
The Coming of Age of Biosimilars: A Personal Perspective


Abstract
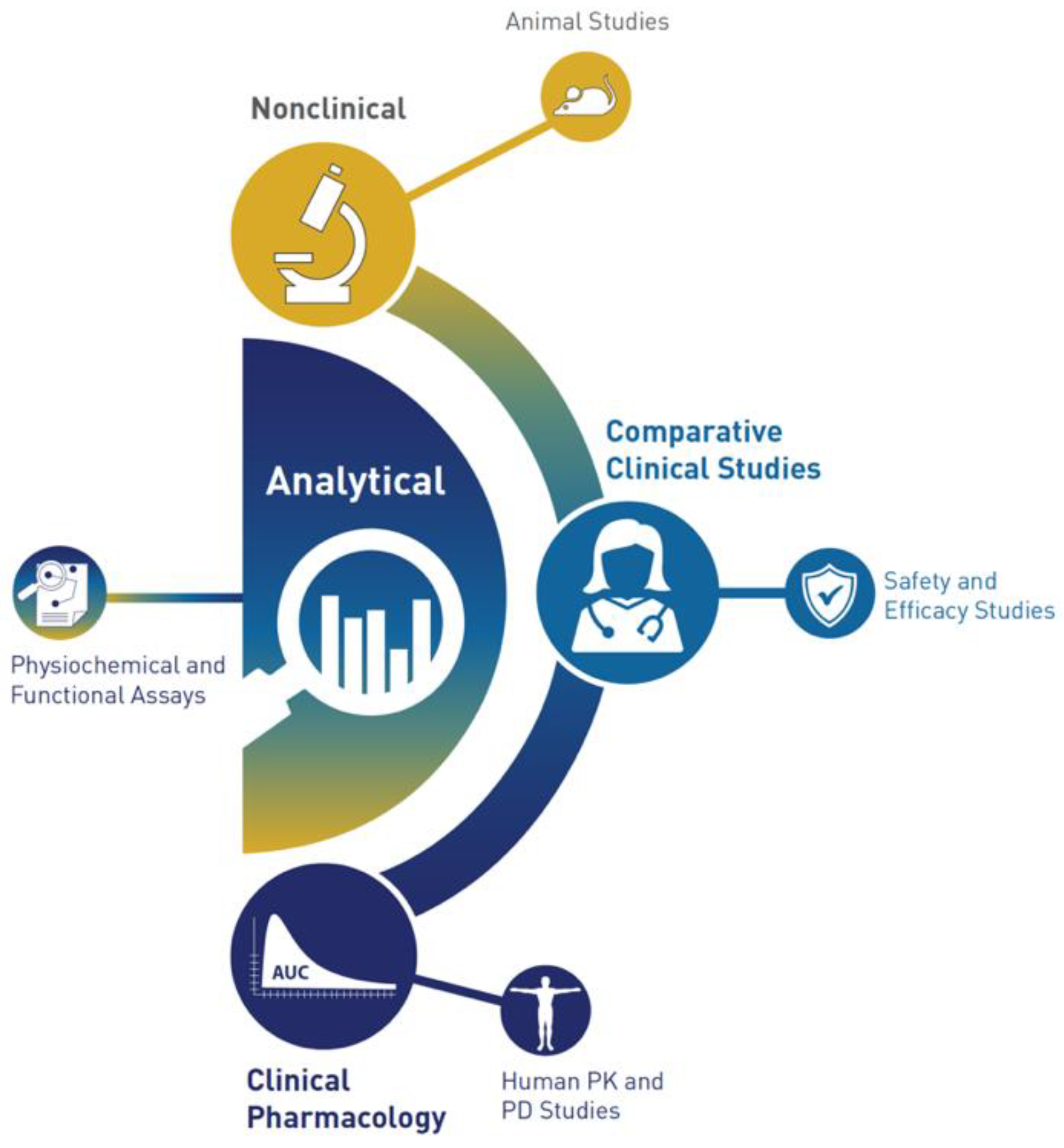
:1. Introduction
2. Target Molecules
3. Patent Litigation
4. Biosimilar Adoption
4.1. The US Scene
- “Anti-competitive practices, such as making false or misleading statements comparing biological reference products and biosimilars, may be slowing progress and hampering the uptake of these important therapies”, quoted from an FDA and Federal Trade Commission (FTC) joint statement [22] made in February 2020.
- The FDA also agreed to “take appropriate steps to address companies making false or misleading communications about biologics, including biosimilars and interchangeable products, which will help deter anti-competitive behavior in the biologics market and lead to the use of all available biological products”, according to the statement. In a news release dated 20 July 2021, the FDA stated that Amgen is making false claims regarding its Neulasta medicine being more effective in its new delivery system Onpro, citing this joint statement [23].
- President Biden signed an executive order titled “Promoting Competition in the American Economy” [24], which directs the Federal Trade Commission to issue rules to prevent “unfair anticompetitive conduct or agreements in the prescription drug industries, such as agreements to delay the market entry of generic drugs or biosimilars”. The order also directs the FDA to address several issues affecting biosimilars, including: (1) “improving and clarifying interchangeability standards for biological products”; (2) “supporting biosimilar product adoption by providing effective educational materials and communications to improve understanding of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”; and (3) “facilitating the development (by sponsors) and approval (acceptance) of biosimilar and interchangeable products among healthcare providers, patients, and caregivers”. Status: enacted.
- A new law, the “Advancing Education on Biosimilars Act” [25], now calls for the government to provide educational materials to healthcare providers, patients, and the general public to increase awareness, knowledge, and confidence in the safety and efficacy of approved biosimilars. Status: enacted.
- The “Star Rating for Biosimilars Act” [26], recently presented, adds a qualification system to Medicare plans. Status: introduced.
- The “Bolstering Innovative Options to Save Immediately on Medicines” (BIOSIM) Act [27] intends to lower biologic drug prices by temporarily increasing reimbursement to ASP plus 8% (from ASP plus 6% previously) for providers that employ a biosimilar that is less expensive than the reference product. Status: introduced.
- The “Preserve Access to Affordable Generics and Biosimilars Act” [28] changes the Federal Trade Commission Act to presumptively render anticompetitive “pay-for-delay” (also known as “reverse-payment”) settlement agreements that prohibit or delay the introduction of generic pharmaceuticals or biosimilars. Status: enacted.
4.2. The European Scene
5. Regulatory Pathway
5.1. The US Scene
- The FDA has created two new guidelines, the extension of the Q&A presentations [34] and the third revised draft guidance [35] titled “New and Revised Draft Q&As on Biosimilar Development and the BPCI Act”. The details refer to fulfilling pediatric assessment or PREA requirements, post-approval filing, and the assertion that the 351(k) cannot have a different route or dosage form. However, the strength issue was delayed, adding new indications and orphan exclusivity. The FDA also updated The Purple Book FAQ section [36].
- FDA has also published new fact sheets [37] to provide additional educational materials on biosimilar and interchangeable products and the biosimilar regulatory review and approval process. The BPCIA states [38] that the “Secretary may determine, in the Secretary’s discretion, that an element described in clause (i) (I) [the biosimilar testing] is unnecessary in an application submitted under this subsection”. The FDA has subtly implemented this change in its new biosimilar guidance [39].
- The BPCIA text [38] states that “an application submitted under this subsection shall include information demonstrating that the biological product is biosimilar to a reference product based upon data derived from analytical studies, animal studies, and clinical studies”. The new education material includes the phrase “in addition to analytical studies, other studies that may be needed”, not shall be, as stated in the BPCIA.
- Animal studies are now described as unnecessary for providing toxicology or pharmacology information about a biosimilar.
- Clinical pharmacology studies show that the proposed biosimilar passes through the body the same way as the reference product and has the same effects. This could include an immunogenicity test to see how the biosimilar affects a patient’s immune system.
