
Kidney Biopsy in a Patient with Cardiorenal Metabolic Syndrome—How to Interpret Histopathology



Abstract
1. Background
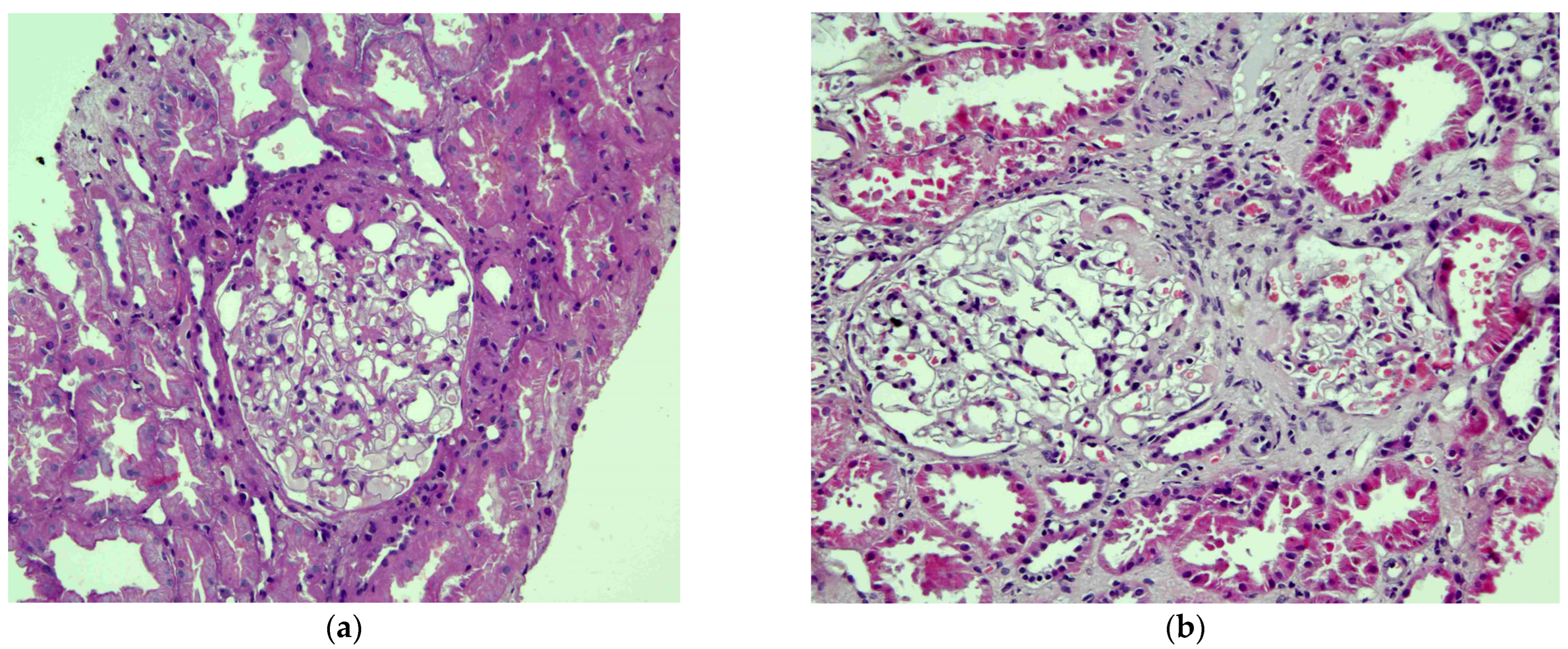
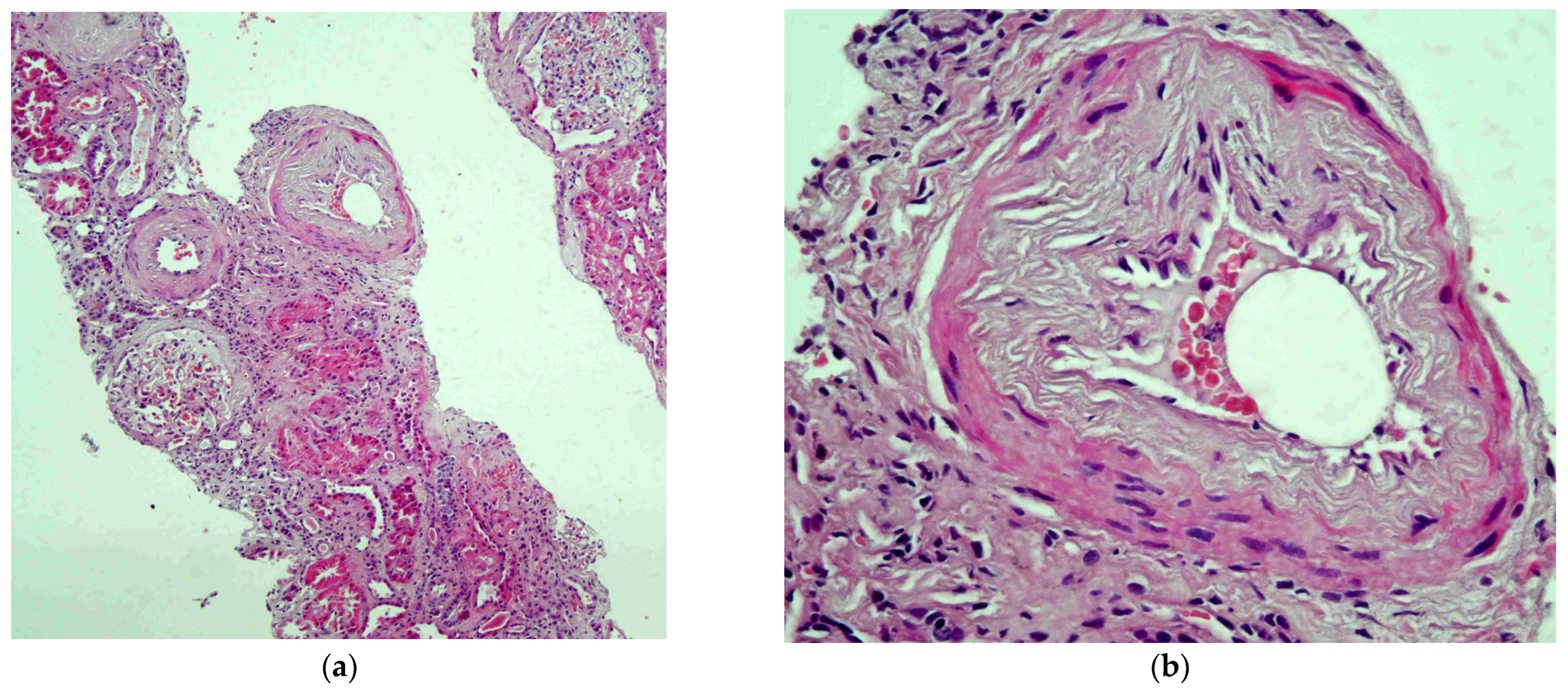
2. The Case
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whaley-Connell, A.; Sowers, J.R. Basic science: Pathophysiology: The cardiorenal metabolic syndrome. J. Am. Soc. Hypertens 2014, 8, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Rangaswami, J.; Tuttle, K.; Vaduganathan, M. Cardio-Renal-Metabolic Care Models. Toward Achieving Effective Interdisciplinary Care. Circ. Cardiovasc. Qual. Outcomes 2020, 13, e007264. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Maegawa, H.; Watada, H.; Yabe, D.; Node, K.; Murohara, T.; Wada, J. Interconnection between cardiovascular, renal and metabolic disorders: A narrative review with a focus on Japan. Diabetes Obes. Metab. 2022, 24, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Mattina, A.; Argano, C.; Brunori, G.; Lupo, U.; Raspanti, M.; Monaco, M.L.; Bocchio, R.M.; Natoli, G.; Giusti, M.A.; Corrao, S. Clinical complexity and diabetes: A multidimensional approach for the management of cardiorenal metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 2730–2738. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, Y.; Anderson, J.E.; Bakris, G.L.; Ballantyne, C.M.; Beckman, J.A.; Bhatt, D.L.; Bloomgarden, Z.T.; Bozkurt, B.; Budoff, M.J.; Butler, J.; et al. DCRM Multispecialty Practice Recommendations for the management of diabetes, cardiorenal, and metabolic diseases. J Diabetes Complicat. 2022, 36, 108101. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.P.; Patel, T.V.; Farag, Y.M.; Florez, A.; Rennke, H.G.; Singh, A.K. Kidney pathological changes in metabolic syndrome: A cross-sectional study. Am. J. Kidney Dis. 2009, 53, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Joyce, T.; Chirino, Y.I.; Natalia, M.T.; Jose, P.C. Renal damage in the metabolic syndrome (MetSx): Disorders implicated. Eur. J. Pharmacol. 