high efficiency. Lignin can easily be dissolved in an IL at a lower temperature and for short periods using this method [142]. Additionally, 1-butyl-3-methylimidazolium hydrogen sulfate (an IL) not only performs as an efficient catalyst for lignin depolymerization, but can also be recycled five times without loss of catalytic activity [143].
3.6. Depolymerization of Lignin Using Sub- and Supercritical Fluids
Developing an alternative green technology with minimal environmental impact is important to produce specific desired products from lignin. Therefore, the reduction of energy consumption, the use of less toxic materials, and the efficient conversion of lignin are crucial requirements [144,145]. Supercritical solvent-based techniques have been considered an efficient, greener technology to depolymerize lignin to successfully prevent condensation reactions [146,147]. Sub- and supercritical fluids are formed under extremely high temperatures and pressures. Supercritical fluids exhibit distinct properties, such as low viscosity, low dielectric constant, and high diffusivity, which penetrate the structure of the lignin and easily solubilize the depolymerized products [148].
Supercritical methods have been used since the 1990s, and during the initial stage of their use, KL and OL were treated with supercritical methanol [73]. Different types of sub- and supercritical fluids, such as water [149], ethanol [150], and carbon dioxide [151], have been used. Condensation is very common in depolymerization, but supercritical depolymerization reactions are highly efficient at preventing condensation. Studies have shown that intermediates formed from the lignin can couple with the hydrogen radicals generated from the supercritical fluids and prevent the condensation reaction [51,60]. The reaction conditions of sub- and supercritical depolymerization of lignin are summarized in Table 7.
Table 7.
Sub- and supercritical depolymerization of lignin.
Wahyudiono et al. successfully conducted supercritical water depolymerization of lignin at 350–400 °C and 25–40 MPa [160]. They showed that the formation of heavier compounds increased with temperature, and the formation of heavier compounds decreased with reaction time. Catechol, m,p-cresol, o-cresol, and phenol were identified as the major products, which indicated that water is an outstanding solvent for converting lignin into monomers. Another study was performed by Numan-Al-Mobin et al. on the depolymerization of lignin with a mixture of water and subcritical CO2 (ScCO2) [151]. Subcritical CO2 is a naturally available compound known for its catalytic properties, sustainability, and non-flammable nature. The combined use of water and subcritical CO2 was an efficient, greener method for synthesizing phenolic monomers.
Depolymerization of lignin has also been performed using sub- and supercritical ethanol, methanol, and combined with water. This study showed that ethanol can significantly enhance the depolymerization of lignin and decrease char formation [150]. This result may be due to the formation of hydrogen radicals from the ethanol, which is coupled with the intermediates from lignin and inhibits the repolymerization reaction intermediates from forming char. In contrast, supercritical methanol has some interesting properties (e.g., hydrogen donation ability, high heat transfer, high dispersity, and improvement of catalytic activity), which favor the depolymerization of lignin [157]. The depolymerization of lignin into phenolic compounds can also be performed by mixing sub- and supercritical ethanol and water [152]. Supercritical ethanol is more effective than methanol at liquefying the woody biomass into crude products. Additionally, supercritical ethanol is expected to dissolve and stabilize the liquid products more easily. The following schematic diagram summarizes different depolymerization methods (Figure 5).
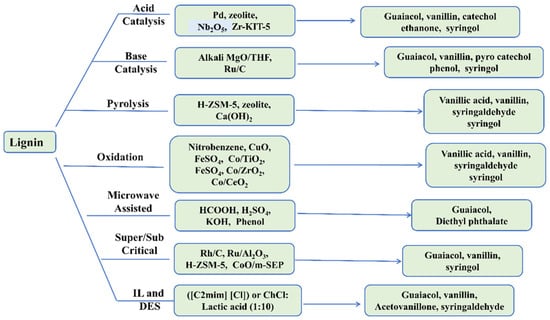
Figure 5.
Schematic diagram of different depolymerization methods.
3.7. Depolymerization of Lignin by Ionic Liquids and Deep Eutectic Solvents
The previously discussed methods are very effective for depolymerizing lignin into aromatic compounds. However, using corrosive chemicals, extreme reaction conditions, and harmful waste production increases the processing cost. Therefore, a sustainable green method is needed to depolymerize lignin into aromatic monomers to reduce production waste, avoid harming the environment, and decrease processing costs. Currently, ILs are considered a green technology for depolymerizing lignin through the cleavage of β-O-4 bonds. ILs show numerous characteristics desirable for a green medium, such as low cost of formulation, non-volatile, chemical inertness, low viscosity, acidity, and excellent miscibility [161,162]. In addition, since they are liquid over a wide range of temperatures and show thermal, chemical, and electrochemical stability, ILs have numerous applications and are suitable environmentally friendly alternative solvents for the depolymerization of lignin [163].
Recent studies have reported that ILs are an excellent solvent for fractionating lignocellulose into its basic components and the solubilization of lignin, since they are long-range, noncovalent, interaction-based liquid formulations. The interactions among their cations and anions help break the bonds of lignin and help them to keep their state in a liquid phase, typically below 100 °C [162,164,165]. ILs can potentially control the degree of oxidative depolymerization and cooperate with different catalysts [166]. Consequently, ILs have gained much attention from the scientific community as a method of depolymerizing lignin into value-added products. Numerous combinations of ionic liquids with metal catalysts, such as Co, Cu, and Mn, have been studied to depolymerize lignin. Stark et al. have converted lignin with combinations of different ionic liquids and the catalyst Mn(NO3)2. They have reported that the combination of Mn(NO3)2 and 1-ethyl-3-methylimidazolium trifluoromethanesulfonate [EMIM][CF3SO3] was the most effective reaction medium for lignin depolymerization. They also found that more than 63% of organosolv beach lignin can be selectively converted into phenolic monomers with 11.5 wt% of 2,6-dimethoxy-1,4-benzoquinone at 100 °C for 24 h [87]. Li et al. converted organosolv bagasse lignin into phenolic monomers using cooperative [bmim][CF3SO3]/[bSmim][HSO4] and water [167]. They reported that the conversion of lignin reached 66.7% with 14.5 wt% of phenolic compounds and negligible char formation at milder reaction conditions (250 °C, 30 min). At 250 °C, water is subcritical, which converted 36.4% of the lignin into phenolic monomers by self-catalytic activity due to the improved dissociation of H+. Most lignin is converted to unwanted char product at this temperature due to the carbocation mechanism [168]. Nevertheless, [bmim][CF3SO3] is an excellent hydrogen bond donor, where the conversion of lignin increased to 45.2% without any char formation by facilitating the cleavage of the hydrogen-bonded linkages [169]. After the addition of an acidic IL [bSmim][HSO4], the conversion of lignin reached 60.1%, and when the concentration of IL [bSmim][HSO4] increased from 2.0 to 3.0 mmol, the conversion reached 66.7%.
ILs can act as acids, bases, and nucleophiles because of their high structural flexibility. Therefore, ILs can mimic the acid- or base-catalyzed depolymerization of lignin without using any corrosive chemicals. The efficient depolymerization of lignin by ILs depends significantly on the cations and anions of the ILs [170]. Recent studies have shown that the anions primarily affect the integrity of the lignin structure, while cations act like spectators [171]. Another study showed that this anionic activity is affected by the strength of the corresponding coordination interaction with the hydrogen in the hydroxyl group of the lignin backbone and the lignin-like structures [172]. Figure 6 shows that the coordination with hydrogen directs the nucleophilic attack toward the carbon double bond by stabilizing the electronic environment of the compounds. Tolesa et al. used an ammonium-based IL to selectively depolymerize the lignin into phenolic monomers. Two different types of ionic liquids, equimolar diisopropylethylamine (DIPEA) and acetic acid (A) or octanoic acid (O), were used as a solvent for the selective depolymerization of lignin. They have shown that ILs not only act as a solvent, but also as a catalyst [173]. Cox et al. investigated how the anion of an acidic IL can stabilize the hydroxyl groups of lignin compounds, resulting in more rapid cleavage of the C–O bonds of lignin [174].