- Additional clinical studies can sometimes be conducted after other studies to address any remaining uncertainty about whether the proposed biosimilar has clinically meaningful differences from the reference product.
5.2. The European Scene
6. Analytical Assessment
6.1. Product-Related Attributes
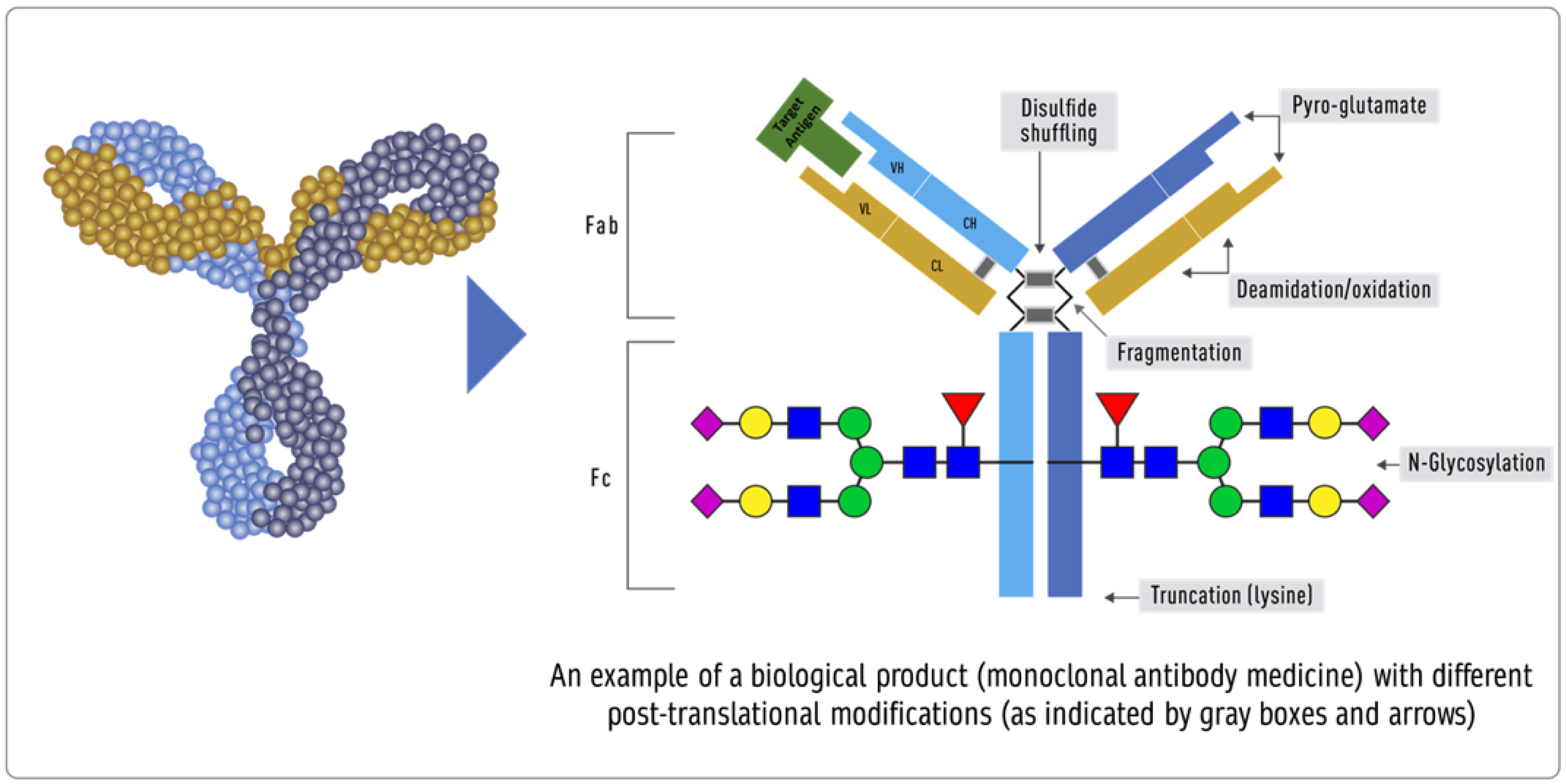
- Peptide mapping (LC-MS), peptide mass fingerprinting (MALDI-MS), MALDI TOF, and MS amino acid sequencing are all examples of primary structure sequencing.
- Higher-order structures can be confirmed using thermodynamic DSC, NMR, SPR, ELISA, fluorescence, far and near UV CD, DSC, NMR, SPR, and ELISA. While process-related testing is straightforward and well-established, testing product-related attributes can be improved by testing the UV and fluorescence spectra under various stress conditions, temperature surfactants, electrolytes, and pH [53]. Newer and more sensitive methods are always needed.
- Cell-based assays, SPR, and ELISA, to test receptor binding.
- Forced degradation: degradation is forced to match intramolecular bond strength as a structural similarity measure.
6.2. Process-Related Attributes
- Protein content. Biological products label potency of 100 IU/mL for insulin in vials. Based on shared experience, the protein content cannot always be the same due to filling variability, concentration testing variability, and many other unpredictable factors. For this reason, most products are allowed an acceptable practical range of variability of ±5% [57]. However, this quality attribute is controversial, as the first FDA guideline required this attribute to be tested for equivalence. The 95% CI of the biosimilar product cannot go beyond 1.5*SD of the reference product in an equivalence test. This range was established entirely arbitrarily. If the SD of the reference product turns out to be small, all batches of the biosimilar product will fail despite being within the release specification of ±5%. This means that a biosimilar product might be acceptable for patients but not for approval by the FDA. This situation arose when the first biosimilar EP2006 required the testing of 50 lots to match the equivalence criteria of Amgen’s Neupogen, despite all lots meeting the release specifications [58]. We can use this as an example to remove the comparative testing of the protein content from side-by-side testing. However, if a biosimilar product has a higher variability, this must be confirmed with the variability in the reference product lots.
- Post-translation modifications, aggregates, and isomers should be tested in a range model, wherein 90% of the values of the biosimilar lots should fall within 3 × SD of the reference product to establish analytical similarity and the specification should include a range of no more than 3 × SD of the reference product.
- Bioassay limits are calculated as specified in the statistical analysis of biological assays and test results. They are typically expressed as an acceptable range for the estimated potency (e.g., 80–125 percent of the stated potency) and an acceptable range for the confidence limits of the estimated potency (e.g., 64–156 percent of the stated potency) [59].
- Impurities in biological products, also known as residuals, are of much greater importance than in chemical drugs. Impurities can be either process- or product-related. Process-related impurities are derived from the manufacturing process—for example, cell culture, downstream, or cell substrates. In contrast, product-related impurities are non-active molecular variants of the biologic and are formed during expression, manufacture, or storage. Understanding these impurities is essential to developing control strategies to reduce or remove them from the final product. The impurities caused by the upstream process may include cell culture reagents, antifoams, growth modifiers (insulin), antibiotics, protein a, solubilizers, residual solvents, chelating agents, extractable extracts, and leachable. The downstream-derived impurities may include detergent, protein a, process additives, chromatographic resins, extractable, and leachable. Cell-derived impurities include host cell DNA and host cell proteins. Product-related impurities include truncated forms such as fragments; modified forms such as disulfide, oxidation, deamidation, and glycosylation; and aggregates including multimers and subvisible particles. When present in a substantial quantity, these impurities may reduce the product’s potency and, worse, induce immunogenic responses or alter the product’s pharmacokinetics. While process-related impurities can be readily isolated, product-related impurities are often difficult to separate because of their close structural similarity to the active molecules. As a result, a biosimilar product must not have any unmatched impurity. There is also no analytical method or biological test that can ensure the safety of an unmatched impurity since any testing of immunogenicity in an animal species may not match the immune response in humans. In some cases, an unmatched impurity may be acceptable if the same regulatory agency has approved an identical structure or there is sufficient published proof of its safety. Since matched impurities can reduce efficacy if they are not as efficacious, a variation of 3% is generally allowed as a legacy attribute. Additionally, the 3% variation must not include more than 1% of any single impurity. However, these acceptance criteria can also be established by profiling the reference product.