2018, 5, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Tan, W.; Pan, X.; Tian, E.; Wu, Z.; Yang, J. Metabolic Syndrome-Related Kidney Injury: A Review and Update. Front. Endocrinol. 2022, 13, 904001. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.B.; Miot, H.A.; Domingues, M.A.C.; Oliveira, C.C. Autopsy Patients with Obesity or Metabolic Syndrome as Basic Cause of Death: Are There Pathological Differences Between These Groups? Clin. Med. Insights Pathol. 2018, 11, 1179555718791575. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Li, Y.; Yu, Y.; Xu, M.; Chen, H.; Li, L.; Peng, T.; Zhao, K.; Zhuang, Y. Obesity-Related Glomerulopathy: From Mechanism to Therapeutic Target. Diabetes Metab. Syndr. Obes. 2021, 28, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Cao, C.; Deng, T.; Zhou, Z. Obesity-Related Glomerulopathy: A Latent Change in Obesity Requiring More Attention. Kidney Blood Press Res. 2020, 45, 510–522. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; DD'Agati, V.; Lamb, H.J.; Barlovic, D.P.; Hojs, R.; et al. ERA-EDTA Working Group Diabesity. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Allford, S.L.; Hunt, B.J.; Rose, P.; Machin, S.J. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br. J. Haematol. 2003, 120, 556–573. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Hunt, B.J.; Benjamin, S.; Liesner, R.; Rose, P.; Peyvandi, F.; Cheung, B.; Machin, S.J.; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br. J. Haematol. 