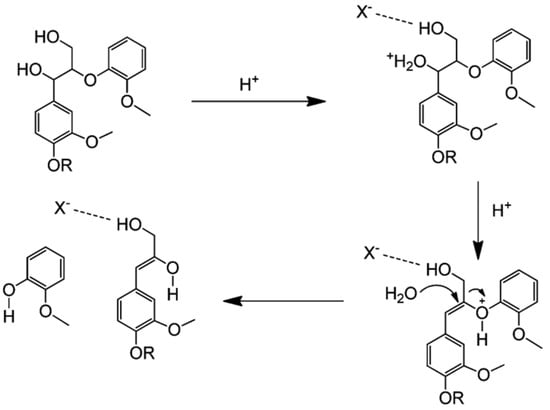
Figure 6.
Schematic reaction mechanism of depolymerization of lignin under acidic conditions showing the effect of IL anions; adopted from [170].
DES-based depolymerization is one of the newest approaches in lignin bond depolymerization. Electrochemical catalysis, microwave-irradiated, and typical heating methods using deep eutectic solvents are the most common methods used so far [133,175,176,177]. The efficacy of these methods compared to the conventional methods has been characterized and monitored using 13C-NMR, 1H-NMR, two-dimensional-heteronuclear single quantum correlation (2D-HSQC) NMR, 31P NMR, cyclic voltammetry (CV), gel permeation chromatography (GPC), scanning electron microscopy (SEM) techniques, and gas chromatography−mass spectrometry (GC−MS) [133,175,176,177]. The following methods and their significant outcomes are tabulated in Table 8.
Table 8.
Ionic liquid and deep eutectic solvent-based depolymerization of lignin.
The most common solvent used to dissolve KL is a NaOH solution. This basic solution is suitable for the electrochemical reaction due to its high conductivity. Typical interlinkages, such as C–C and C–O bonds in lignin, can be cleaved by catalytic properties using an appropriate electrode potential [177]. The major shortcomings of this process are selectivity, low yields at mild conditions, and consecutive oxidations of the products producing unwanted CO2 and organic acids. The high pH also limits the choice of electrode materials to a few metals [177].
After the DES-based depolymerization, lignin recovery is also an important step. Recovery can be made by diluting the reaction mixture with a diluted (0.01 M) H2SO4 solution, followed by the precipitation of insoluble lignin [177]. Then, the solution is further dissolved in 1 M NaOH for size-exclusion chromatography analysis. The soluble part of lignin is extracted from the supernatant using the liquid–liquid extraction (LLE) technique, where ethyl acetate is preferable as an organic solvent because it is greener. The aromatic compounds produced in the depolymerization step can be extracted with methyl isobutyl ketone (MIBK) and the LLE technique for further analysis. Both EA and MIBK phases should be well-mixed for a few hours [177].
In the case of alkali lignin depolymerization, ChCl (choline chloride), more specifically chloride ions, works as a bridge to form hydrogen bonds with the -OH groups of lignin and methanol [23,182]. This mechanism could be responsible for the enhanced dissolution and mass transfer efficiency of the lignin depolymerization reaction [175].
Two-dimension-HSQC NMR spectra analysis indicates that the addition of water can break the interactions among hydrogen bond acceptors (HBAs) and hydrogen bond donors (HBDs) as a deep eutectic solvent (DES) is formulated by mixing HBA and HBD molecules under simple heating and stirring approaches [175]. Thus, the DES system decreases the solubility of lignin, and condensation and depolymerization of the side chains of the alkali lignin can occur simultaneously [175]. If the side chains are longer, they might inhibit acetic acid formation, and higher amounts of guaiacyl moieties might increase the yield of acetovanillone. DES could enhance the catalytic activities of metal-based catalysts by improving their redox potential [175].
The selective cleavage of H-moieties can be achieved using a metal-based DES, such as 1:2 ChCl:FeCl3. This DES can selectively cleave the ester or β-O-4 bonds of the lignin structure, which eventually produces p-coumaric acid (pCA) units [181]. Careful modification of pCA produces desirable methyl p-hydroxycinnamate (MPC). The metal-based DES catalyst is recyclable, easy to prepare, and economical. This method is even effective for corncob lignin and can produce 116.6 mg/g of MPC [181].
The efficacy of depolymerization techniques can be further improved by applying microwave heating, which significantly reduces the reaction time [133]. NMR, GPC, and molecular dynamics simulations show that microwave heating selectively cleaves the target C–C and ether bonds, providing a narrower MW distribution [133]. Microwave irradiation stretches specific lignin bonds, increasing the probability of the bond breaking.
Overall, it has been shown that DES-based lignin depolymerization provides more selectivity, higher yield, uses greener and recyclable chemicals, and requires lower temperatures and less time. Comparative advantages and disadvantages of different greener depolymerization techniques has been summarized in the following table (Table 9).
Table 9.
Comparative advantages and disadvantages of greener depolymerization of lignin.
4. Conclusions and Future Challenges
This review summarizes acid catalyst, base catalyst, oxidative, pyrolysis, microwave, sub- and supercritical fluids, ionic and deep eutectic liquid-based lignin depolymerization, and their advantages and disadvantages, which were compared in terms of green chemistry perspectives. Acid and base catalyst depolymerization are very straightforward but are less selective and require hazardous chemicals and high temperatures and pressures, making them economically and environmentally unfeasible. Pyrolytic methods also require very high temperatures and pressures and special instruments to perform the reaction, making this method very expensive. In contrast, the microwave-assisted method is fast, highly efficient, uniform, selective, cost-effective, and eco-friendly. However, it is only selective for polar organic compounds, making it less versatile and less selective than other methods. The oxidative method is highly selective and promising for lignin depolymerization. The main purpose of this method is to selectively break the ether bonds between the aromatic units to keep them intact. Strong oxidants can also break the aromatic moieties, producing less desirable lower molecular weight compounds, making this method unsuccessful. In contrast, milder oxidants break the ether bond, keeping the aromatic unit intact. As a result, oxidative depolymerization with milder oxidants shows higher selectivity to produce aromatic monomers through the successful decomposition of lignin.
Greener depolymerization methods, such as sub- and supercritical, ionic liquids, and deep eutectic solvent-based depolymerization, show excellent performance and decrease the use of harsh and corrosive chemicals. Although the sub- and supercritical fluids method requires special and costly instruments, it also requires less toxic, recyclable, and easily recoverable solvents, making it a promising method for the depolymerization of lignin from greener perspectives. IL is also considered a greener solvent, but its high viscosity, toxicity, high cost, and difficult formulation make it less promising as a green method. DESs are analogous to IL but easier to formulate, less toxic, and environmentally friendly, making it a more favorable and safer method. DESs can act as both solvent and catalyst and therefore do not require additional chemicals or compounds as a catalyst. Their easily recoverable and recyclable properties make them more economically feasible than other methods.
To further decrease the cost of the depolymerization method requires the minimization of the process conditions and equipment costs. Therefore, several studies have been conducted to improve the efficiency of the greener methods by using fast and available techniques, such as microwave and ultrasound irradiation, and greener solvents, such as ethanol and dimethyl sulfoxide. As ILs and DESs act as both the solvent and catalyst, more studies should be conducted to improve their catalytic activity performance during depolymerization. In addition, those green solvents can be used as co-solvents in acid and base catalyst depolymerization reactions to minimize the processing conditions and use of harsh chemicals. For instance, a study showed that an ionic liquid (e.g., 1-ethyl-3-methylimidazolium chloride) successfully depolymerizes lignin at a comparatively lower temperature (110–150 °C) [183]. Another study showed that the acidic depolymerization of lignin with ethanol occurred quickly and efficiently. Moreover, the microwave-assisted depolymerization of black-liquor lignin was performed at a lower temperature (110–180 °C) and shorter reaction time (5–90 min) [56].