- Particle size (subvisible), residual DNA, fill volume, and sterility standards are well defined in several official compendia, and these should be acceptable.
- Physical properties. If the formulation is the same, then the formulation’s physical properties, such as surfactants, osmolality, and pH, should fall within three standard deviations of that of the reference product. However, when the formulation is different, the release specifications will be based on testing multiple lots of biosimilar products. The BPCIA allows a biosimilar product to have a different formulation; however, using the same formulation as the reference product reduces the risk of higher immunogenicity, especially if the inactive component(s) are used in another biological product and have the same route of administration. This is in contrast to the WHO’s suggestion that “relevant differences in formulation (for example, use of excipients in the biosimilar that are not widely used in medicinal products)” can be tested using animal models [60], despite experience gained from the incidence of immunological reactions induced by erythropoietin formulations that used a different formulation [61]. No animal testing can establish the safety of inactive components when used in a biological drug formulation.
7. Nonclinical Pharmacology
8. Clinical Pharmacology
9. Clinical Efficacy and Safety
10. Interchangeability
11. Development Perspective
12. Recommendations
- (a)
- Since 60% of all new drugs are biologics, there will be a long list of eligible biosimilars for the future.
- (b)
- More than 100 biological products have expired patents and expired exclusivity waiting for biosimilar candidacy.
- (c)
- Veterinary biological products are additional choices for biosimilars that have been neglected.
- (d)
- It will take a price drop of 70% or more across all biological products to make biosimilars accessible to all. However, many countries have already reached this stage.
- (e)
- The COGs of all antibodies are between USD 95 and 200 per gram, and they are priced at 100×; despite the price drop, there will still be high profit margins.
- (f)
- The adoption of biosimilars will require taking stakeholders into confidence, particularly prescribers and patients.
- (g)
- Countries where forced switching and alternating are doing just as well despite restrictions.
- (h)
- Global markets will require approval from the EU and US. Both agencies offer fee-free advice. Design studies are acceptable in both the EU and US. US protocols will likely be acceptable to the EMA, but not the other way round.
- (i)
- Regulatory guidelines are neither binding on the agencies nor the developers. Therefore, we need to question them, challenge them, and create a rational development plan that does not originate from the agencies.
- (j)
- Biosimilars and interchangeable product guidelines will undergo substantial revision, reducing the burden of testing and replacing it with advance testing tools.
- (k)
- An analytical assessment is most pivotal to approval; we need to adopt newer technologies and plans, not redundant testing. We can reduce testing by limiting product-related attributes. We can outsource analytical assessments to avoid delays in regulatory approval.
- (l)
- Do not offer to conduct any animal testing; it is not the role of regulatory agencies to tell companies what not to do.
- (m)
- Design creative clinical pharmacology protocols to reduce the size of studies and secure all data from one study.
- (n)
- Do not offer to conduct clinical efficacy testing and challenge the suggestion made by the regulatory agencies to identify the “residual uncertainty”.
- (o)
- If a clinical efficacy test must be conducted, choose an indication where markers are better defined to reduce the study size, such as using psoriasis to test adalimumab.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The US. Omnitrope Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021426s000TOC.cfm (accessed on 23 March 2022).
- Zarxio Drug Approval Package. US. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=125553 (accessed on 23 March 2022).
- FDA Approves Cyltezo, the First Interchangeable Biosimilar to Humira. Second Interchangeable Biosimilar Product Approved by FDA. US. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-cyltezo-first-interchangeable-biosimilar-humira (accessed on 23 March 2022).
- JDSUPRA. How the, U.S. Compares to Europe on Biosimilar Approvals and Products In the Pipeline (Updated). Available online: https://www.jdsupra.com/legalnews/how-the-u-s-compares-to-europe-on-6251301/ (accessed on 23 March 2022).
- IQVIA. The Impact of Biosimilar Competition in Europe December 2021. Available online: https://www.iqvia.com/-/media/iqvia/pdfs/library/white-papers/the-impact-of-biosimilar-competition-in-europe-2021.pdf (accessed on 23 March 2022).
- Kansteiner, F. JP Morgan 2022: Jan 11, 2022. Amgen Dials up Biosimilar Ambitions, with at Least $4B in Expected Sales by 2030. Available online: https://www.fiercepharma.com/pharma/jpm-2022-amgen-aims-to-double-biosimilar-sales-by-2030 (accessed on 23 March 2022).
- Medicare Part B Drug Average Sales Price, Center for Medicare and Medicaid Services. Available online: https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice (accessed on 23 March 2022).
- Chen, Y.; Monnard, A.; da Silva, J.S. An Inflection Point for Biosimilars. McKinsey & Co. 7 June 2021. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/an-inflection-point-for-biosimilars (accessed on 23 March 2022).
- Biosimilars Approved in Europe (Updated 28 January 2022), Generic and Biosimilar Initiative. Available online: https://gabionline.net/biosimilars/general/biosimilars-approved-in-europe (accessed on 23 March 2022).
- Biosimilar Product Information. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 4 April 2022).
- Amgen Biosimilar Trend Report. Amgen Biosimilars. Available online: https://www.amgenbiosimilars.com/-/media/Themes/Amgen/amgenbiosimilars-com/Amgenbiosimilars-com/pdf/USA-CBU-80961_Amgen-Biosimilars-Trend-Report.pdf (accessed on 23 March 2022).
- Generic Enoxaparin Questions and Answers. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/generic-enoxaparin-questions-and-answers (accessed on 23 March 2022).
- FDA Statement: Insulin Gains New Pathway to Increased Competition. Available online: https://www.fda.gov/news-events/press-announcements/insulin-gains-new-pathway-increased-competition (accessed on 23 March 2022).
- IQVIA. Biosimilars to Continue Rapid Growth over the Next Decade. Available online: https://www.iqvia.com/blogs/2021/12/biosimilars-to-continue-rapid-growth-over-the-next-decade (accessed on 23 March 2022).
- Drug Patent Watch. Available online: https://www.drugpatentwatch.com (accessed on 23 March 2022).
- Biologics Market Dynamics: Setting the Stage for Biosimilars. Available online: https://www.ftc.gov/system/files/documents/public_events/1568297/aitken_-_biologics_market_dynamics_setting_the_stage_for_biosimilars_slides.pdf (accessed on 23 March 2022).
- Biologics Market--Growth, Trends, COVID-19 Impact, and Forecasts. (2022–2027). Mordor Intelligence. Available online: https://www.mordorintelligence.com/industry-reports/biologics-market#:~:text=The%20biologics%20market%20was%20valued,forecast%20period%2C%202021%2D2026 (accessed on 23 March 2022).
- BPCIA Litigation. Big Molecule Watch. Available online: https://www.bigmoleculewatch.com/bpcia-patent-litigations/ (accessed on 23 March 2022).
- Kracov, D.; Marsh, D.; Tabas, M.; Ho, T. FDA Seeks to Deepen Engagement with USPTO. 27 September 2021. Available online: https://www.arnoldporter.com/en/perspectives/publications/2021/09/fda-seeks-to-deepen-engagement-with-uspto (accessed on 23 March 2022).
- Biosimilars Year in Review 2021, Fish & Richardson. Available online: https://www.fr.com/biosimilars-2021-year-in-review/ (accessed on 23 March 2022).
- Association for Accessible Medicines. 2021 U.S. Generic and Biosimilar Medicines Savings Report. Retrieved from Association for Accessible Medicines: Generics & Biosimilars. October 2021. Available online: https://accessiblemeds.org/sites/default/files/2021-10/AAM-2021-US-Generic-Biosimilar-Medicines-Savings-Report-web.pdf (accessed on 4 April 2022).
