
Guillain–Barré Syndrome in COVID-19—The Potential Role of NCAM-1 and Immunotherapy

 ,
,  , and
, and
Abstract
:1. Introduction
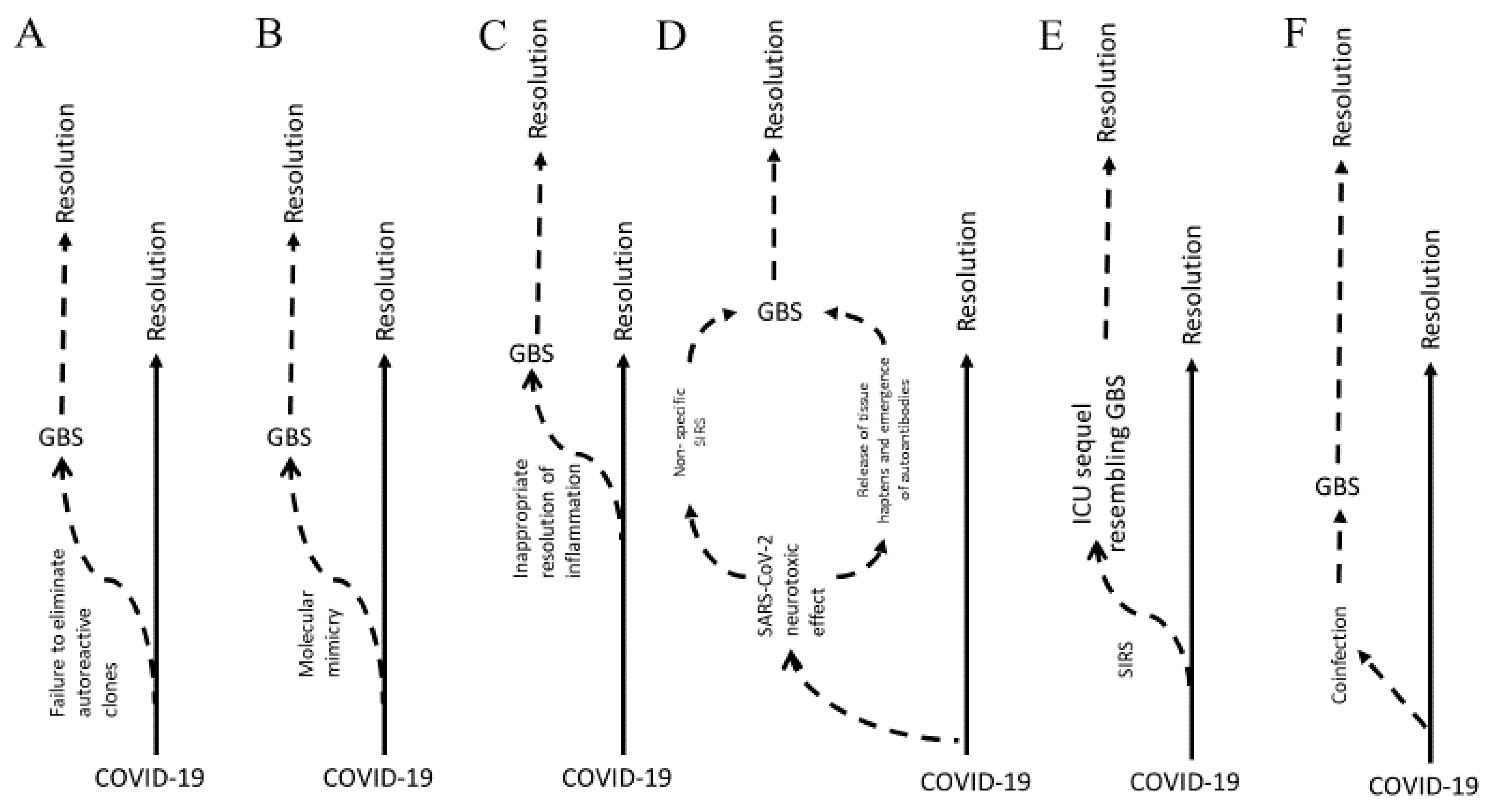
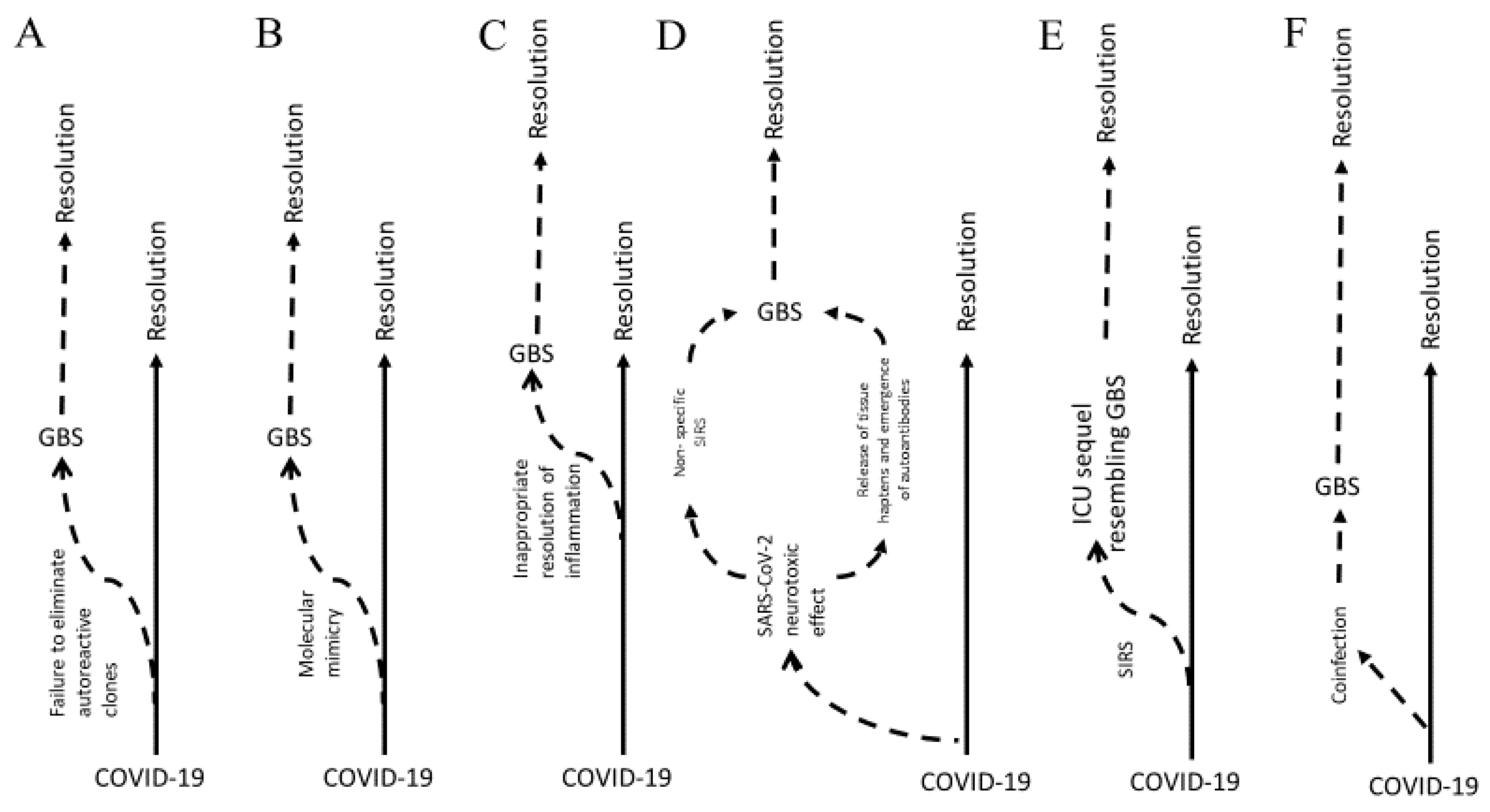
1.1. Models of Pathophysiology
1.1.1. Molecular Mimicry
1.1.2. Inflammation
1.2. Correlation with COVID-19
2. Case Presentation
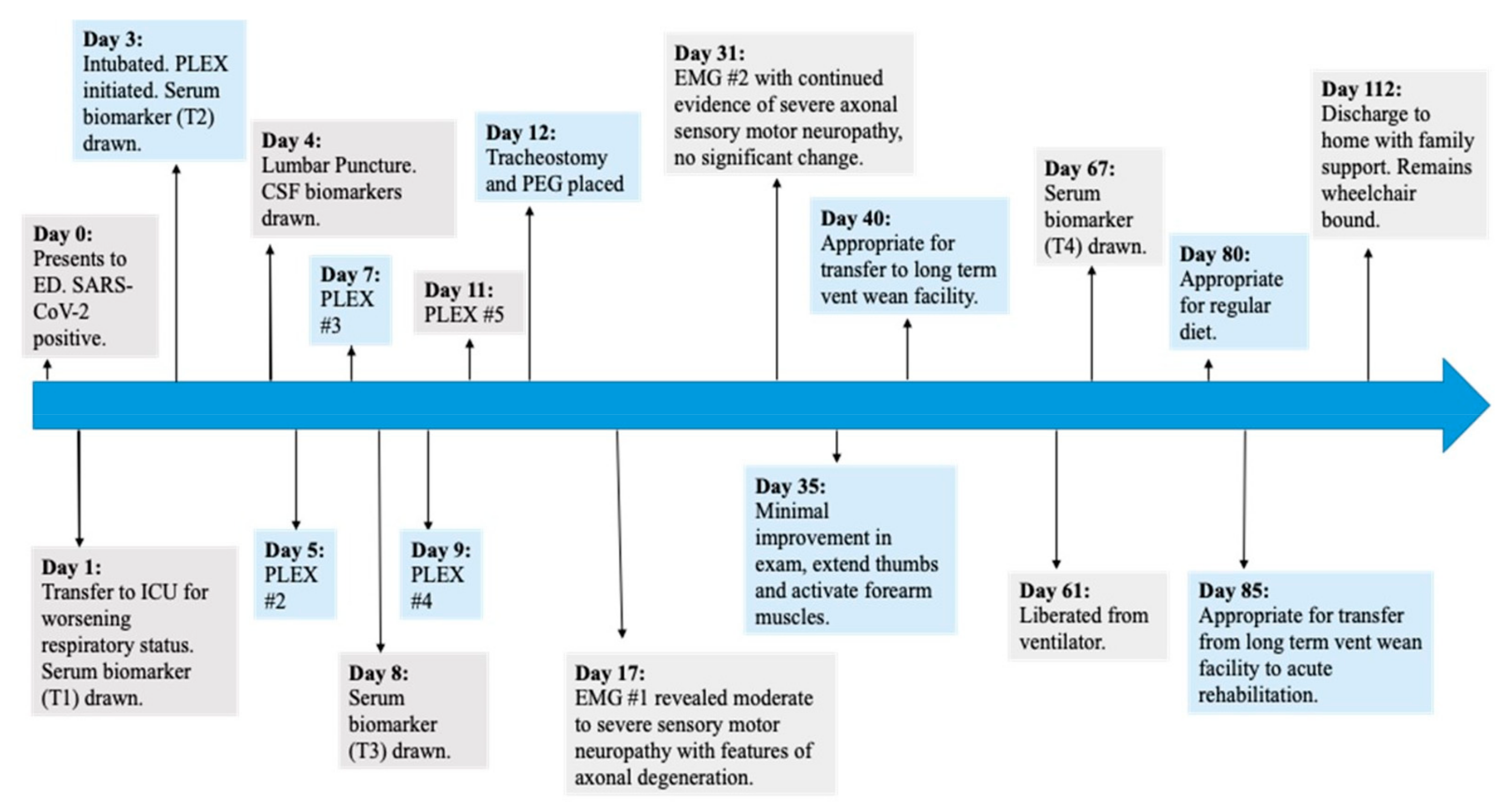
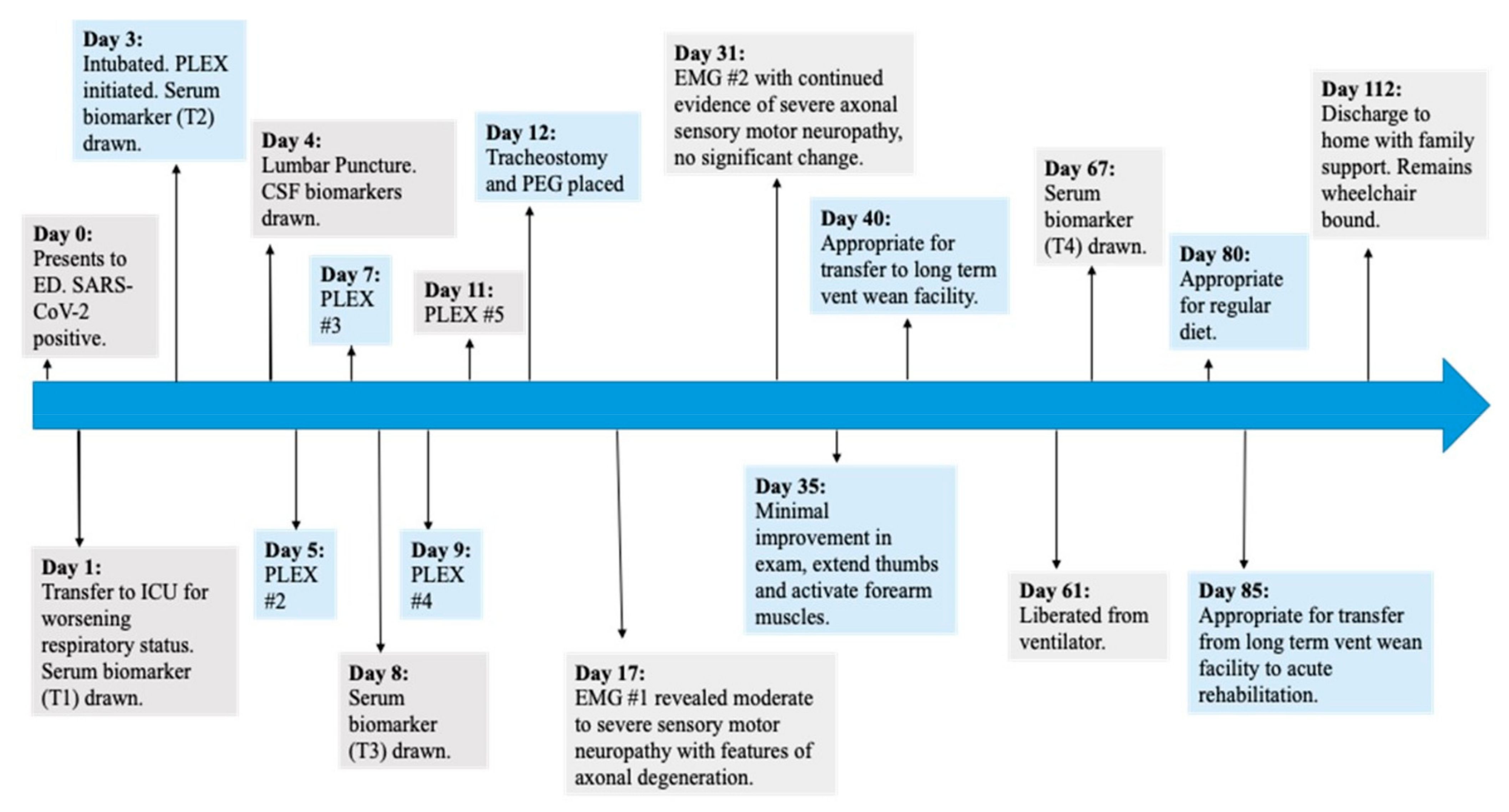
2.1. Methods
2.2. Case Report
2.3. Clinical Presentation Discussion
3. Discussion
NCAM-1 as a Potential Link between GBS and COVID-19
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| KLK6 | Kallikrein-6 |
| TOT TAU | Total tau protein |
| NCAM-1 | Neural cell adhesion molecule 1 |
| TDP43 | TAR DNA-binding protein 43 |
| NFL | Neurofilament light chain |
| NRGN | Neurogranin |
| FGF21 | Fibroblast growth factor 21 |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| BLC | B lymphocyte chemoattractant |
| YKL40/CHI3L1 | Chitinase-3-like protein 1 |
| RAGE | Receptor for advanced glycation endproducts |
| TNF-α | Tumor necrosis factor alpha |
| AMAN | Acute motor axonal neuropathy |
| AIDP | Acute inflammatory demyelinating polyneuropathy |
| COVID-19 | Coronavirus disease 2019 |
| GBS | Guillain–Barré syndrome |
| CSF | Cerebrospinal fluid |
| ApoE | Apolipoprotein E |
| IL-37 | Interleukin 37 |
| IL-17A | Interleukin 17A |
| IFN-γ | Interferon gamma |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| CRP | C-reactive protein |
| RT-PCR | Reverse transcription polymerase chain reaction |
| PLEX | Plasmapheresis |
| EMG | Electromyography |
| IgG | Immunoglobulin G |
| IL-6 | Interleukin 6 |
| MS | Multiple sclerosis |
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| ALS | Amyotrophic lateral sclerosis |
| FTD | Frontotemporal dementia |
References
- Dimachkie, M.M.; Barohn, R.J. Guillain-Barré syndrome and variants. Neurol. Clin. 2013, 31, 491–510. [Google Scholar] [CrossRef] [Green Version]
- Diez-Porras, L.; Vergés, E.; Gil, F.; Vidal, M.J.; Masson, J.; Arboix, A. Guillain-Barré-Strohl syndrome and COVID-19: Case report and literature review. Neuromuscul. Disord. 2020, 30, 859–861. [Google Scholar] [CrossRef]
- Saporta, M.A.; Shy, M.E. Chapter 12—Peripheral neuropathies. In Neurobiology of Brain Disorders; Zigmond, M.J., Rowland, L.P., Coyle, J.T., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 167–188. [Google Scholar]
- Eldar, A.H.; Chapman, J. Guillain Barré syndrome and other immune mediated neuropathies: Diagnosis and classification. Autoimmun. Rev. 2014, 13, 525–530. [Google Scholar] [CrossRef]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef]