Finally, greener methods are more cost-effective, eco-friendly, and sustainable than traditional methods. Deep eutectic solvent-based depolymerization seems to be a more viable and promising method among the greener methods. Before commercialization, these methods must be studied at a pilot plant scale for consistency and reproducibility.
Author Contributions
R.R., M.S.R., T.A.A., and B.J. conceived this study. R.R. and M.S.R. prepared the framework, drawing figures, and wrote first draft of all sections equally and revised. T.A.A. and B.J. helped by collecting data and editing equally. All authors critically reviewed this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
Authors declare they have no competing interests.
References
- Nashawi, I.S.; Malallah, A.; Al-Bisharah, M. Forecasting world crude oil production using multicyclic Hubbert model. Energy Fuels 2010, 24, 1788–1800. [Google Scholar] [CrossRef]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, P.F.; Hackmann, M.M.; Bos, H.L. Green building blocks for bio-based plastics. Biofuels Bioprod. Biorefining 2014, 8, 306–324. [Google Scholar] [CrossRef]
- Menon, V.; Rao, M. Trends in bioconversion of lignocellulose: Biofuels, platform chemicals & biorefinery concept. Prog. Energy Combust. Sci. 2012, 38, 522–550. [Google Scholar]
- Mood, S.H.; Golfeshan, A.H.; Tabatabaei, M.; Jouzani, G.S.; Najafi, G.H.; Gholami, M.; Ardjmand, M. Lignocellulosic biomass to bioethanol, a comprehensive review with a focus on pretreatment. Renew. Sustain. Energy Rev. 2013, 27, 77–93. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, J.; Erdocia, X.; Sánchez, C.; Alriols, M.G.; Labidi, J. Lignin depolymerization for phenolic monomers production by sustainable processes. J. Energy Chem. 2017, 26, 622–631. [Google Scholar] [CrossRef]
- Gosselink, R.; deJong, E.; Guran, B.; Abacherli, A. Coordination network for lignin—3. standardisation, production and applications adapted to market requirements. Eurolignin Ind. Crops Prod N 2004, 20, 121–129. [Google Scholar] [CrossRef]
- Roy, R.; Jadhav, B.; Rahman, M.S.; Raynie, D.E. Characterization of residue from catalytic hydrothermal depolymerization of lignin. Curr. Res. Green Sustain. Chem. 2021, 4, 100052. [Google Scholar] [CrossRef]
- Roy, R. Characterization of Naturally Derived Polymer by Oxidative, Thermal, and Spectrometric Methods; South Dakota State University: Brookings, SD, USA, 2020. [Google Scholar]
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol–formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef]
- Li, T.; Takkellapati, S. The current and emerging sources of technical lignins and their applications. Biofuels Bioprod. Biorefining 2018, 12, 756–787. [Google Scholar] [CrossRef]
- Ouyang, X.; Zhu, G.; Huang, X.; Qiu, X. Microwave assisted liquefaction of wheat straw alkali lignin for the production of monophenolic compounds. J. Energy Chem. 2015, 24, 72–76. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Romero, R.A.; Redondo, A.; Gnanakaran, S. Theoretical study of the remarkably diverse linkages in lignin. J. Phys. Chem. Lett. 2011, 2, 2660–2666. [Google Scholar] [CrossRef]
- Brittain, A.D.; Chrisandina, N.J.; Cooper, R.E.; Buchanan, M.; Cort, J.R.; Olarte, M.V.; Sievers, C. Quenching of reactive intermediates during mechanochemical depolymerization of lignin. Catal. Today 2018, 302, 180–189. [Google Scholar] [CrossRef]
- Lundquist, K. Low-molecular weight lignin hydrolysis products. In Applied Polymer Symposium; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1976; pp. 1393–1407. [Google Scholar]
- Long, J.; Xu, Y.; Wang, T.; Yuan, Z.; Shu, R.; Zhang, Q.; Ma, L. Efficient base-catalyzed decomposition and in situ hydrogenolysis process for lignin depolymerization and char elimination. Appl. Energy 2015, 141, 70–79. [Google Scholar] [CrossRef]
- Wu, G.; Heitz, M. Catalytic mechanism of Cu2+ and Fe3+ in alkaline O2 oxidation of lignin. J. Wood Chem. Technol. 1995, 15, 189–202. [Google Scholar] [CrossRef]
- Mora-Pale, M.; Meli, L.; Doherty, T.V.; Linhardt, R.J.; Dordick, J.S. Room temperature ionic liquids as emerging solvents for the pretreatment of lignocellulosic biomass. Biotechnol. Bioeng. 2011, 108, 1229–1245. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, H. Lignin pyrolysis reactions. J. Wood Sci. 2017, 63, 117–132. [Google Scholar] [CrossRef]
- Roman, K.; Barwicki, J.; Hryniewicz, M.; Szadkowska, D.; Szadkowski, J. Production of electricity and heat from biomass wastes using a converted aircraft turbine AI-20. Processes 2021, 9, 364. [Google Scholar] [CrossRef]
- Yunpu, W.; Leilei, D.; Liangliang, F.; Shaoqi, S.; Yuhuan, L.; Roger, R. Review of microwave-assisted lignin conversion for renewable fuels and chemicals. J. Anal. Appl. Pyrolysis 2016, 119, 104–113. [Google Scholar] [CrossRef]
- Yan, L.; Ma, R.; Li, L.; Fu, J. Hot water pretreatment of lignocellulosic biomass: An effective and environmentally friendly approach to enhance biofuel production. Chem. Eng. Technol. 2016, 39, 1759–1770. [Google Scholar] [CrossRef]
- Roy, R.; Rahman, M.S.; Raynie, D.E. Recent Advances of Greener Pretreatment Technologies of Lignocellulose. Curr. Res. Green Sustain. Chem. 2020, 3, 100035. [Google Scholar] [CrossRef]
- Gujjula, P.; Kumar, N.; Lynam, J.G. Pretreatment of Loblolly Pine Tree Needles Using Deep Eutectic Solvents. Biomass 2021, 1, 1–10. [Google Scholar] [CrossRef]
- Scelsi, E.; Angelini, A.; Pastore, C. Deep eutectic solvents for the valorisation of lignocellulosic biomasses towards Fine Chemicals. Biomass 2021, 1, 29–59. [Google Scholar] [CrossRef]
- Houasni, A.; Grigorakis, S.; Kellil, A.; Makris, D.P. Organosolv treatment/polyphenol extraction from olive leaves (Olea europaea L.) using glycerol and glycerol-based deep eutectic solvents: Effect on metabolite stability. Biomass 2022, 2, 46–61. [Google Scholar] [CrossRef]
- McCarthy, J.L.; Islam, A. Lignin chemistry, technology, and utilization: A brief history. ACS Symp. Ser. 2000, 742, 2–99. [Google Scholar]
- Charani, P.R.; Dehghani-Firouzabadi, M.; Afra, E.; Blademo, Å.; Naderi, A.; Lindström, T. Production of microfibrillated cellulose from unbleached kraft pulp of Kenaf and Scotch Pine and its effect on the properties of hardwood kraft: Microfibrillated cellulose paper. Cellulose 2013, 20, 2559–2567. [Google Scholar] [CrossRef]