- FDA Press Announcement: FDA and FTC Announce New Efforts to Further Deter Anticompetitive Business Practices. Available online: https://www.fda.gov/news-events/press-announcements/fda-and-ftc-announce-new-efforts-further-deter-anti-competitive-business-practices-support (accessed on 23 March 2022).
- FDA Notifies Amgen of Branding its Product Neulasta. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-notifies-amgen-misbranding-its-biological-product-neulasta-due-false-or-misleading-promotional (accessed on 4 April 2022).
- Presidential Order on Promoting Competition in the American Economy. Available online: https://www.whitehouse.gov/briefing-room/presidential-actions/2021/07/09/executive-order-on-promoting-competition-in-the-american-economy/ (accessed on 23 March 2022).
- US Senate: Advancing Education on Biosimilars Act. Available online: https://www.cassidy.senate.gov/imo/media/doc/AdvancingEducationonBiosimilarsAct.pdf (accessed on 23 March 2022).
- US Congress. Star Rating for Biosimilars Act. Available online: https://www.congress.gov/bill/117th-congress/house-bill/2855?q=%7B%22search%22%3A%5B%22Star+Rating+for+Biosimilars+Act%22%2C%22Star%22%2C%22Rating%22%2C%22for%22%2C%22Biosimilars%22%2C%22Act%22%5D%7D&s=1&r=1 (accessed on 4 April 2022).
- US Congress. H.R.2815—BIOSIM Act. Available online: https://www.congress.gov/bill/117th-congress/house-bill/2815/text?r=1&s=1 (accessed on 23 March 2022).
- US Congress. Preserve Access to Af3 Fordable Generics and Biosimilars Act. Available online: https://www.congress.gov/117/bills/s1428/BILLS-117s1428rs.pdf (accessed on 23 March 2022).
- Rémuzat, C.; Kapuśniak, A.; Caban, A.; Ionescu, D.; Radière, G.; Mendoza, C.; Toumi, M. Supply-side and demand-side policies for biosimilars: An overview in 10 European member states. J. Mark. Access Health Policy 2017, 5, 1307315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorkens, E.; Vulto, A.G.; Huys, I.; Dylst, P.; Godman, B.; Keuerleber, S.; Claus, B.; Dimitrova, M.; Petrova, G.; Sović-Brkičić, L.; et al. Policies for biosimilar uptake in Europe: An overview. PLoS ONE 2017, 12, e0190147. [Google Scholar]
- Love, B. US Plays catch-up with Europe over Biosimilar Patents, Financial Times. 16 June 2021. Available online: https://www.ft.com/content/3f7ca3f4-8256-4570-a6a3-b255e185f162 (accessed on 23 March 2022).
- Moorkens, E.; Vulto, A.G.; Huys, I. An overview of patents on therapeutic monoclonal antibodies in Europe: Are they a hurdle to biosimilar market entry? MAbs 2020, 12, 1743517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA-TRACK: Center for Drug Evaluation & Research—Pre-Approval Safety Review—Biosimilars Dashboard. Available online: https://www.fda.gov/about-fda/fda-track-agency-wide-program-performance/fda-track-center-drug-evaluation-research-pre-approval-safety-review-biosimilars-dashboard (accessed on 23 March 2022).
- Questions and Answers on Biosimilar Development and the BPCI Act Guidance for Industry. September 2021. Available online: https://www.fda.gov/media/119258/download (accessed on 23 March 2022).
- New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 3) Guidance for Industry. September 2021. Available online: https://www.fda.gov/media/119278/download (accessed on 23 March 2022).
- FDA. Purple Book. Available online: https://purplebooksearch.fda.gov/faqs#5 (accessed on 4 April 2022).
- FDA. Healthcare Providers Materials. Available online: https://www.fda.gov/drugs/biosimilars/health-care-provider-materials?utm_medium=email&utm_source=govdelivery#fact (accessed on 23 March 2022).
- US Congress. Title VII—Improving Access to Innovative Medical Therapies Subtitle A—Biologics Price Competition and Innovation. Available online: https://www.fda.gov/media/78946/download (accessed on 23 March 2022).
- FDA. Biological Regulatory Review and Approval. Available online: https://www.fda.gov/media/151061/download (accessed on 4 April 2022).
- FDA. Level 1 Biosimilar and Interchangeable Products Foundational Concepts. Available online: https://www.fda.gov/drugs/biosimilars/curriculum-materials-health-care-degree-programs-biosimilars (accessed on 23 March 2022).
- EMA. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use Official Journal L–311, 28/11/2004, pp. 67–128. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/directive-2001/83/ec-european-parliament-council-6-november-2001-community-code-relating-medicinal-products-human-use_en.pdf (accessed on 23 March 2022).
- European Medicines Agency. Human Regulatory. Biosimilars. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar#-product-specific-biosimilar-guidelines-section (accessed on 23 March 2022).
- Wolff-Holz, E.; Tiitso, K.; Vleminckx, C.; Weise, M. Evolution of the EU Biosimilar Framework: Past and Future. BioDrugs 2019, 33, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. EPAR Biosimilars. Available online: https://www.ema.europa.eu/en/search/search/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar?search_api_views_fulltext=biosimilar%20monographs (accessed on 23 March 2022).
- FDA. Freedom of Information Act. Available online: https://www.fda.gov/regulatory-information/freedom-information (accessed on 23 March 2022).
- FDA. Guideline on Statistical Approaches to Evaluate Analytical Similarity Guidance for Industry. 2017. Available online: https://www.pbwt.com/content/uploads/2018/06/UCM576786.pdf (accessed on 23 March 2022).
- FDA Withdraws Draft Guidance for Industry: Statistical Approaches to Evaluate Analytical Similarity. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-withdraws-draft-guidance-industry-statistical-approaches-evaluate-analytical-similarity (accessed on 23 March 2022).
- FDA. Development of Therapeutic Protein Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-quality (accessed on 23 March 2022).
- Forbes Magazine. One Man’s Mission to Fix the FDA’s Biosimilar Problem. Available online: https://www.forbes.com/sites/nicolefisher/2018/07/25/one-mans-mission-to-fix-the-fdas-biosimilar-problem/?sh=1843e1723808 (accessed on 23 March 2022).
- Niazi, S. Analysis of FDA-Licensed Biosimilars: Time for a Paradigm Shift. AJMC, Center for Biosimilars. Available online: https://www.centerforbiosimilars.com/view/analysis-of-fda-licensed-biosimilars-time-for-a-paradigm-shift (accessed on 23 March 2022).
- European Medicines Agency Biotechnology Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf (accessed on 23 March 2022).
- Vandekerckhove, K.; Seidl, A.; Gutka, H.; Kumar, M.; Gratzl, G.; Keire, D.; Coffey, T.; Kuehne, H. Rational Selection, Criticality Assessment, and Tiering of Quality Attributes and Test Methods for Analytical Similarity Evaluation of Biosimilars. AAPS J. 2018, 20, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niazi, S. Methods for Comparing a Structure of a First Biomolecule and a Second Biomolecule. U.S. Patent 20,140,356,968, 4 December 2014. Available online: https://tinyurl.com/h58tdjnr (accessed on 23 March 2022).
- European Directorate for the Quality of Medicines & HealthCare. Biotherapeutics Monographs. Available online: https://www.edqm.eu/sites/default/files/medias/bio_tab_portfolio_january_2022.pdf (accessed on 4 April 2022).
- USP. Statement on Monographs for Biologics. US Pharmacopoeia. Available online: https://www.usp.org/news/statement-on-monographs-for-biologics (accessed on 23 March 2022).