- Yuki, N.; Hartung, H.P. Guillain–Barré syndrome. N. Engl. J. Med. 2012, 366, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J.; Goodyear, C.S. Glycolipid antigens and autoantibodies in autoimmune neuropathies. Trends Immunol. 2013, 34, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Devaux, J.J.; Odaka, M.; Yuki, N. Nodal proteins are target antigens in Guillain-Barré syndrome. J. Peripher. Nerv. Syst. 2012, 17, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Selvan, S.R. Hapten-induced contact hypersensitivity, autoimmune reactions, and tumor regression: Plausibility of mediating antitumor immunity. J. Immunol. Res. 2014, 2014, 1755265. [Google Scholar] [CrossRef]
- Yalcin Kehribar, D.; Cihangiroglu, M.; Sehmen, E.; Avci, B.; Capraz, A.; Yildrim Gilgin, A.; Gunaydin, C.; Ozgen, M. The receptor for advanced glycation end product (RAGE) pathway in COVID-19. Biomarkers 2021, 26, 114–118. [Google Scholar] [CrossRef]
- Inoue, S.; Hatakeyama, J.; Kondo, Y.; Hifumi, T.; Sakuramoto, H.; Kawasaki, T.; Taito, S.; Nakamura, K.; Unoki, T.; Kawai, Y.; et al. Post-intensive care syndrome: Its pathophysiology, prevention, and future directions. Acute Med. Surg. 2019, 6, 233–246. [Google Scholar] [CrossRef] [Green Version]
- Delmont, E.; Manso, C.; Querol, L.; Cortese, A.; Berardinelli, A.; Lozza, A.; Belghazi, M.; Malissart, P.; Labauge, P.; Taieb, G.; et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017, 140, 1851–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, A.; Frontera, J.; Placantonakis, D.G.; Lighter, J.; Galetta, S.; Balcer, L.; Melmed, K.R. Cerebrospinal fluid in COVID-19: A systematic review of the literature. J. Neurol. Sci. 2021, 421, 117316. [Google Scholar] [CrossRef] [PubMed]
- Guadarrama-Ortiz, P.; Choreño-Parra, J.A.; Sánchez-Martínez, C.M.; Pacheco-Sánchez, F.J.; Rodríguez-Nava, A.I.; García-Quintero, G. Neurological Aspects of SARS-CoV-2 Infection: Mechanisms and Manifestations. Front. Neurol. 2020, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- Kajumba, M.M.; Kolls, B.J.; Koltai, D.C.; Kaddumukasa, M.; Kaddumukasa, M.; Laskowitz, D.T. COVID-19-Associated Guillain-Barre Syndrome: Atypical Para-infectious Profile, Symptom Overlap, and Increased Risk of Severe Neurological Complications. SN Compr. Med. 2020, 1–13. [Google Scholar] [CrossRef]
- Wang, X.K.; Zhang, H.L.; Meng, F.H.; Chang, M.; Wang, Y.Z.; Jin, T.; Mix, E.; Zhu, J. Elevated levels of S100B, tau and pNFH in cerebrospinal fluid are correlated with subtypes of Guillain–Barré syndrome. Neurol. Sci. 2013, 34, 655–661. [Google Scholar] [CrossRef]
- Gigli, G.L.; Bax, F.; Marini, A.; Pellitteri, G.; Scalise, A.; Surcinelli, A.; Valente, M. Guillain-Barré syndrome in the COVID-19 era: Just an occasional cluster? J. Neurol. 2021, 268, 1195–1197. [Google Scholar] [CrossRef]
- Shang, P.; Feng, J.; Wu, W.; Zhang, H.L. Intensive Care and Treatment of Severe Guillain-Barré Syndrome. Front. Pharmacol. 2021, 12, 608130. [Google Scholar] [CrossRef]
- Jin, T.; Hu, L.S.; Chang, M.; Wu, J.; Winblad, B.; Zhu, J. Proteomic identification of potential protein markers in cerebrospinal fluid of GBS patients. Eur. J. Neurol. 2007, 14, 563–568. [Google Scholar] [CrossRef]
