
The Pathophysiological Basis of Diabetic Kidney Protection by Inhibition of SGLT2 and SGLT1

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Physiology of SGLT1 and SGLT2 in the Kidney
3. Potential Upregulation of SGLT2 Expression in the Diabetic Kidney
4. SGLT2 Inhibitors Protect Kidney Function in Patients with and without T2DM
5. The Metabolic Signature of SGLT2 Inhibition
6. SGLT2 Inhibition Acutely Lowers GFR to Preserve It in the Long-Term
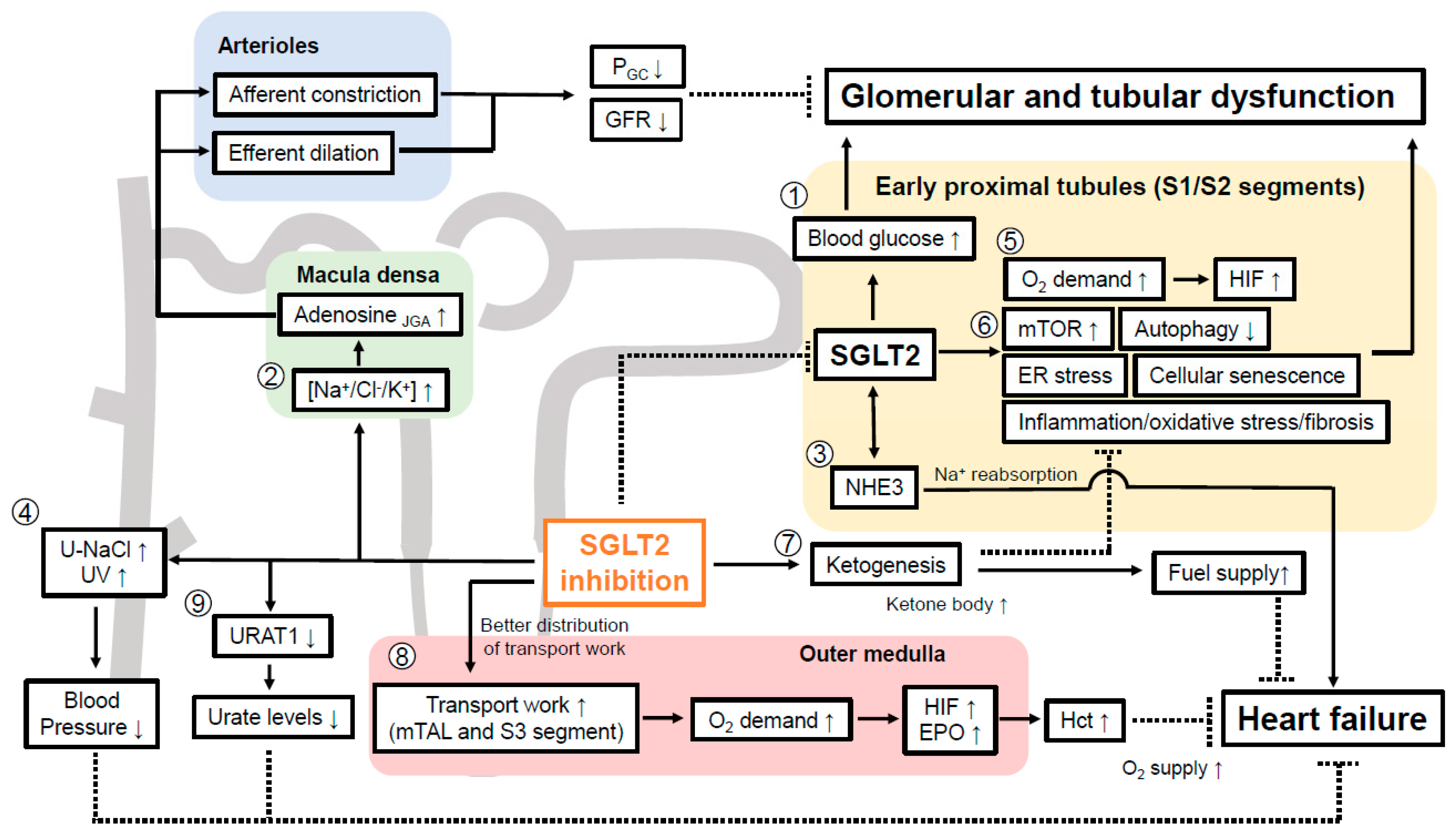
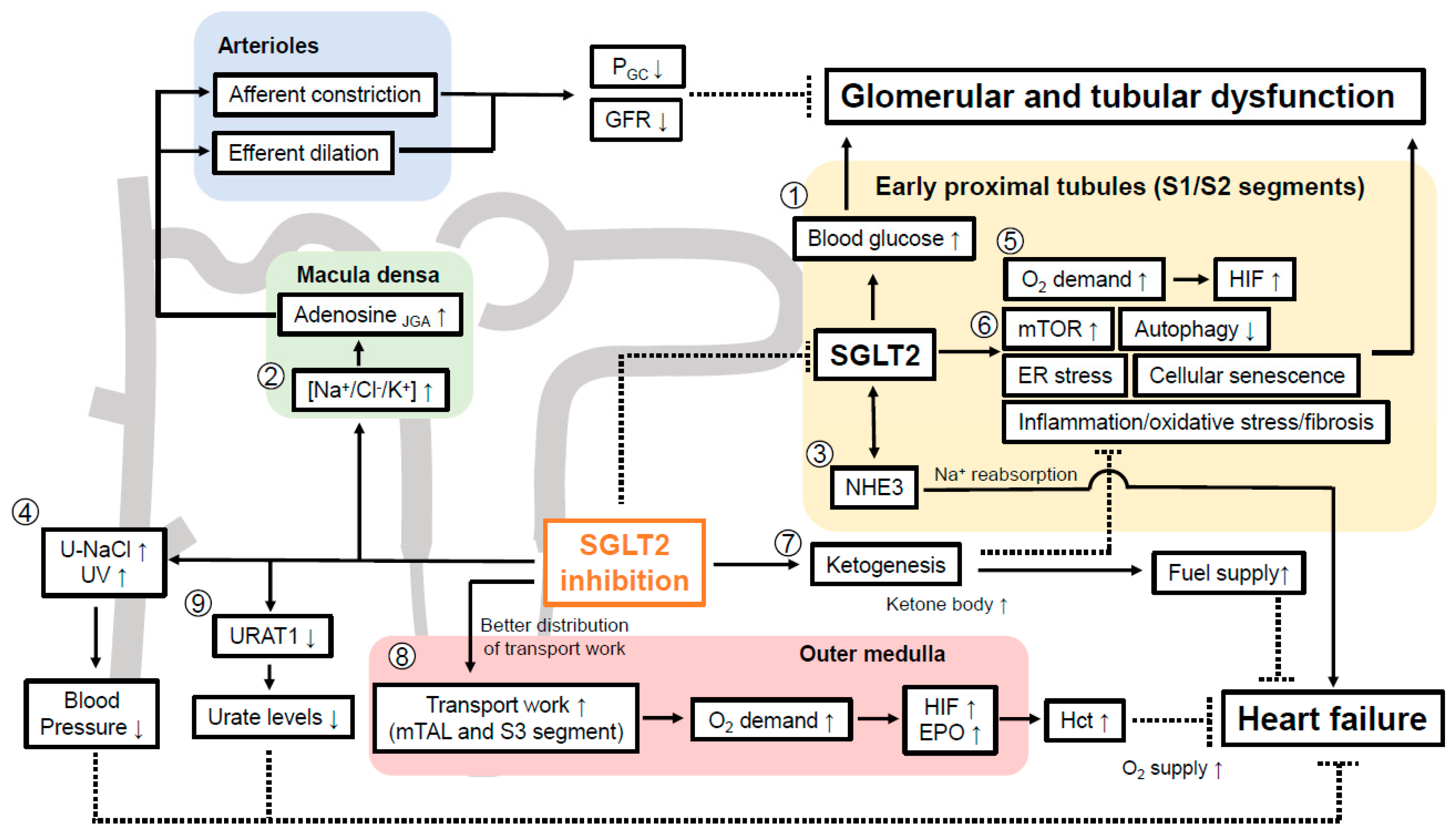
7. How can SGLT2 Inhibitors Preserve Kidney Function?
7.1. SGLT2 Inhibition and Oxygen Handling in the Kidney Cortex
7.2. Renal Transport Work Is More Equal Distributed by SGLT2 Inhibition and Potential Mimicking of Systemic Hypoxia at the Oxygen Sensor in the Kidney
7.3. SGLT2 Inhibitors Promote Mitochondrial Metabolism in the Kidney
7.4. SGLT2 Inhibition Suppresses Tubular mTOR Activity via Enhancing Ketogenesis
7.5. SGLT2 Inhibition Increases Tubular Autophagy
7.6. SGLT2 Inhibition Attenuates Cellular Senescence in Renal Tubular Cells
7.7. Evidence That SGLT2 Inhibitor Alleviates ER Stress in Kidney
7.8. SGLT2 Inhibitors Reduce Inflammation and Oxidative Stress in DKD
7.9. Evidence That SGLT2 Inhibition May Affect EMT to Facilitate Renal Fibrosis
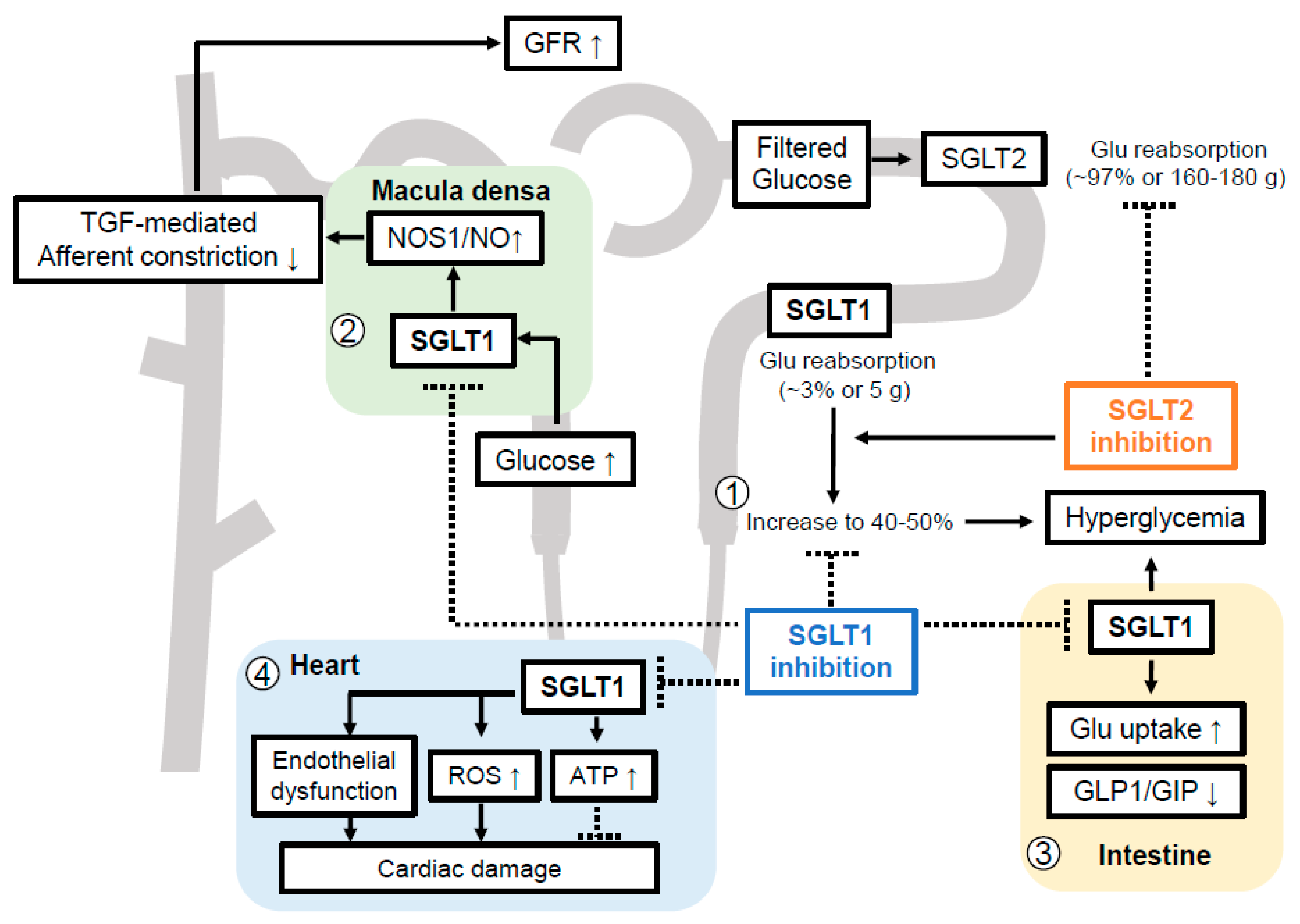
8. The Pathophysiological Basis for Inhibiting SGLT1 in DKD
8.1. Compensatory Glucose Uptake by Tubular SGLT1 When SGLT2 Is Inhibited
8.2. Macula Densa SGLT1-NOS1 Pathway Determines Glomerular Hyperfiltration and Blood Pressure Regulation in Diabetic Setting