- Phillips, M. The Chemistry of Lignin. Chem. Rev. 1934, 14, 103–170. [Google Scholar] [CrossRef]
- Howard, R.; Abotsi, E.; Van Rensburg, E.J.; Howard, S. Lignocellulose biotechnology: Issues of bioconversion and enzyme production. Afr. J. Biotechnol. 2003, 2, 602–619. [Google Scholar] [CrossRef]
- Demirbaş, A. Relationships between lignin contents and fixed carbon contents of biomass samples. Energy Convers. Manag. 2003, 44, 1481–1486. [Google Scholar] [CrossRef]
- Brudecki, G.; Cybulska, I.; Rosentrater, K.; Julson, J. Optimization of clean fractionation processing as a pre-treatment technology for prairie cordgrass. Bioresour. Technol. 2012, 107, 494–504. [Google Scholar] [CrossRef]
- Huang, F.; Singh, P.M.; Ragauskas, A.J. Characterization of milled wood lignin (MWL) in loblolly pine stem wood, residue, and bark. J. Agric. Food Chem. 2011, 59, 12910–12916. [Google Scholar] [CrossRef] [PubMed]
- Demirbaş, A. Effect of lignin content on aqueous liquefaction products of biomass. Energy Convers. Manag. 2000, 41, 1601–1607. [Google Scholar] [CrossRef]
- Adapa, P.; Tabil, L.; Schoenau, G. Compaction characteristics of barley, canola, oat and wheat straw. Biosyst. Eng. 2009, 104, 335–344. [Google Scholar] [CrossRef]
- Amit, T.A.; Roy, R.; Raynie, D.E. Thermal and structural characterization of two commercially available technical lignins for high-value applications. Curr. Res. Green Sustain. Chem. 2021, 4, 100106. [Google Scholar] [CrossRef]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Kaiser, K.; Benner, R. Characterization of lignin by gas chromatography and mass spectrometry using a simplified CuO oxidation method. Anal. Chem. 2011, 84, 459–464. [Google Scholar] [CrossRef]
- Guo, D.; Chen, F.; Inoue, K.; Blount, J.W.; Dixon, R.A. Downregulation of caffeic acid 3-O-methyltransferase and caffeoyl CoA 3-O-methyltransferase in transgenic alfalfa: Impacts on lignin structure and implications for the biosynthesis of G and S lignin. Plant Cell 2001, 13, 73–88. [Google Scholar] [CrossRef]
- Chen, J.D.; Cui, C.; Li, Y.Q.; Zhou, L.; Ou, Q.D.; Li, C.; Li, Y.; Tang, J.X. Single-junction polymer solar cells exceeding 10% power conversion efficiency. Adv. Mater. 2015, 27, 1035–1041. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Gosselink, R.J.; van Dam, J.E.; de Jong, E.; Scott, E.L.; Sanders, J.P.; Li, J.; Gellerstedt, G. Fractionation, analysis, and PCA modeling of properties of four technical lignins for prediction of their application potential in binders. Holzforschung 2010, 64, 193–200. [Google Scholar] [CrossRef]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Zakzeski, J.; Jongerius, A.L.; Weckhuysen, B.M. Transition metal catalyzed oxidation of Alcell lignin, soda lignin, and lignin model compounds in ionic liquids. Green Chem. 2010, 12, 1225–1236. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Biswas, B.; Saini, K.; Kumar, A.; Kumar, J.; Krishna, B.B.; Bhaskar, T. Effect of hydrogen peroxide on the depolymerization of prot lignin. Ind. Crops Prod. 2020, 150, 112355. [Google Scholar] [CrossRef]
- Pu, Y.; Zhang, D.; Singh, P.M.; Ragauskas, A.J. The new forestry biofuels sector. Biofuels Bioprod. Biorefining Innov. Sustain. Econ. 2008, 2, 58–73. [Google Scholar] [CrossRef]
- Chen, Z.; Wan, C. Biological valorization strategies for converting lignin into fuels and chemicals. Renew. Sustain. Energy Rev. 2017, 73, 610–621. [Google Scholar] [CrossRef]
- Reiter, J.; Strittmatter, H.; Wiemann, L.O.; Schieder, D.; Sieber, V. Enzymatic cleavage of lignin β-O-4 aryl ether bonds via net internal hydrogen transfer. Green Chem. 2013, 15, 1373–1381. [Google Scholar] [CrossRef]
- Brebu, M.; Vasile, C. Thermal degradation of lignin—A review. Cellul. Chem. Technol. 2010, 44, 353. [Google Scholar]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Hydrolytic depolymerization of hydrolysis lignin: Effects of catalysts and solvents. Bioresour. Technol. 2015, 190, 416–419. [Google Scholar] [CrossRef]
- Hewson, W.B.; Hibbert, H. Studies on lignin and related compounds. LXV. Re-ethanolysis of isolated lignins 1. J. Am. Chem. Soc. 1943, 65, 1173–1176. [Google Scholar] [CrossRef]
- Chio, C.; Sain, M.; Qin, W. Lignin utilization: A review of lignin depolymerization from various aspects. Renew. Sustain. Energy Rev. 2019, 107, 232–249. [Google Scholar] [CrossRef]
- Gasson, J.R.; Forchheim, D.; Sutter, T.; Hornung, U.; Kruse, A.; Barth, T. Modeling the lignin degradation kinetics in an ethanol/formic acid solvolysis approach. Part 1. Kinetic model development. Ind. Eng. Chem. Res. 2012, 51, 10595–10606. [Google Scholar] [CrossRef]
- Forchheim, D.; Gasson, J.R.; Hornung, U.; Kruse, A.; Barth, T. Modeling the lignin degradation kinetics in a ethanol/formic acid solvolysis approach. Part 2. validation and transfer to variable conditions. Ind. Eng. Chem. Res. 2012, 51, 15053–15063. [Google Scholar] [CrossRef]
- Dong, C.; Feng, C.; Liu, Q.; Shen, D.; Xiao, R. Mechanism on microwave-assisted acidic solvolysis of black-liquor lignin. Bioresour. Technol. 2014, 162, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, B.; Sharypov, V.; Chesnokov, N.; Beregovtsova, N.; Baryshnikov, S.; Lavrenov, A.; Vosmerikov, A.; Agabekov, V. Lignin conversion in supercritical ethanol in the presence of solid acid catalysts. Kinet. Catal. 2015, 56, 434–441. [Google Scholar] [CrossRef]
- Deepa, A.K.; Dhepe, P.L. Lignin depolymerization into aromatic monomers over solid acid catalysts. ACS Catal. 2015, 5, 365–379. [Google Scholar] [CrossRef]
- Wanmolee, W.; Daorattanachai, P.; Laosiripojana, N. Depolymerization of organosolv lignin to valuable chemicals over homogeneous and heterogeneous acid catalysts. Energy Procedia 2016, 100, 173–177. [Google Scholar] [CrossRef]
- Güvenatam, B.; Heeres, E.H.; Pidko, E.A.; Hensen, E.J. Lewis acid-catalyzed depolymerization of soda lignin in supercritical ethanol/water mixtures. Catal. Today 2016, 269, 9–20. [Google Scholar] [CrossRef]
- Ma, R.; Guo, M.; Lin, K.t.; Hebert, V.R.; Zhang, J.; Wolcott, M.P.; Quintero, M.; Ramasamy, K.K.; Chen, X.; Zhang, X. Peracetic acid depolymerization of biorefinery lignin for production of selective monomeric phenolic compounds. Chem. A Eur. J. 2016, 22, 10884–10891. [Google Scholar] [CrossRef]