- Li, C.; Zhao, P.; Sun, X.; Che, Y.; Jiang, Y. Elevated levels of cerebrospinal fluid and plasma interleukin-37 in patients with Guillain-Barré syndrome. Mediat. Inflamm. 2013, 2013, 639712. [Google Scholar] [CrossRef] [Green Version]
- Mella, C.; Figueroa, C.D.; Otth, C.; Ehrenfeld, P. Involvement of Kallikrein-Related Peptidases in Nervous System Disorders. Front. Cell. Neurosci. 2020, 14, 166. [Google Scholar] [CrossRef]
- Lue, L.F.; Guerra, A.; Walker, D.G. Amyloid Beta and Tau as Alzheimer’s Disease Blood Biomarkers: Promise from New Technologies. Neurol. Ther. 2017, 6, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Insel, P.S.; Palmqvist, S.; Portelius, E.; Zetterberg, H.; Weiner, M.; Blennow, K.; Hansson, O. Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol. Med. 2016, 8, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.; Chang, B.; Yue, J.K.; Chiu, A.; Winkler, E.A.; Puccio, A.M.; Diaz-Arrastia, R.; Yuh, E.L.; Mukherjee, P.; Valadka, A.B.; et al. Comparing Plasma Phospho Tau, Total Tau, and Phospho Tau-Total Tau Ratio as Acute and Chronic Traumatic Brain Injury Biomarkers. JAMA Neurol. 2017, 74, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Asbury, A. CSF tau protein: A new prognostic marker for Guillain-Barre syndrome. Neurology 2007, 68, 1438–1439. [Google Scholar] [CrossRef]
- Jin, K.; Takeda, A.; Shiga, Y.; Sato, S.; Ohnuma, A.; Nomura, H.; Arai, H.; Kusunoki, S.; Ikeda, M.; Itoyama, Y. CSF tau protein: A new prognostic marker for Guillain-Barré syndrome. Neurology 2006, 67, 1470–1472. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G.; Flöel, A.; Stahl, B. Cross-Reactivity as a Mechanism Linking Infections to Stroke. Front. Neurol. 2019, 10, 469. [Google Scholar] [CrossRef] [PubMed]
- Ziliotto, N.; Zivadinov, R.; Jakimovski, D.; Baroni, M.; Tisato, V.; Secchiero, P.; Bergsland, N.; Ramasamy, D.P.; Weinstock-Guttman, B.; Bernardi, F.; et al. Plasma levels of soluble NCAM in multiple sclerosis. J. Neurol. Sci. 2019, 396, 36–41. [Google Scholar] [CrossRef]
- Yang, X.; Zou, M.; Pang, X.; Liang, S.; Sun, C.; Wang, J.; Fan, L.; Xia, W.; Wu, L. The association between NCAM1 levels and behavioral phenotypes in children with autism spectrum disorder. Behav. Brain Res. 2019, 359, 234–238. [Google Scholar] [CrossRef]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef]
- Gaetani, L. Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef]
- Gaiottino, J.; Norgren, N.; Dobson, R.; Topping, J.; Nissim, A.; Malaspina, A.; Bestwick, J.P.; Monsch, A.U.; Regeniter, A.; Lindberg, R.; et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS ONE 2013, 8, e75091. [Google Scholar]