- FDA-USP Clash over Biologics Monographs. Available online: https://www.raps.org/news-and-articles/news-articles/2019/6/fda-usp-clash-over-biologics-monographs (accessed on 4 April 2022).
- Goyal, P.; Pai, H.V.; Kodali, P.; Vats, B.; Vajpai, N.; Annegowda, S.; Mane, K.; Mohan, S.; Saxena, S.; Veerabhadraia, A.B.; et al. Physicochemical and functional characterization of MYL-1501D, a proposed biosimilar to insulin glargine. PLoS ONE 2021, 16, e0253168. [Google Scholar] [CrossRef] [PubMed]
- FDA. Oncology Briefing Document. 15 January 2015. Available online: https://patentdocs.typepad.com/files/briefing-document.pdf (accessed on 23 March 2022).
- European Directorate for the Quality of Medicines & HealthCare. Synthetic Peptides and rDNA Products. Available online: https://www.edqm.eu/sites/default/files/guide_ph_eur_synthetic_peptides_and_rdna_proteins_2018.pdf (accessed on 23 March 2022).
- World Health Organization. WHO Guidelines on Evaluation of Biosimilars. Available online: https://cdn.who.int/media/docs/default-source/biologicals/who-guidelines-on-evaluation-of-biosimilars---4-nov-2021.pdf?sfvrsn=f17799ae_5 (accessed on 23 March 2022).
- McKoy, J.M.; Stonecash, R.E.; Cournoyer, D.; Rossert, J.; Nissenson, A.R.; Raisch, D.W.; Casadevall, N.; Bennett, C.L. Epoetin-associated pure red cell aplasia: Past, present, and future considerations. Transfusion 2008, 48, 1754–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajar, R. Animal testing and medicine. Heart Views. 2011, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S. Debate over Animal Toxicology Studies. AJMC. Center for Biosimilars. Available online: https://www.centerforbiosimilars.com/view/opinion-the-debate-over-animal-toxicology-studies (accessed on 23 March 2022).
- FDA. Drug Approval Package: Mvasi (bevacizumab-awwb). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761028Orig1s000TOC.cfm (accessed on 23 March 2022).
- FDA. Drug approval package: Trazimera (trastuzumab-qyyp). Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=761081 (accessed on 4 April 2022).
- FDA. Drug Approval Package: Herzuma (tratuzumabn-pkrb). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761091Orig1s000TOC.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package: Renflexis (Infliximab-abda). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761054Orig1s000TOC.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package Inflectra (infliximab-dyyb) for injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/125544Orig1s000TOC.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package: Eticovo. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761066Orig1s000TOC.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package: Erelzi (etanercept-szzs). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761042Orig1_toc.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package: Retacrit (epoetin alfa-epbx). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/125545Orig1s000TOC.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package: Udenyca. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761039Orig1s000TOC.cfm (accessed on 4 April 2022).
- FDA. Drug Approval Package: Fulphila. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761075Orig1s000TOC.cfm (accessed on 23 March 2022).
- Pipalava, P.; Patel, R.; Mehta, M.; Dahiya, M.; Singh, I.; Jose, V. An update on the animal studies conducted for biosimilar approvals—Regulatory requirement vs. actual scenario. Regul. Toxicol. Pharmacol. 2019, 107, 104415. [Google Scholar] [CrossRef] [PubMed]
- CDSCI India. Guidelines on Similar Biologics. Available online: https://birac.nic.in/webcontent/Guidelines_on_Similar_Biologics_06_10_2017.pdf (accessed on 23 March 2022).
- van Aerts, L.A.; De Smet, K.; Reichmann, G.; van der Laan, J.W.; Schneider, C.K. Biosimilars entering the clinic without animal studies, a paradigm shift in the European Union. MAbs 2014, 6, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Bailey, J. Chapter 19: Genetic Modification of Animals: Scientific and Ethical Issues. In Animal Experimentation: Working towards a Paradigm Change; Brill: Leiden, The Netherlands, 2019; pp. 443–479. ISBN 9789004391192. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. EMA Implements New Measures to Minimise Testing During Medicines Development. Available online: https://www.ema.europa.eu/en/news/ema-implements-new-measures-minimise-animal-testing-during-medicines-development (accessed on 23 March 2022).
- FDA. Advancing Alternate Methods at FDA. Available online: https://www.fda.gov/science-research/about-science-research-fda/advancing-alternative-methods-fda (accessed on 23 March 2022).
- European Medicines Agency. Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes Text with EEA Relevance. Directive 2010/63/EU. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32010L0063 (accessed on 23 March 2022).
- European Medicines Agency. Regulatory Science Strategy. Available online: https://www.ema.europa.eu/en/about-us/how-we-work/regulatory-science-strategy#regulatory-science-strategy-to-2025-section (accessed on 23 March 2022).
- Schiestl, M.; Ranganna, G.; Watson, K.; Jung, B.; Roth, K.; Capsius, B.; Trieb, M.; Bias, P.; Maréchal-Jamil, J. The Path Towards a Tailored Clinical Biosimilar Development. BioDrugs 2020, 34, 297–306. [Google Scholar] [CrossRef] [Green Version]
- FDA. Evaluating Inclusion and Exclusion Criteria in Clinical Trials. Available online: https://www.fda.gov/media/134754/download (accessed on 23 March 2022).
- Niazi, S. Testimony to the US. Available online: https://www.regulations.gov/document/FDA-2019-P-1236-0003 (accessed on 4 April 2022).
- FDA. Immunogenicity of Protein-Based Therapeutics. June 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/immunogenicity-protein-based-therapeutics (accessed on 23 March 2022).
- FDA. Biosimilars Action Plan. Available online: https://www.fda.gov/media/114574/download (accessed on 4 April 2022).
- Keown, A. FDA Allows Waiver of Clinical Trials for Insulin Biosimilars as Recommended in Niazi Citizen Petition. BioSpace. Published 3 December 2019. Available online: https://www.biospace.com/article/releases/fda-allows-waiver-of-clinical-trials-for-insulin-biosimilars-as-recommended-in-niazi-citizen-petition/ (accessed on 23 March 2022).
- FDA. Clinical Immunogenicity Considerations for Biosimilars and Interchangeable Insulin Products. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-immunogenicity-considerations-biosimilar-and-interchangeable-insulin-products (accessed on 23 March 2022).
- FDA. Guidance: Clinical Pharmacology Data to Demonstrate Biosimilarity to a Reference Product. 2016. Available online: https://www.fda.gov/media/88622/download (accessed on 23 March 2022).
- European Medicines Agency. Tailored Scientific Advice for Biosimilars Development. Available online: https://www.ema.europa.eu/en/documents/report/tailored-scientific-advice-biosimilar-development-report-experience-pilot-2017-2020_en.pdf (accessed on 23 March 2022).
- Moore, T.J.; Mouslim, M.C.; Blunt, J.L.; Alexander, G.C.; Shermock, K.M. Assessment of Availability, Clinical Testing, and US Review of Biosimilar Biologic Products. JAMA Intern. Med. 2021, 181, 52–60. [Google Scholar] [CrossRef]
- Biosimilars Clinical Testing Registered. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=biosimilar&cntry=&state=&city=&dist= (accessed on 23 March 2022).
- Brennan, J.; Regulatory Affairs Professionals Society. FDA’s Woodcock Says the Clinical Trial System Is Broken. Available online: https://www.raps.org/regulatory-focus%E2%84%A2/news-articles/2017/9/fda-s-woodcock-the-clinical-trials-system-is-broken (accessed on 23 March 2022).
- FDA. 21st Century Cures Act. Available online: https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act (accessed on 23 March 2022).