- Wang, Q.; Guan, S.; Shen, D. Experimental and kinetic study on lignin depolymerization in water/formic acid system. Int. J. Mol. Sci. 2017, 18, 2082. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Danby, A.M.; Ramanathan, A.; Chaudhari, R.V.; Subramaniam, B. Dual Function Lewis Acid Catalyzed Depolymerization of Industrial Corn Stover Lignin into Stable Monomeric Phenols. ACS Sustain. Chem. Eng. 2018, 7, 1362–1371. [Google Scholar] [CrossRef]
- Du, B.; Liu, B.; Yang, Y.; Wang, X.; Zhou, J. A Phosphotungstic Acid Catalyst for Depolymerization in Bulrush Lignin. Catalysts 2019, 9, 399. [Google Scholar] [CrossRef]
- Asawaworarit, P.; Daorattanachai, P.; Laosiripojana, W.; Sakdaronnarong, C.; Shotipruk, A.; Laosiripojana, N. Catalytic depolymerization of organosolv lignin from bagasse by carbonaceous solid acids derived from hydrothermal of lignocellulosic compounds. Chem. Eng. J. 2019, 356, 461–471. [Google Scholar] [CrossRef]
- Yuan, Z.; Cheng, S.; Leitch, M.; Xu, C.C. Hydrolytic degradation of alkaline lignin in hot-compressed water and ethanol. Bioresour. Technol. 2010, 101, 9308–9313. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, W.G.; Garrett, M.D.; Hardacre, C.; Nieuwenhuyzen, M.; Sheldrake, G.N. An efficient and flexible synthesis of model lignin oligomers. Green Chem. 2013, 15, 3031–3038. [Google Scholar] [CrossRef]
- Thring, R. Alkaline degradation of ALCELL® lignin. Biomass Bioenergy 1994, 7, 125–130. [Google Scholar] [CrossRef]
- Evans, L.; Littlewolf, A.; Lopez, M.; Miller, J. Batch Microreactor Studies of Base Catalyzed Ligin Depolymerization in Alcohol Solvents; Sandia National Laboratories: Albuquerque, NM, USA; Livermore, CA, USA, 1999. [Google Scholar]
- Lavoie, J.-M.; Baré, W.; Bilodeau, M. Depolymerization of steam-treated lignin for the production of green chemicals. Bioresour. Technol. 2011, 102, 4917–4920. [Google Scholar] [CrossRef]
- Roberts, V.M.; Stein, V.; Reiner, T.; Lemonidou, A.; Li, X.; Lercher, J.A. Towards quantitative catalytic lignin depolymerization. Chem. A Eur. J. 2011, 17, 5939–5948. [Google Scholar] [CrossRef]
- Beauchet, R.; Monteil-Rivera, F.; Lavoie, J. Conversion of lignin to aromatic-based chemicals (L-chems) and biofuels (L-fuels). Bioresour. Technol. 2012, 121, 328–334. [Google Scholar] [CrossRef]
- Miller, J.; Evans, L.; Littlewolf, A.; Trudell, D. Batch microreactor studies of lignin and lignin model compound depolymerization by bases in alcohol solvents. Fuel 1999, 78, 1363–1366. [Google Scholar] [CrossRef]
- McFarland, E.W.; Metiu, H. Catalysis by doped oxides. Chem. Rev. 2013, 113, 4391–4427. [Google Scholar] [CrossRef] [PubMed]
- Katahira, R.; Mittal, A.; McKinney, K.; Chen, X.; Tucker, M.P.; Johnson, D.K.; Beckham, G.T. Base-catalyzed depolymerization of biorefinery lignins. ACS Sustain. Chem. Eng. 2016, 4, 1474–1486. [Google Scholar] [CrossRef]
- Hidajat, M.J.; Riaz, A.; Park, J.; Insyani, R.; Verma, D.; Kim, J. Depolymerization of concentrated sulfuric acid hydrolysis lignin to high-yield aromatic monomers in basic sub-and supercritical fluids. Chem. Eng. J. 2017, 317, 9–19. [Google Scholar] [CrossRef]
- Chaudhary, R.; Dhepe, P.L. Solid base catalyzed depolymerization of lignin into low molecular weight products. Green Chem. 2017, 19, 778–788. [Google Scholar] [CrossRef]
- Solt, P.; Rößiger, B.; Konnerth, J.; Van Herwijnen, H.W. Lignin phenol formaldehyde resoles using base-catalysed depolymerized Kraft lignin. Polymers 2018, 10, 1162. [Google Scholar] [CrossRef]
- Chaudhary, R.; Dhepe, P.L. Depolymerization of lignin using a solid base catalyst. Energy Fuels 2019, 33, 4369–4377. [Google Scholar] [CrossRef]
- Paananen, H.; Eronen, E.; Mäkinen, M.; Jänis, J.; Suvanto, M.; Pakkanen, T.T. Base-catalyzed oxidative depolymerization of softwood kraft lignin. Ind. Crops Prod. 2020, 152, 112473. [Google Scholar] [CrossRef]
- Bernhardt, J.J.; Rößiger, B.; Hahn, T.; Pufky-Heinrich, D. Kinetic modeling of the continuous hydrothermal base catalyzed depolymerization of pine wood based kraft lignin in pilot scale. Ind. Crops Prod. 2021, 159, 113119. [Google Scholar] [CrossRef]
- Kudo, S.; Honda, E.; Nishioka, S.; Hayashi, J.-i. Formation of p-Unsubstituted Phenols in Base-catalyzed Lignin Depolymerization. In Proceedings of the MATEC Web of Conferences, 18th Asian Pacific Confederation of Chemical Engineering Congress (APCChE 2019), Sapporo, Japan, 23–27 September 2019. [Google Scholar]
- Knill, C.J.; Kennedy, J.F. Degradation of cellulose under alkaline conditions. Carbohydr. Polym. 2003, 51, 281–300. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. A route for lignin and bio-oil conversion: Dehydroxylation of phenols into arenes by catalytic tandem reactions. Angew. Chem. Int. Ed. 2013, 52, 11499–11503. [Google Scholar] [CrossRef]
- Ferrini, P.; Rinaldi, R. Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions. Angew. Chem. Int. Ed. 2014, 53, 8634–8639. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zhang, Q.; Wang, T.; Zhang, X.; Xu, Y.; Ma, L. An efficient and economical process for lignin depolymerization in biomass-derived solvent tetrahydrofuran. Bioresour. Technol. 2014, 154, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Stärk, K.; Taccardi, N.; Bösmann, A.; Wasserscheid, P. Oxidative depolymerization of lignin in ionic liquids. ChemSusChem 2010, 3, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Kadla, J.F.; Chang, H.-m.; Jameel, H. The reactions of lignin model compounds with hydrogen peroxide at low pH. Holzforschung 2003, 57, 52–88. [Google Scholar] [CrossRef]
- Smith, C.; Utley, J.H.; Petrescu, M.; Viertler, H. Biomass electrochemistry: Anodic oxidation of an organo-solv lignin in the presence of nitroaromatics. J. Appl. Electrochem. 1989, 19, 535–539. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef]
- Ngadi, N.; Halim, N.A.A.; Ibrahim, M.N.M. Isolation and characterization of vanillin from coconut husk lignin via alkaline nitrobenzene oxidation. J. Teknol. 2014, 67, 4. [Google Scholar] [CrossRef][Green Version]
- Ouyang, X.-P.; Tan, Y.-D.; Qiu, X.-Q. Oxidative degradation of lignin for producing monophenolic compounds. J. Fuel Chem. Technol. 2014, 42, 677–682. [Google Scholar] [CrossRef]
- Lyu, G.; Yoo, C.G.; Pan, X. Alkaline oxidative cracking for effective depolymerization of biorefining lignin to mono-aromatic compounds and organic acids with molecular oxygen. Biomass Bioenergy 2018, 108, 7–14. [Google Scholar] [CrossRef]