- Aamodt, A.H.; Høgestøl, E.A.; Popperud, T.H.; Holter, J.C.; Dyrhol-Riise, A.M.; Tonby, K.; Stiksrud, B.; Quist-Paulsen, E.; Berge, T.; Barratt-Due, A.; et al. Blood neurofilament light concentration at admittance: A potential prognostic marker in COVID-19. J. Neurol. 2021, 1–10. [Google Scholar] [CrossRef]
- Chen, S.; Chen, S.T.; Sun, Y.; Xu, Z.; Wang, Y.; Yao, S.Y.; Yao, W.B.; Gao, X.D. Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer’s disease. Redox Biol. 2019, 22, 101133. [Google Scholar] [CrossRef]
- Desikan, R.S.; Thompson, W.K.; Holland, D.; Hess, C.P.; Brewer, J.B.; Zetterberg, H.; Blennow, K.; Andreassen, O.A.; McEvoy, L.K.; Hyman, B.T.; et al. The Role of Clusterin in Amyloid-β–Associated Neurodegeneration. JAMA Neurol. 2014, 71, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.; Ubogu, E.E. The role of chemokines in Guillain-Barré syndrome. Muscle Nerve 2013, 48, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoneveld, L.; Ladang, A.; Henket, M.; Frix, A.N.; Cavalier, E.; Guiot, J. COVID-19 Clinical Investigators of the CHU de Liège. YKL-40 as a new promising prognostic marker of severity in COVID infection. Crit. Care 2021, 25, 66. [Google Scholar] [CrossRef]
- Baldacci, F.; Lista, S.; Cavedo, E.; Bonuccelli, U.; Hampel, H. Diagnostic function of the neuroinflammatory biomarker YKL-40 in Alzheimer’s disease and other neurodegenerative diseases. Expert Rev. Proteom. 2017, 14, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.; Candan, S.A.; Soysal Tomruk, M.; Elibol, N.; Dada, O.; Truijen, S.; Saeys, W. Is Guillain-Barré Syndrome Associated with COVID-19 Infection? A Systemic Review of the Evidence. Front. Neurol. 2021, 11, 566308. [Google Scholar] [CrossRef]
- Zhang, D.Q.; Wang, R.; Li, T.; Zhou, J.P.; Chang, G.Q.; Zhao, N.; Yang, L.N.; Zhai, H.; Yang, L. Reduced soluble RAGE is associated with disease severity of axonal Guillain-Barré syndrome. Sci. Rep. 2016, 6, 21890. [Google Scholar] [CrossRef]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Cheng, L.; Li, H.; Li, L.; Liu, C.; Yan, S.; Chen, H.; Li, Y. Ferritin in the coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. J. Clin. Lab. Anal. 2020, 34, e23618. [Google Scholar] [CrossRef] [PubMed]
- Namaste, S.M.; Rohner, F.; Huang, J.; Bhushan, N.L.; Flores-Ayala, R.; Kupka, R.; Mei, Z.; Rawat, R.; Williams, A.M.; Raiten, D.J.; et al. Adjusting ferritin concentrations for inflammation: Biomarkers Reflecting Inflammation and Nutritional Determinants of Anemia (BRINDA) project. Am. J. Clin. Nutr. 2017, 106, 359s–371s. [Google Scholar] [PubMed]
- Gagarkin, D.A.; Dombrowski, K.E.; Thakar, K.B.; DePetrillo, J.C. Acute inflammatory demyelinating polyneuropathy or Guillain-Barré syndrome associated with COVID-19: A case report. J. Med. Case Rep. 2021, 15, 219. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.L.; Liu, C.H.; Wang, N.; Xu, J.; Zhu, X.Y.; Su, C.S.; Li, H.L.; Zhang, H.F.; Li, Z.Z.; Li, H.L.; et al. Elevated Plasma D-Dimer in Adult Community-Acquired Pneumonia Patients is Associated with an Increased Inflammatory Reaction and Lower Survival. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Lu, M.O.; Zhu, J. The role of cytokines in Guillain-Barré syndrome. J. Neurol. 2020, 258, 533–548. [Google Scholar] [CrossRef]