- Li, J.; Florian, J.; Campbell, E.; Schrieber, S.J.; Bai, J.P.; Weaver, J.L.; Hyland, P.L.; Thway, T.M.; Matta, M.K.; Lankapalli, R.H.; et al. Advancing Biosimilar Development Using Pharmacodynamic Biomarkers in Clinical Pharmacology Studies. Clin. Pharmacol. Ther. 2020, 107, 40–42. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Guidance: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. 2015. Available online: https://www.fda.gov/media/82647/download (accessed on 23 March 2022).
- Li, L.; Ma, L.; Schrieber, S.J.; Rahman, N.A.; Deisseroth, A.; Farrell, A.T.; Wang, Y.; Sinha, V.; Marathe, A. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G-CSF in breast cancer patients. Clin. Pharmacol. Ther. 2018, 104, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Mbuaki, A.; Simoens, S.; Declerck, P.; Vulto, A.G.; Huys, I. Regulatory Information and Guidance on Biosimilars and Their Use Across Europe: A Call for Strengthened One Voice Messaging. Available online: https://www.frontiersin.org/articles/10.3389/fmed.2022.820755/full (accessed on 23 March 2022).
- Glintborg, B.; Loft, A.G.; Omerovic, E.; Hendricks, O.; Linauskas, A.; Espesen, J.; Danebod, K.; Jensen, D.V.; Nordin, H.; Dalgaard, E.B.; et al. To switch or not to switch: Results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis. 2019, 78, 192–200. [Google Scholar] [CrossRef] [PubMed]
- European Pharmaceutical Review. Top 10 Drugs by Annual Revenue in 2025. Available online: https://www.europeanpharmaceuticalreview.com/article/102539/top-10-drugs-by-annual-revenue-in-2025/ (accessed on 4 April 2022).
- WHO. Call for Consultant on Monoclonal Antibodies for Infectious Diseases. Available online: https://www.who.int/news-room/articles-detail/call-for-consultant-on-monoclonal-antibodies-for-infectious-diseases (accessed on 23 March 2022).
- Hendrikx, J.J.M.A.; Haanen, J.B.A.G.; Voest, E.E.; Schellens, J.H.M.; Huitema, A.D.R.; Beijnen, J.H. Fixed Dosing of Monoclonal Antibodies in Oncology. Oncologist 2017, 22, 1212–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price of Rituxan. Available online: https://www.webmd.com/rx/drug-prices/rituxan (accessed on 23 March 2022).