- Bjelić, S.; Garbuio, L.; Arturi, K.R.; van Bokhoven, J.A.; Jeschke, G.; Vogel, F. Oxidative Biphasic Depolymerization (BPD) of Kraft Lignin at Low pH. ChemistrySelect 2018, 3, 11680–11686. [Google Scholar] [CrossRef]
- Abdelaziz, O.Y.; Meier, S.; Prothmann, J.; Turner, C.; Riisager, A.; Hulteberg, C.P. Oxidative Depolymerisation of Lignosulphonate Lignin into Low-Molecular-Weight Products with Cu–Mn/δ-Al2O3. Top. Catal. 2019, 62, 639–648. [Google Scholar] [CrossRef]
- Kumar, A.; Biswas, B.; Bhaskar, T. Effect of cobalt on titania, ceria and zirconia oxide supported catalysts on the oxidative depolymerization of prot and alkali lignin. Bioresour. Technol. 2020, 299, 122589. [Google Scholar] [CrossRef] [PubMed]
- Irmak, S.; Kang, J.; Wilkins, M. Depolymerization of lignin by wet air oxidation. Bioresour. Technol. Rep. 2020, 9, 100377. [Google Scholar] [CrossRef]
- Crestini, C.; Caponi, M.C.; Argyropoulos, D.S.; Saladino, R. Immobilized methyltrioxo rhenium (MTO)/H2O2 systems for the oxidation of lignin and lignin model compounds. Bioorganic Med. Chem. 2006, 14, 5292–5302. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, I.; Inoue, Y.; Muranaka, Y.; Yasukawa, T.; Mae, K. Selective production of organic acids and depolymerization of lignin by hydrothermal oxidation with diluted hydrogen peroxide. Energy Fuels 2011, 25, 791–796. [Google Scholar] [CrossRef]
- Jennings, J.A.; Parkin, S.; Munson, E.; Delaney, S.P.; Calahan, J.L.; Isaacs, M.; Hong, K.; Crocker, M. Regioselective Baeyer–Villiger oxidation of lignin model compounds with tin beta zeolite catalyst and hydrogen peroxide. RSC Adv. 2017, 7, 25987–25997. [Google Scholar] [CrossRef]
- Zhang, C.; Li, H.; Lu, J.; Zhang, X.; MacArthur, K.E.; Heggen, M.; Wang, F. Promoting lignin depolymerization and restraining the condensation via an oxidation− hydrogenation strategy. ACS Catal. 2017, 7, 3419–3429. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Xu, F.; Wang, B.; Yang, D.; Ming, X.; Jiang, Y.; Hao, J.; Qiao, Y.; Tian, Y. TG-FTIR and Py-GC/MS study on pyrolysis mechanism and products distribution of waste bicycle tire. Energy Convers. Manag. 2018, 175, 288–297. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Understanding the fast pyrolysis of lignin. ChemSusChem 2011, 4, 1629–1636. [Google Scholar] [CrossRef]
- Serio, M.A.; Charpenay, S.; Bassilakis, R.; Solomon, P.R. Measurement and modeling of lignin pyrolysis. Biomass Bioenergy 1994, 7, 107–124. [Google Scholar] [CrossRef]
- Gardner, D.J.; Schultz, T.P.; McGinnis, G.D. The pyrolytic behavior of selected lignin preparations. J. Wood Chem. Technol. 1985, 5, 85–110. [Google Scholar] [CrossRef]
- Faix, O.; Meier, D.; Grobe, I. Studies on isolated lignins and lignins in woody materials by pyrolysis-gas chromatography-mass spectrometry and off-line pyrolysis-gas chromatography with flame ionization detection. J. Anal. Appl. Pyrolysis 1987, 11, 403–416. [Google Scholar] [CrossRef]
- Evans, R.J.; Milne, T.A. Molecular characterization of the pyrolysis of biomass. Energy Fuels 1987, 1, 123–137. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, G.; Yue, J.; Xu, G. Pyrolysis of lignin for phenols with alkaline additive. Fuel Processing Technol. 2014, 124, 212–221. [Google Scholar] [CrossRef]
- Zhou, S.; Brown, R.C.; Bai, X. The use of calcium hydroxide pretreatment to overcome agglomeration of technical lignin during fast pyrolysis. Green Chem. 2015, 17, 4748–4759. [Google Scholar] [CrossRef]
- Lou, R.; Wu, S.; Lyu, G. Quantified monophenols in the bio-oil derived from lignin fast pyrolysis. J. Anal. Appl. Pyrolysis 2015, 111, 27–32. [Google Scholar] [CrossRef]
- Chu, J.; Jiang, W.; Wu, S. Depolymerization characteristics during the pyrolysis of two industrial lignins. BioResources 2017, 12, 7241–7254. [Google Scholar]
- Junior, J.S.; Carvalho, W.; Ataíde, C. Catalytic effect of ZSM-5 zeolite and HY-340 niobic acid on the pyrolysis of industrial kraft lignins. Ind. Crops Prod. 2018, 111, 126–132. [Google Scholar]
- Wu, Z.; Zhu, X.; Guo, H.; Jiang, Y.; Gu, X. A kinetic study of lignin pyrolysis over base catalyst during steam exploded depolymerization. Catal. Today 2019, 327, 226–234. [Google Scholar] [CrossRef]
- Wulandari, Y.R.; Chen, S.S.; Hermosa, G.C.; Hossain, M.S.A.; Yamauchi, Y.; Ahamad, T.; Alshehri, S.M.; Wu, K.C.; Wu, H.-S. Effect of N2 flow rate on kinetic investigation of lignin pyrolysis. Environ. Res. 2020, 190, 109976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Choi, C.; Machida, H.; Huo, Z.; Norinaga, K. Catalytic hydrotreatment of alkaline lignin and its consequent influences on fast pyrolysis. Carbon Resour. Convers. 2021, 4, 219–229. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Catalytic pyrolysis-GC/MS of lignin from several sources. Fuel Processing Technol. 2010, 91, 1446–1458. [Google Scholar] [CrossRef]
- Asmadi, M.; Kawamoto, H.; Saka, S. Gas-and solid/liquid-phase reactions during pyrolysis of softwood and hardwood lignins. J. Anal. Appl. Pyrolysis 2011, 92, 417–425. [Google Scholar] [CrossRef]
- Asmadi, M.; Kawamoto, H.; Saka, S. Thermal reactions of guaiacol and syringol as lignin model aromatic nuclei. J. Anal. Appl. Pyrolysis 2011, 92, 88–98. [Google Scholar] [CrossRef]
- Ledesma, E.B.; Marsh, N.D.; Sandrowitz, A.K.; Wornat, M.J. An experimental study on the thermal decomposition of catechol. Proc. Combust. Inst. 2002, 29, 2299–2306. [Google Scholar] [CrossRef]
- Ma, Z.; van Bokhoven, J.A. Deactivation and regeneration of H-USY zeolite during lignin catalytic fast pyrolysis. ChemCatChem 2012, 4, 2036–2044. [Google Scholar] [CrossRef]
- Ma, Z.; Custodis, V.; van Bokhoven, J.A. Selective deoxygenation of lignin during catalytic fast pyrolysis. Catal. Sci. Technol. 2014, 4, 766–772. [Google Scholar] [CrossRef]
- Beneroso, D.; Monti, T.; Kostas, E.; Robinson, J. Microwave pyrolysis of biomass for bio-oil production: Scalable processing concepts. Chem. Eng. J. 2017, 316, 481–498. [Google Scholar] [CrossRef]
- Lam, S.S.; Mahari, W.A.W.; Jusoh, A.; Chong, C.T.; Lee, C.L.; Chase, H.A. Pyrolysis using microwave absorbents as reaction bed: An improved approach to transform used frying oil into biofuel product with desirable properties. J. Clean. Prod. 2017, 147, 263–272. [Google Scholar] [CrossRef]