- Ahmadpoor, P.; Rostaing, L. Why the immune system fails to mount an adaptive immune response to a COVID-19 infection. Transpl. Int. 2020, 33, 824–825. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr. Pharm. Des. 2008, 14, 1555–1565. [Google Scholar] [CrossRef]
- Tsygan, N.V.; Trashkov, A.P.; Litvinenko, I.V.; Yakovleva, V.A.; Ryabtsev, A.V.; Vasiliev, A.G.; Churilov, L.P. Autoimmunity in acute ischemic stroke and the role of blood-brain barrier: The dark side or the light one? Front. Med. 2019, 13, 420–426. [Google Scholar] [CrossRef]
- Javidi, E.; Magnus, T. Autoimmunity after Ischemic Stroke and Brain Injury. Front. Immunol. 2019, 10, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliao Caamaño, D.S.; Alonso Beato, R. Facial diplegia, a possible atypical variant of Guillain-Barré Syndrome as a rare neurological complication of SARS-CoV-2. J. Clin. Neurosci. 2020, 77, 230–232. [Google Scholar] [CrossRef]
- Toscano, G.; Palmerini, F.; Ravaglia, S.; Ruiz, L.; Invernizzi, P.; Cuzzoni, M.G.; Franciotta, D.; Baldanti, F.; Daturi, R.; Postorino, P.; et al. Guillain-Barré Syndrome Associated with SARS-CoV-2. N. Engl. J. Med. 2020, 382, 2574–2576. [Google Scholar] [CrossRef]
- Marta-Enguita, J.; Rubio-Baines, I.; Gastón-Zubimendi, I. Fatal Guillain-Barre syndrome after infection with SARS-CoV-2. Neurologia 2020, 35, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Scheidl, E.; Canseco, D.D.; Hadji-Naumov, A.; Bereznai, B. Guillain-Barré syndrome during SARS-CoV-2 pandemic: A case report and review of recent literature. J. Peripher. Nerv. Syst. 2020, 25, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Cotti Piccinelli, S.; Gazzina, S.; Foresti, C.; Frigeni, B.; Servalli, M.C.; Sessa, M.; Cosentino, G.; Marchioni, E.; Ravaglia, S.; et al. Guillain-Barré syndrome and COVID-19: An observational multicentre study from two Italian hotspot regions. J. Neurol. Neurosurg. Psychiatry 2021, 92, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, S. Coronavirus Disease 2019 and the Risk of Guillain-Barré Syndrome. Ann. Neurol. 2021, 89, 846. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barré Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Y. Hepatitis E virus-associated Guillain-Barre syndrome: Revision of the literature. Brain Behav. 2020, 10, e01496. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.C.; O’Brien, S.J.; Petersen, I.; Islam, A.; Hayward, A.; Rodrigues, L.C. Guillain-Barré syndrome and preceding infection with campylobacter, influenza and Epstein-Barr virus in the general practice research database. PLoS ONE 2007, 2, e344. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, E.P.; Altomare, N.J.; Parekh, Y.H.; Gowda, R.C.; Parikh, P.D.; Lazar, M.H.; Blaser, M.J. COVID-19 as a Trigger of Recurrent Guillain-Barré Syndrome. Pathogens 2020, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Padroni, M.; Mastrangelo, V.; Asioli, G.M.; Pavolucci, L.; Abu-Rumeileh, S.; Piscaglia, M.G.; Querzani, P.; Callegarini, C.; Foschi, M. Guillain-Barré syndrome following COVID-19: New infection, old complication? J. Neurol. 2020, 267, 1877–1879. [Google Scholar] [CrossRef] [Green Version]
- Jawa, R.S.; Anillo, S.; Huntoon, K.; Baumann, H.; Kulaylat, M. Interleukin-6 in surgery, trauma, and critical care part II: Clinical implications. J. Intensive Care Med. 2011, 26, 73–87. [Google Scholar] [CrossRef]
- Grifoni, E.; Valoriani, A.; Cei, F.; Lamanna, R.; Gelli, A.; Ciambotti, B.; Vannucchi, V.; Moroni, F.; Pelagatti, L.; Tarquini, R.; et al. Interleukin-6 as prognosticator in patients with COVID-19. J. Infect. 2020, 81, 452–482. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Faccini, L.; Ribeiro, E.M.; Feitosa, I.M.; Horovitz, D.D.; Cavalcanti, D.P.; Pessoa, A.; Doriqui, M.J.; Neri, J.I.; Neto, J.M.; Wanderley, H.Y.; et al. Possible Association between Zika Virus Infection and Microcephaly—Brazil, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Charlins, P.; Veselinovic, M.; Kinner-Bibeau, L.; Hu, S.; Curlin, J.; Remling-Mulder, L.; Olson, K.E.; Aboellail, T.; Akkina, R. Zika viral infection and neutralizing human antibody response in a BLT humanized mouse model. Virology 2018, 515, 235–242. [Google Scholar] [CrossRef]
- Santos, F.R.S.; Nunes, D.A.F.; Lima, W.G.; Davyt, D.; Santos, L.L.; Taranto, A.G.; Ferreira, J.M.S. Identification of Zika Virus NS2B-NS3 Protease Inhibitors by Structure-Based Virtual Screening and Drug Repurposing Approaches. J. Chem. Inf. Model. 2020, 60, 731–737. [Google Scholar] [CrossRef]
- Morsy, S. NCAM protein and SARS-COV-2 surface proteins: In-silico hypothetical evidence for the immunopathogenesis of Guillain-Barré syndrome. Med. Hypotheses 2020, 145, 110342. [Google Scholar] [CrossRef] [PubMed]
- Paladino, L.; Vitale, A.M.; Caruso Bavisotto, C.; Conway de Macario, E.; Cappello, F.; Macario, A.; Gammazza, A.M. The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19. J. Clin. Med. 2020, 9, 3518. [Google Scholar] [CrossRef]
- Lucchese, G.; Flöel, A. Molecular mimicry between SARS-CoV-2 and respiratory pacemaker neurons. Autoimmun. Rev. 2020, 19, 102556. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. On the molecular determinants of the SARS-CoV-2 attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 2020, 217, 108480. [Google Scholar] [CrossRef]
- Guadarrama-Ortiz, P.; Choreño-Parra, J.A.; Pacheco-Sánchez, F.J.; Ponce-Sánchez, J.M.; García-Quintero, G.; Rodríguez-Muñoz, P.E.; Prieto-Rivera, Á.D. Chronic subclinical spondylotic myelopathy exacerbated by COVID-19: A case report. Interdiscip. Neurosurg. 2021, 23, 100896. [Google Scholar] [CrossRef]