2012, 158, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.C.; D’agati, V.D.; Nester, C.M.; Smith, R.J.; Haas, M.; Appel, G.B.; Alpers, C.E.; Bajema, I.M.; Bedrosian, C.; Braun, M.; et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013, 84, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Nester, C.M.; Smith, R.J. Treatment options for C3 glomerulopathy. Curr. Opin. Nephrol. Hypertens 2013, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Schena, F.P.; Esposito, P.; Rossini, M. A Narrative Review on C3 Glomerulopathy: A Rare Renal Disease. Int. J. Mol. Sci. 2020, 21, 525. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| 10.2014 | 11.2019 | 01.2020 | 11.2021 | 02.2022 | |
|---|---|---|---|---|---|
| SCr µmol/L (53–115) | 203 | 198 | 225 | 255 | 254 |
| eGFR CKD-EPI mL/min/1.73 m2 | 35 | 35 | 30 | 25 | 25 |
| UA µmol/L (155–428) | 527 | 604 | 621 | 690 | 661 |
| TCh mmol/L (1.3–5.3) | 6.03 | 6.33 | 6.01 | 7.90 | 7.24 |
| HDL mmol/L (1.0–1.55) | 0.76 | 0.94 | 1.09 | ||
| LDL mmol/L (1.9–3.3) | 3.74 | 3.93 | 5.09 | ||
| TG mmol/L (0.6–2.3) | 3.46 | 4.97 | 6.47 | ||
| AC (2.28–3.02) | 7.0 | 5.4 | 6.3 | ||
| FG mmol/L (3.5–5.56) | 5.3 | 8.2 | 6.0 | 6.8 | 7.1 |
| PU g/24 h (0.0–0.15) | 0.8 | 2.7 | 4.0 | 0.9 | 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharova, E.; Vorobyeva, O. Kidney Biopsy in a Patient with Cardiorenal Metabolic Syndrome—How to Interpret Histopathology. Kidney Dial. 2023, 3, 171-177. https://doi.org/10.3390/kidneydial3020015
Zakharova E, Vorobyeva O. Kidney Biopsy in a Patient with Cardiorenal Metabolic Syndrome—How to Interpret Histopathology. Kidney and Dialysis. 2023; 3(2):171-177. https://doi.org/10.3390/kidneydial3020015
Chicago/Turabian StyleZakharova, Elena, and Olga Vorobyeva. 2023. "Kidney Biopsy in a Patient with Cardiorenal Metabolic Syndrome—How to Interpret Histopathology" Kidney and Dialysis 3, no. 2: 171-177. https://doi.org/10.3390/kidneydial3020015
APA StyleZakharova, E., & Vorobyeva, O. (2023). Kidney Biopsy in a Patient with Cardiorenal Metabolic Syndrome—How to Interpret Histopathology. Kidney and Dialysis, 3(2), 171-177. https://doi.org/10.3390/kidneydial3020015