- Liew, R.K.; Chai, C.; Yek, P.N.Y.; Phang, X.Y.; Chong, M.Y.; Nam, W.L.; Su, M.H.; Lam, W.H.; Ma, N.L.; Lam, S.S. Innovative production of highly porous carbon for industrial effluent remediation via microwave vacuum pyrolysis plus sodium-potassium hydroxide mixture activation. J. Clean. Prod. 2019, 208, 1436–1445. [Google Scholar] [CrossRef]
- Farag, S.; Fu, D.; Jessop, P.G.; Chaouki, J. Detailed compositional analysis and structural investigation of a bio-oil from microwave pyrolysis of kraft lignin. J. Anal. Appl. Pyrolysis 2014, 109, 249–257. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Pineda, A.; Romero, A.A.; Luque, R.; Labidi, J. Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: Catalyst screening. Appl. Catal. B Environ. 2014, 145, 43–55. [Google Scholar] [CrossRef]
- Fu, D.; Farag, S.; Chaouki, J.; Jessop, P.G. Extraction of phenols from lignin microwave-pyrolysis oil using a switchable hydrophilicity solvent. Bioresour. Technol. 2014, 154, 101–108. [Google Scholar] [CrossRef]
- Shen, D.; Liu, N.; Dong, C.; Xiao, R.; Gu, S. Catalytic solvolysis of lignin with the modified HUSYs in formic acid assisted by microwave heating. Chem. Eng. J. 2015, 270, 641–647. [Google Scholar] [CrossRef]
- Zhu, G.; Qiu, X.; Zhao, Y.; Qian, Y.; Pang, Y.; Ouyang, X. Depolymerization of lignin by microwave-assisted methylation of benzylic alcohols. Bioresour. Technol. 2016, 218, 718–722. [Google Scholar] [CrossRef]
- Liu, Q.; Li, P.; Liu, N.; Shen, D. Lignin depolymerization to aromatic monomers and oligomers in isopropanol assisted by microwave heating. Polym. Degrad. Stab. 2017, 135, 54–60. [Google Scholar] [CrossRef]
- Dai, J.; Styles, G.N.; Patti, A.F.; Saito, K. CuSO4/H2O2-catalyzed lignin depolymerization under the irradiation of microwaves. ACS Omega 2018, 3, 10433–10441. [Google Scholar] [CrossRef]
- Muley, P.D.; Mobley, J.K.; Tong, X.; Novak, B.; Stevens, J.; Moldovan, D.; Shi, J.; Boldor, D. Rapid microwave-assisted biomass delignification and lignin depolymerization in deep eutectic solvents. Energy Convers. Manag. 2019, 196, 1080–1088. [Google Scholar] [CrossRef]
- Liu, X.; Bouxin, F.P.; Fan, J.; Budarin, V.L.; Hu, C.; Clark, J.H. Microwave-assisted catalytic depolymerization of lignin from birch sawdust to produce phenolic monomers utilizing a hydrogen-free strategy. J. Hazard. Mater. 2021, 402, 123490. [Google Scholar] [CrossRef]
- Agarwal, A.; Jo, Y.-T.; Park, J.-H. Hybrid microwave-ultrasound assisted catalyst-free depolymerization of Kraft lignin to bio-oil. Ind. Crops Prod. 2021, 162, 113300. [Google Scholar] [CrossRef]
- Li, M.-F.; Sun, S.-N.; Xu, F.; Sun, R.-C. Microwave-assisted organic acid extraction of lignin from bamboo: Structure and antioxidant activity investigation. Food Chem. 2012, 134, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Zlotorzynski, A. The application of microwave radiation to analytical and environmental chemistry. Crit. Rev. Anal. Chem. 1995, 25, 43–76. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Watheyb, B.; Westmana, J. Microwave assisted organic synthesisÐa review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Gedye, R.N.; Smith, F.E.; Westaway, K.C. The rapid synthesis of organic compounds in microwave ovens. Can. J. Chem. 1988, 66, 17–26. [Google Scholar] [CrossRef]
- Zhu, G.; Jin, D.; Zhao, L.; Ouyang, X.; Chen, C.; Qiu, X. Microwave-assisted selective cleavage of CαCβ bond for lignin depolymerization. Fuel Processing Technol. 2017, 161, 155–161. [Google Scholar] [CrossRef]
- Xu, W.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid. ChemSusChem 2012, 5, 667–675. [Google Scholar] [CrossRef]
- Merino, O.; Fundora-Galano, G.; Luque, R.; Martínez-Palou, R. Understanding microwave-assisted lignin solubilization in protic ionic liquids with multiaromatic imidazolium cations. ACS Sustain. Chem. Eng. 2018, 6, 4122–4129. [Google Scholar] [CrossRef]
- Pan, J.; Fu, J.; Deng, S.; Lu, X. Microwave-assisted degradation of lignin model compounds in imidazolium-based ionic liquids. Energy Fuels 2014, 28, 1380–1386. [Google Scholar] [CrossRef]
- Reverchon, E. Micro and nano-particles produced by supercritical fluid assisted techniques: Present status and perspectives. Chem Eng Trans 2002, 2, 1–10. [Google Scholar]
- Cansell, F.; Aymonier, C.; Loppinet-Serani, A. Review on materials science and supercritical fluids. Curr. Opin. Solid State Mater. Sci. 2003, 7, 331–340. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Park, J.; Hwang, H.; Kim, J.K.; Song, I.K.; Choi, J.W. Catalytic depolymerization of lignin macromolecule to alkylated phenols over various metal catalysts in supercritical tert-butanol. J. Anal. Appl. Pyrolysis 2015, 113, 99–106. [Google Scholar] [CrossRef]
- Gosselink, R.J.; Teunissen, W.; Van Dam, J.E.; De Jong, E.; Gellerstedt, G.; Scott, E.L.; Sanders, J.P. Lignin depolymerisation in supercritical carbon dioxide/acetone/water fluid for the production of aromatic chemicals. Bioresour. Technol. 2012, 106, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Cocero, M.J.; Cabeza, A.; Abad, N.; Adamovic, T.; Vaquerizo, L.; Martinez, C.M.; Pazo-Cepeda, M.V. Understanding biomass fractionation in subcritical & supercritical water. J. Supercrit. Fluids 2018, 133, 550–565. [Google Scholar]
- Yong, T.L.-K.; Yukihiko, M. Kinetic analysis of guaiacol conversion in sub-and supercritical water. Ind. Eng. Chem. Res. 2013, 52, 9048–9059. [Google Scholar] [CrossRef]
- Guo, D.; Liu, B.; Tang, Y.; Zhang, J.; Xia, X.; Tong, S. Catalytic Depolymerization of Alkali Lignin in Sub-and Super-critical Ethanol. BioResources 2017, 12, 5001–5016. [Google Scholar] [CrossRef]
- Numan-Al-Mobin, A.M.; Kolla, P.; Dixon, D.; Smirnova, A. Effect of water–carbon dioxide ratio on the selectivity of phenolic compounds produced from alkali lignin in sub-and supercritical fluid mixtures. Fuel 2016, 185, 26–33. [Google Scholar] [CrossRef]
- Cheng, S.; Wilks, C.; Yuan, Z.; Leitch, M.; Xu, C.C. Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water–ethanol co-solvent. Polym. Degrad. Stab. 2012, 97, 839–848. [Google Scholar] [CrossRef]
- Yong, T.L.-K.; Matsumura, Y. Kinetic analysis of lignin hydrothermal conversion in sub-and supercritical water. Ind. Eng. Chem. Res. 2013, 52, 5626–5639. [Google Scholar] [CrossRef]