- Lucchese, G.; Flöel, A.; Stahl, B. A Peptide Link between Human Cytomegalovirus Infection, Neuronal Migration, and Psychosis. Front. Psychiatry 2020, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Dariolli, R.; Campana, C.; Gutierrez, A.; Sobie, E.A. In vitro and In silico Models to Study SARS-CoV-2 Infection: Integrating Experimental and Computational Tools to Mimic “COVID-19 Cardiomyocyte”. Front. Physiol. 2021, 12, 624185. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
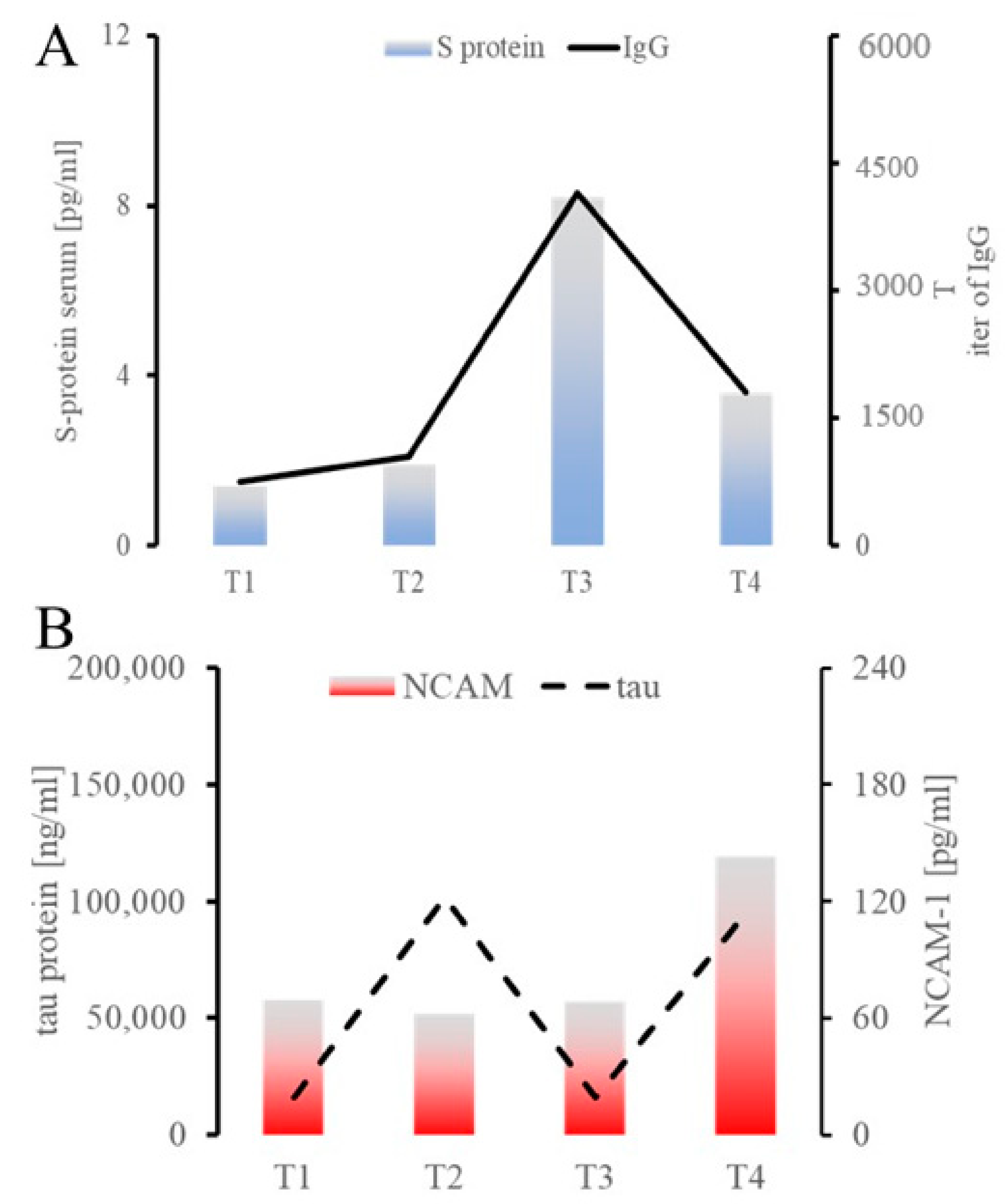{kind=link}
| Biomarker | Blood | CSF | Reference Ranges | General Description | |||
|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T4 | Relevance to COVID-19 | |||
| KLK6 | 923.7 | 3953.6 | 3442.7 | 465.2 | 14,849.4 | unknown | Serum protease, neuroinflammation, linked to multiple sclerosis (MS), elevated in Alzheimer’s disease (AD) when tau also elevated, decreased in Parkinson’s disease (PD), degraded in respiratory disease, regulates myelin volume. No association with GBS or COVID-19 [21] |
| Amyloid B1-41 | BDL | BDL | BDL | BDL | BDL | unknown | Was not found in the literature [22] |
| Amyloid B1-42 | 4.2 | 7.8 | 12.6 | 16.1 | 29.3 | 0–400 pg/mL (SIMOA) | Lower in AD, no relation to COVID-19 found [22] |
| TOT TAU | BDL | 121.7 | BDL | 114.1 | BDL | 100–300 in GBS, 0–360 pg(SIMOA) | Higher values associated with worse prognosis in GBS [22,23,24,25,26] |
| NCAM-1 | 57,758.5 | 51,593.3 | 57,231.2 | 119,018.3 | 22,356.6 | 246.03 pg/mL control, 153 in autism | Also called CD56, helps synapse formation, found to be low in autism, high in MS, hypothetically associated with COVID-19 pathogenesis [27,28,29,30] |
| TAU PT181 | BDL | BDL | BDL | BDL | BDL | unknown | Another marker for tau, no association found with GBS or COVID-19 [23,24] |
| TDP43 | ND | 4045.3 | 1620.1 | 5583.8 | BDL | unknown | Elevated in amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), not GBS. No association with COVID-19 seen [5] |
| NFL | 41.85 | 93.15 | 38.61 | 192.9 | unknown | Elevated in AD, GBS, and ALS, in addition to non-survivors of COVID-19 [31,32,33] | |
| NRGN | 6052.2 | 11,867.6 | 5743.6 | 11,040.7 | 1048.9 | unknown | Neurogranin: decreased in schizophrenia, increased in AD, no association found with COVID-19 or GBS [23] |
| FGF21 | BDL | BDL | BDL | BDL | BDL | unknown | Neuroprotectant, involved in carbohydrate metabolism, no link with GBS or COVID [34] |
| CLUSTERIN | 11,039.5 | 1673.9 | 28,787,864.31 | 56,444,777.02 | 29,957.99667 | unknown | Increased in AD, no evidence for role in GBS or COVID-19 [35] |
| TREM2 | 109.5 | 144.6 | 286.51 | 1833.17 | 728.849106 | unknown | Associated with a risk for AD, can suppress inflammation but also trigger it, no clear association with GBS or COVID-19 [36] |