- FDA. Biosimilar User Fee Act (BsUFA) Research Grant (U01) Clinical Trials Optional. Available online: https://grants.nih.gov/grants/guide/rfa-files/RFA-FD-22-026.html (accessed on 13 April 2022).
- Niazi, S.K. Biosimilars: A futuristic fast-to-market advice to developers. Expert Opin. Biol. Ther. 2022, 22, 149–155. Available online: https://www.tandfonline.com/doi/citedby/10.1080/14712598.2022.2020241?scroll=top&needAccess=true (accessed on 13 April 2022). [CrossRef] [PubMed]
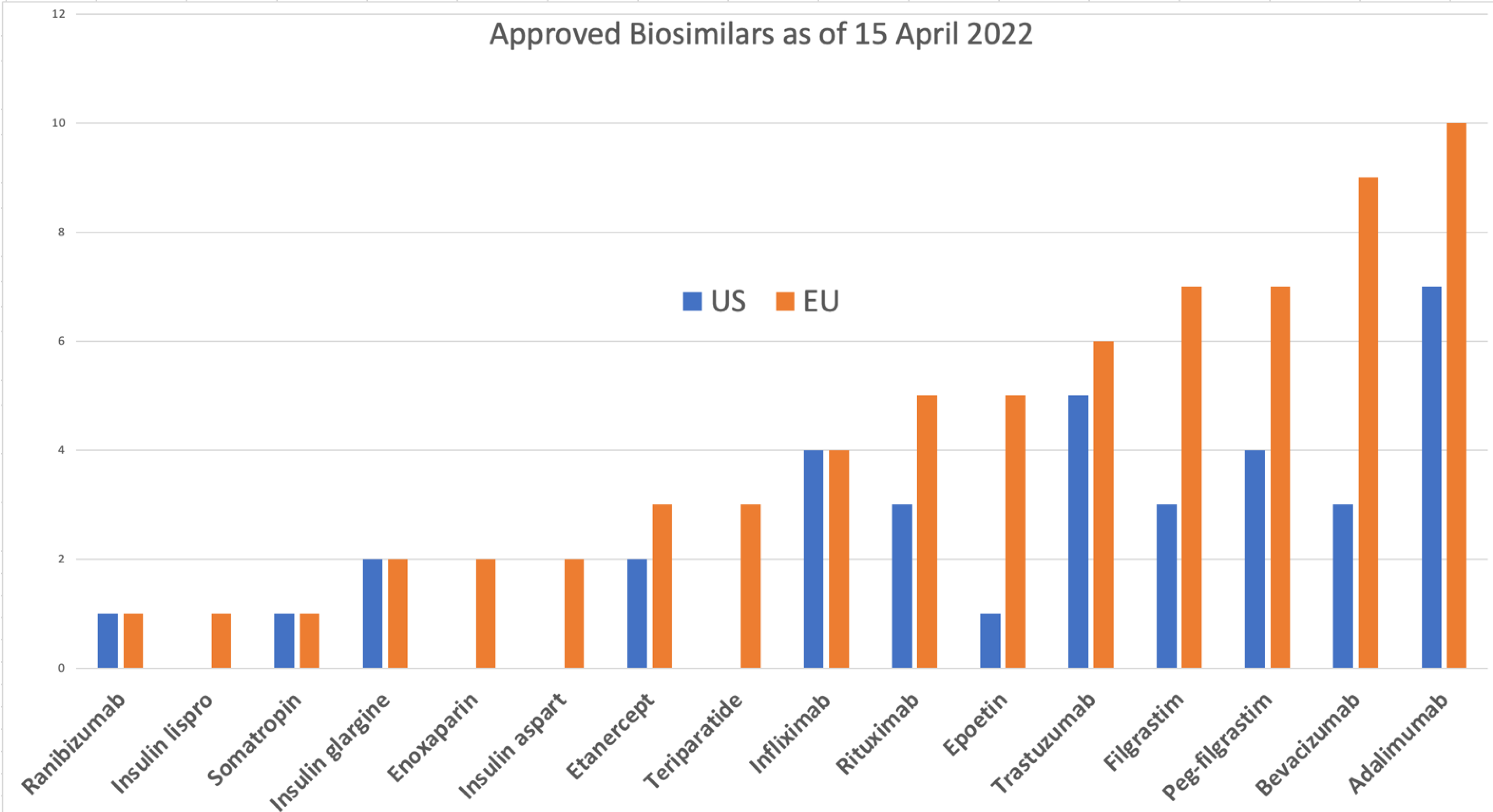
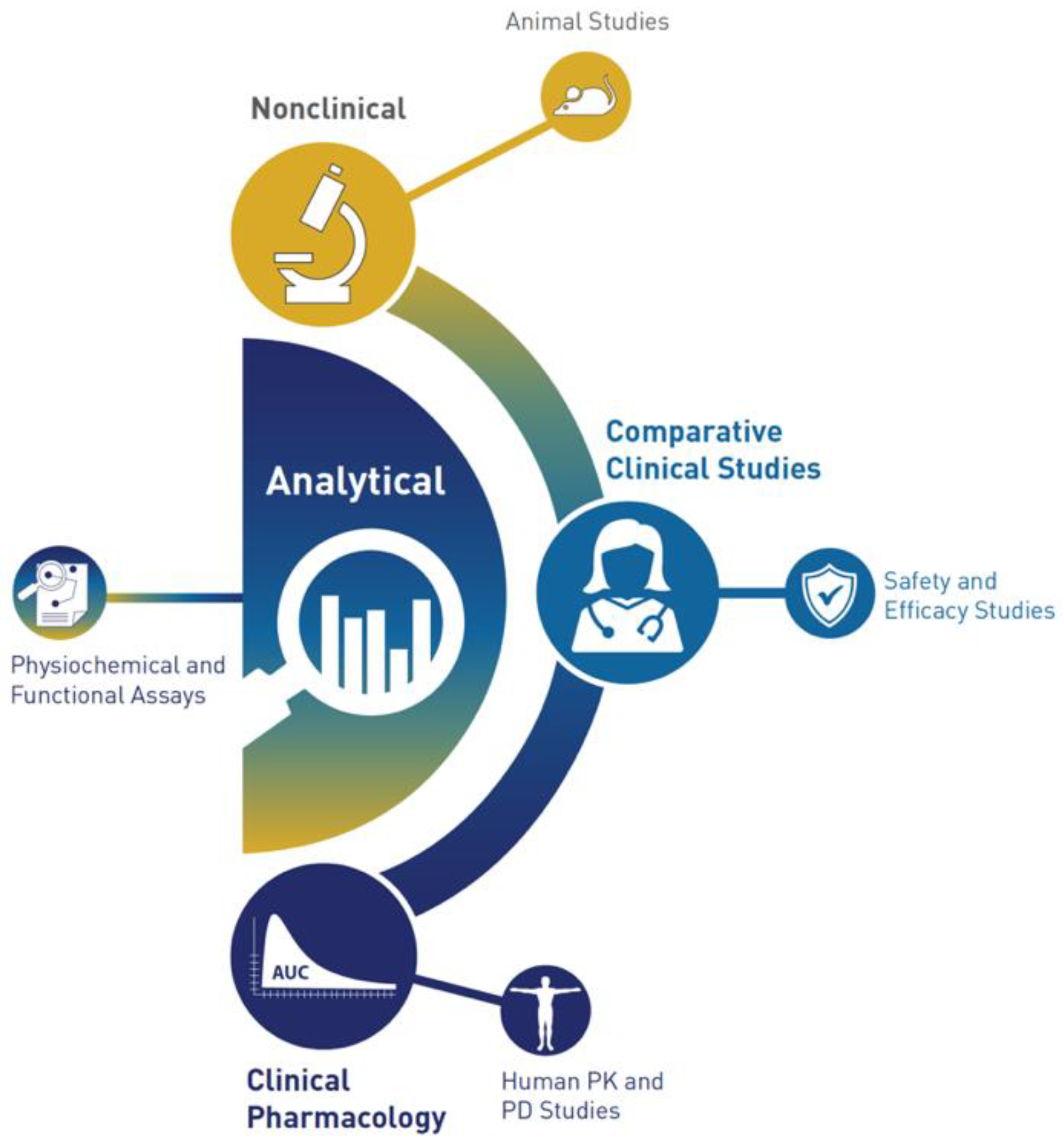
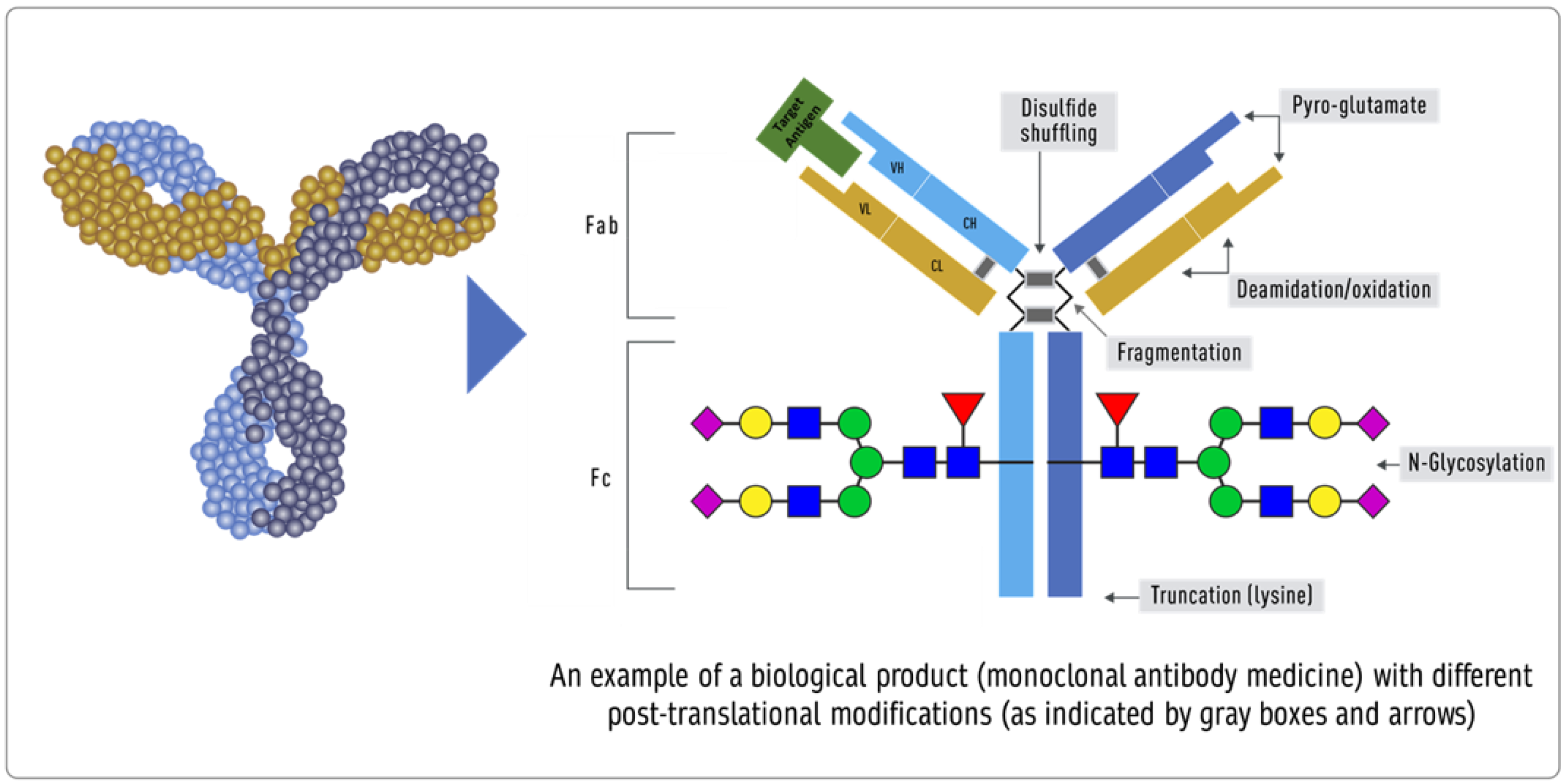

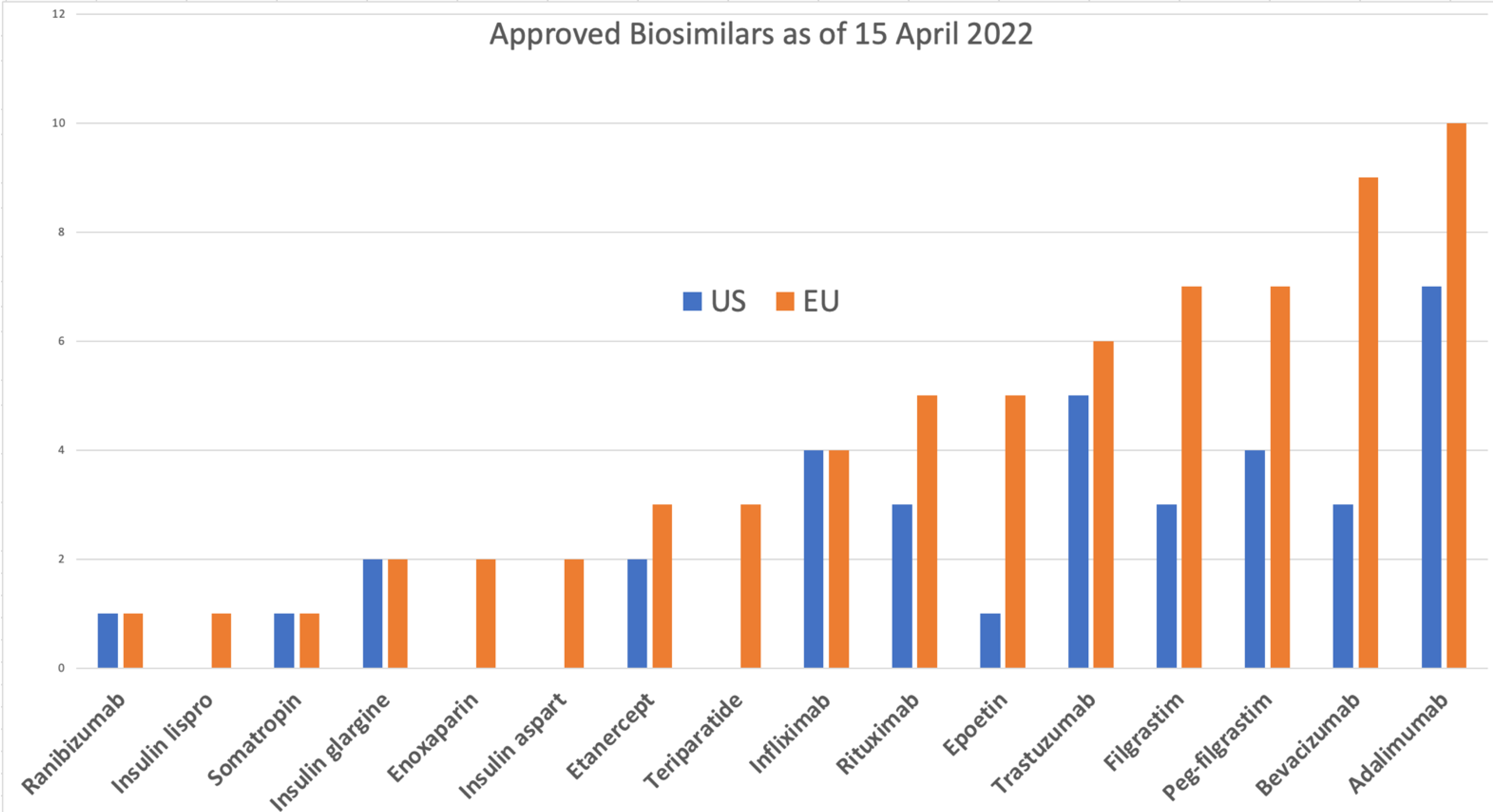{kind=link}
{kind=link}
{kind=link}
| Biological Products | |||
|---|---|---|---|
| Abatacept | Abciximab | Aflibercept | Alemtuzumab |
| Alirocumab | Atezolizumab | Avelumab | Basiliximab |
| Bedinvetman (V) | Belimumab | Benralizumab | Bevacizumab |
| Bezlotoxumab | Blinatumomab | Blood factors | Brentuximab vedotin |
| Brodalumab | Brolucizumab | Burosumab | Canakinumab |
| Caplacizumab | Cemiplimab | Certolizumab pegol | Cetuximab |
| Crizanlizumab | Daclizumab | Daratumumab | Darbepoetin alfa |
| Denosumab | Dinutuximab | Dupilumab | Durvalumab |
| Eculizumab | Elotuzumab | Emapalumab | Emicizumab |
| Erenumab | Etanercept | Evolocumab | Follitropin alfa |
| Fremanezumab | Frunevetmab (V) | Galcanezumab | Gemtuzumab ozogamicin |
| Golimumab | Guselkumab | Ibalizumab | Idarucizumab |
| Inotuzumab ozogamicin | Insulin detemir | Insulin lispro | Interferons |
| Ipilimumab | Isatuximab | Ixekizumab | Lanadelumab |
| Lokivetab (V) | Mepolizumab | ogamulizumab | Moxetumomab pasudodox |
| Muromonab-CD3 | Natalizumab | Necitumumab | Nivolumab |
| Obiltoxaximab | Obinutuzumab | Ocrelizumab | Ofatumumab |
| Olaratumab | Omalizumab | Palivizumab | Panitumumab |
| Pembrolizumab | Pertuzumab | Polatuzumab vedotin | Ramucirumab |
| Ranibizumab | Ravulizumab | Raxibacumab | Reslizumab |
| Rilonacept | Risankizumab | Romosozumab | Sacituzumab govitecan-hziy |
| Sarilumab | Secukinumab | Selumetinib | Siltuximab |