- Martin, A.; Patil, P.T.; Armbruster, U. Catalytic Hydroprocessing of Lignin in Supercritical Ethanol. In Proceedings of the III. Iberoamerican Conference Supercritical Fluids Cartagena de Indias, Cartagena de Indias, Colombia, 1–5 April 2013. [Google Scholar]
- Fan, D.; Xie, X.-A.; Li, Y.; Li, L.; Sun, J. Comparative study about catalytic liquefaction of alkali lignin to aromatics by HZSM-5 in sub-and supercritical ethanol. J. Renew. Sustain. Energy 2018, 10, 013106. [Google Scholar] [CrossRef]
- Jadhav, B. Screening of Catalysts for the Subcritical Water Depolymerization of Lignin; South Dakota State University: Brookings, SD, USA, 2020. [Google Scholar]
- Chen, M.; Cao, Y.; Wang, Y.; Yang, Z.; Wang, Q.; Sun, Q.; Wang, J. Depolymerization of lignin over CoO/m-SEP catalyst under supercritical methanol. J. Renew. Sustain. Energy 2019, 11, 013103. [Google Scholar] [CrossRef]
- Yang, T.; Wu, K.; Li, B.; Du, C.; Wang, J.; Li, R. Conversion of lignin into phenolic-rich oil by two-step liquefaction in sub-supercritical ethanol system assisted by carbon dioxide. J. Energy Inst. 2021, 94, 329–336. [Google Scholar] [CrossRef]
- Ciuffi, B.; Loppi, M.; Rizzo, A.M.; Chiaramonti, D.; Rosi, L. Towards a better understanding of the HTL process of lignin-rich feedstock. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Goto, M. Recovery of phenolic compounds through the decomposition of lignin in near and supercritical water. Chem. Eng. Processing Process Intensif. 2008, 47, 1609–1619. [Google Scholar]
- Welton, T. Ionic liquids in green chemistry. Green Chem. 2011, 13, 225. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, J.; Zhang, X.; Xin, J.; Miao, Q.; Wang, J. Ionic liquid-based green processes for energy production. Chem. Soc. Rev. 2014, 43, 7838–7869. [Google Scholar] [CrossRef]
- Marszałł, M.P.; Kaliszan, R. Application of ionic liquids in liquid chromatography. Crit. Rev. Anal. Chem. 2007, 37, 127–140. [Google Scholar] [CrossRef]
- Sun, Y.-C.; Xu, J.-K.; Xu, F.; Sun, R.-C.; Jones, G.L. Dissolution, regeneration and characterisation of formic acid and Alcell lignin in ionic liquid-based systems. Rsc. Adv. 2014, 4, 2743–2755. [Google Scholar] [CrossRef]
- Underkofler, K.A.; Teixeira, R.E.; Pietsch, S.A.; Knapp, K.G.; Raines, R.T. Separation of lignin from corn stover hydrolysate with quantitative recovery of ionic liquid. ACS Sustain. Chem. Eng. 2015, 3, 606–613. [Google Scholar] [CrossRef]
- Wang, H.; Block, L.E.; Rogers, R.D. Catalytic conversion of biomass in ionic liquids. In Catalysis in Ionic Liquids: From Catalyst Synthesis to Application; The Royal Society of Chemistry’s: London, UK, 2014. [Google Scholar]
- Li, Y.; Cai, Z.; Liao, M.; Long, J.; Zhao, W.; Chen, Y.; Li, X. Catalytic depolymerization of organosolv sugarcane bagasse lignin in cooperative ionic liquid pairs. Catal. Today 2017, 298, 168–174. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Jiang, N.; Ragauskas, A.J. Ionic liquid as a green solvent for lignin. J. Wood Chem. Technol. 2007, 27, 23–33. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef]
- George, A.; Tran, K.; Morgan, T.J.; Benke, P.I.; Berrueco, C.; Lorente, E.; Wu, B.C.; Keasling, J.D.; Simmons, B.A.; Holmes, B.M. The effect of ionic liquid cation and anion combinations on the macromolecular structure of lignins. Green Chem. 2011, 13, 3375–3385. [Google Scholar] [CrossRef]
- Cox, B.J.; Jia, S.; Zhang, Z.C.; Ekerdt, J.G. Catalytic degradation of lignin model compounds in acidic imidazolium based ionic liquids: Hammett acidity and anion effects. Polym. Degrad. Stab. 2011, 96, 426–431. [Google Scholar] [CrossRef]
- Tolesa, L.D.; Gupta, B.S.; Lee, M.-J. The chemistry of ammonium-based ionic liquids in depolymerization process of lignin. J. Mol. Liq. 2017, 248, 227–234. [Google Scholar] [CrossRef]
- Casas, A.; Omar, S.; Palomar, J.; Oliet, M.; Alonso, M.V.; Rodriguez, F. Relation between differential solubility of cellulose and lignin in ionic liquids and activity coefficients. RSC Adv. 2013, 3, 3453–3460. [Google Scholar] [CrossRef]
- Yu, Q.; Song, Z.; Chen, X.; Fan, J.; Clark, J.H.; Wang, Z.; Sun, Y.; Yuan, Z. A methanol–choline chloride based deep eutectic solvent enhances the catalytic oxidation of lignin into acetovanillone and acetic acid. Green Chem. 2020, 22, 6415–6423. [Google Scholar] [CrossRef]
- Shen, X.-J.; Chen, T.; Wang, H.-M.; Mei, Q.; Yue, F.; Sun, S.; Wen, J.-L.; Yuan, T.-Q.; Sun, R.-C. Structural and morphological transformations of lignin macromolecules during bio-based deep eutectic solvent (DES) pretreatment. ACS Sustain. Chem. Eng. 2019, 8, 2130–2137. [Google Scholar] [CrossRef]
- Di Marino, D.; Stöckmann, D.; Kriescher, S.; Stiefel, S.; Wessling, M. Electrochemical depolymerisation of lignin in a deep eutectic solvent. Green Chem. 2016, 18, 6021–6028. [Google Scholar] [CrossRef]
- Ogawa, S.; Miyafuji, H. Reaction behavior of milled wood lignin in an ionic liquid, 1-ethyl-3-methylimidazolium chloride. J. Wood Sci. 2015, 61, 285–291. [Google Scholar] [CrossRef]
- De Gregorio, G.F.; Prado, R.; Vriamont, C.; Erdocia, X.; Labidi, J.; Hallett, J.P.; Welton, T. Oxidative depolymerization of lignin using a novel polyoxometalate-protic ionic liquid system. ACS Sustain. Chem. Eng. 2016, 4, 6031–6036. [Google Scholar] [CrossRef]
- Wang, X.; Wang, N.; Nguyen, T.T.; Qian, E.W. Catalytic depolymerization of lignin in ionic liquid using a continuous flow fixed-bed reaction system. Ind. Eng. Chem. Res. 2018, 57, 16995–17002. [Google Scholar] [CrossRef]
- Li, Z.-m.; Long, J.-x.; Zeng, Q.; Wu, Y.-h.; Cao, M.-l.; Liu, S.-j.; Li, X.-h. Production of methyl p-hydroxycinnamate by selective tailoring of herbaceous lignin using metal-based deep eutectic solvents (DES) as catalyst. Ind. Eng. Chem. Res. 2020, 59, 17328–17337. [Google Scholar] [CrossRef]
- Rahman, M.S.; Roy, R.; Jadhav, B.; Hossain, M.N.; Halim, M.A.; Raynie, D.E. Formulation, structure, and applications of therapeutic and amino acid-based deep eutectic solvents: An overview. J. Mol. Liq. 2020, 114745. [Google Scholar] [CrossRef]
- Jia, S.; Cox, B.J.; Guo, X.; Zhang, Z.C.; Ekerdt, J.G. Cleaving the β–O–4 Bonds of Lignin Model Compounds in an Acidic Ionic Liquid, 1-H-3-Methylimidazolium Chloride: An Optional Strategy for the Degradation of Lignin. ChemSusChem 2010, 3, 1078–1084. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).