| BLC | 325.1 | 6462.4 | 155.89 | 200.75 | BDL | unknown | B lymphocyte chemoattractant. Associated with adenocarcinoma. Also called CXCL13 or BCA-1. Other types but not this type associated with GBS [37] |
| YKL40 | 8684.5 | 14,853.5 | 17,258.11 | 18,389.31 | 15,090.6293 | healthy: 80 ng/mL, severe COVID 300 | Also called chitinase-3-like protein 1 (CHI3L1), elevated in AD, not GBS, associated with worse mortality in lung COVID-19 disease [38,39] |
| RAGE | 146.3 | 124.8 | 67.87 | 61.70 | 24.0432183 | serum pg/mL AIDP > 1000, AMAN < 1000 | Binds to pro-inflammatory pathways, higher in neurodegenerative disease, lower in AMAN subtype of GBS, higher in AIDP, higher in asymptomatic COVID-19, lower in severe COVID-19 in enzyme-linked immunosorbent assay in ng/mL [2,10,40,41,42] |
| FERRITIN | 383,593.2 | 207,231.3 | 218,048.4 | 709,040.7 | 51,863.9 | unknown | Increased levels seen in patients that showed inflammation during episode of malaria and COVID-19. Similar results were seen in a patient who presented with GBS associated with COVID-19 [43,44,45] |
| DDIMER | 110,928.0 | 34,814.4 | 25,904.3 | 6,737,038.6 | 720,348.9 | unknown | Elevated d-dimer plasma levels are associated with inflammatory reactions to pneumonia and severe COVID-19. No association with GBS [46] |
| IL-6 | 1.6 | 22.8 | 5.0 | 0.7 | <1.04736328125 | unknown | Uncontrolled inflammation may occur following the increased activation of serum IL-6, similarly to how its role in a “cytokine storm” has been correlated with critical COVID-19 development. It plays both a pro-inflammatory and protective role in GBS [47,48] |
| TNF-α | 0.0 | 0.5 | 0.0 | 0.0 | 0.1 | unknown | Gene polymorphisms prevent efficacious immune response against viruses such as hepatitis B and predict susceptibility to GBS, while decreased levels due to antibiotics (azithromycin specifically) are seen in COVID-19 patients [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudanski, K.; Yakhkind, A.; Restrepo, M.; Draham, L.; Lang, A.E. Guillain–Barré Syndrome in COVID-19—The Potential Role of NCAM-1 and Immunotherapy. BioMed 2021, 1, 80-92. https://doi.org/10.3390/biomed1010006
Laudanski K, Yakhkind A, Restrepo M, Draham L, Lang AE. Guillain–Barré Syndrome in COVID-19—The Potential Role of NCAM-1 and Immunotherapy. BioMed. 2021; 1(1):80-92. https://doi.org/10.3390/biomed1010006
Chicago/Turabian StyleLaudanski, Krzysztof, Aleksandra Yakhkind, Mariana Restrepo, Lindsay Draham, and Adam Edward Lang. 2021. "Guillain–Barré Syndrome in COVID-19—The Potential Role of NCAM-1 and Immunotherapy" BioMed 1, no. 1: 80-92. https://doi.org/10.3390/biomed1010006
APA StyleLaudanski, K., Yakhkind, A., Restrepo, M., Draham, L., & Lang, A. E. (2021). Guillain–Barré Syndrome in COVID-19—The Potential Role of NCAM-1 and Immunotherapy. BioMed, 1(1), 80-92. https://doi.org/10.3390/biomed1010006