| Teprotumumab-trbw | Tildrakizumab | Tocilizumab | Urofollitropin |
| Ustekinumab | Vedolizumab | ||
| No | Product (Brand) Company | Global (Billion USD) Market, 2025 1 [101] | Current Approved US/EU Biosimilars 2 [9,10] | Development Factor 3 |
|---|---|---|---|---|
| 1. | Erythropoietin (Epoetin) Amgen | 18 | 1/3 | 1 (anemia) |
| 2. | Pembrolizumab (Keytruda), Merck | 16 | 0/0 | 5 (oncology) |
| 3. | Nivolumab (Opdivo), BMS | 14 | 0/0 | 5 (oncology) |
| 4. | Adalimumab (Humira) AbbVie | 11 | 7/10 | 2 (TNF) |
| 5. | Etanercept (Enbrel), Amgen | 8 | 2/3 | 2 (TNF) |
| 6. | Infliximab (Remicade), Janssen | 8 | 4/4 | 2 (TNF) |
| 7. | Ustekinumab (Stelara), Janssen | 7.5 | 0/0 | 2 (TNF) |
| 8. | Bevacizumab (Avastin) Roche | 7 | 3/9 | 4 (oncology) |
| 9. | Ocrelizumab (Ocrevis) | 7 | 0/0 | 3 (MS) |
| 10. | Pertuzumab (Perjeta) Roche | 7 | 0/0 | 5 (oncology) |
| 11. | Secukinumab (Cosentyx) | 6 | 0/0 | 2 (TNF) |
| 12. | Aflibercept (Eyelea), Regeneron | 4 | 0/0 | 2 (AMD) |
| 13. | Darbepoetin alfa (Aranesp) Amgen | 4 | 0/0 | 1 (anemia) |
| 14. | Peg-filgrastim (Neulasta), Amgen | 4 | 4/7 | 1 (neutropenia) |
| 15. | Ranibizumab(Lucentis) Novartis | 4 | 1/1 | 2 (AMD) |
| 16. | Trastuzumab (Herceptin), Genentech | 4 | 5/6 | 4 (oncology) |
| 17. | Rituximab (Rituxan) Biogen | 3 | 3/5 | 4 (oncology) |
| 18. | Cetuximab (Erbitux): (Lilly/Merck) | 1 | 0/0 | 5 (oncology) |
| 19. | Eculizumab (Soliris) Alexion | 1 | 0/0 | 3 (hemoglobinuria) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K. The Coming of Age of Biosimilars: A Personal Perspective. Biologics 2022, 2, 107-127. https://doi.org/10.3390/biologics2020009
Niazi SK. The Coming of Age of Biosimilars: A Personal Perspective. Biologics. 2022; 2(2):107-127. https://doi.org/10.3390/biologics2020009
Chicago/Turabian StyleNiazi, Sarfaraz K. 2022. "The Coming of Age of Biosimilars: A Personal Perspective" Biologics 2, no. 2: 107-127. https://doi.org/10.3390/biologics2020009
APA StyleNiazi, S. K. (2022). The Coming of Age of Biosimilars: A Personal Perspective. Biologics, 2(2), 107-127. https://doi.org/10.3390/biologics2020009