8.3. Potential Roles of SGLT1 beyond Intestine and Kidney
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA Consensus Conference. Am. J. Kidney Dis. 2014, 64, 510–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papademetriou, V.; Lovato, L.; Doumas, M.; Nylen, E.; Mottl, A.; Cohen, R.M.; Applegate, W.B.; Puntakee, Z.; Yale, J.F.; Cushman, W.C.; et al. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int. 2015, 87, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef]
- Malek, V.; Suryavanshi, S.V.; Sharma, N.; Kulkarni, Y.A.; Mulay, S.R.; Gaikwad, A.B. Potential of Renin-Angiotensin-Aldosterone System Modulations in Diabetic Kidney Disease: Old Players to New Hope! Rev. Physiol. Biochem. Pharmacol. 2021, 179, 31–71. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Mimura, I.; Tanaka, T.; Nangaku, M. Treatment of Diabetic Kidney Disease: Current and Future. Diabetes Metab. J. 2021, 45, 11–26. [Google Scholar] [CrossRef]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C.; Cavender, M.A.; Fioretto, P.; Fowler, K.J.; Heerspink, H.J.L.; Manley, T.; McGuire, D.K.; Molitch, M.E.; Mottl, A.K.; et al. SGLT2 Inhibition for CKD and Cardiovascular Disease in Type 2 Diabetes: Report of a Scientific Workshop Sponsored by the National Kidney Foundation. Am. J. Kidney Dis. 2021, 77, 94–109. [Google Scholar] [CrossRef]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Almaimani, M.; Sridhar, V.S.; Cherney, D.Z.I. Sodium-glucose cotransporter 2 inhibition in non-diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2021, 30, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Huang, W.; Onishi, A.; Patel, R.; Kim, Y.; van Ginkel, C.; Fu, Y.; Freeman, B.; Koepsell, H.; Thomson, S.C.; et al. Knockout of Na-glucose-cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase-1 in macula densa and glomerular hyperfiltration. Am. J. Physiol.-Ren. Physiol. 2019, 317, F207–F217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wei, J.; Jiang, S.; Xu, L.; Wang, L.; Cheng, F.; Buggs, J.; Koepsell, H.; Vallon, V.; Liu, R. Macula densa SGLT1-NOS1-TGF pathway—A new mechanism for glomerular hyperfiltration during hyperglycemia. J. Am. Soc. Nephrol. 2019, 30, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Gyimesi, G.; Pujol-Gimenez, J.; Kanai, Y.; Hediger, M.A. Sodium-coupled glucose transport, the SLC5 family, and therapeutically relevant inhibitors: From molecular discovery to clinical application. Pflug. Arch. 2020, 472, 1177–1206. [Google Scholar] [CrossRef]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pflug. Arch. 2020, 472, 1345–1370. [Google Scholar] [CrossRef]
- Vallon, V.; Nakagawa, T. Renal Tubular Handling of Glucose and Fructose in Health and Disease. Compr. Physiol. 2021, 12, 2995–3044. [Google Scholar] [CrossRef]
- Song, P.; Onishi, A.; Koepsell, H.; Vallon, V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin. Ther. Targets 2016, 20, 1109–1125. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Madunic, I.V.; Breljak, D.; Karaica, D.; Koepsell, H.; Sabolic, I. Expression profiling and immunolocalization of Na(+)-D-glucose-cotransporter 1 in mice employing knockout mice as specificity control indicate novel locations and differences between mice and rats. Pflug. Arch. 2017, 469, 1545–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrhovac, I.; Balen, E.D.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radovic, N.; Jadrijevic, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflug. Arch. 2015, 467, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol.-Ren. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.; Calado, J. Familial renal glucosuria and SGLT2: From a Mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 2010, 5, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H. Glucose transporters in the small intestine in health and disease. Pflug. Arch. 2020, 472, 1207–1248. [Google Scholar] [CrossRef]
- Martin, M.G.; Turk, E.; Lostao, M.P.; Kerner, C.; Wright, E.M. Defects in Na+/glucose cotransporter (SGLT1) trafficking and function cause glucose-galactose malabsorption. Nat. Genet. 1996, 12, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hummel, C.S.; Lu, C.; Loo, D.D.; Hirayama, B.A.; Voss, A.A.; Wright, E.M. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am. J. Physiol.-Cell Physiol. 2011, 300, C14–C21. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Grahammer, F.; Volkl, H.; Sandu, C.D.; Richter, K.; Rexhepaj, R.; Gerlach, U.; Rong, Q.; Pfeifer, K.; Lang, F. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc. Natl. Acad. Sci. USA 2005, 102, 17864–17869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Grahammer, F.; Richter, K.; Bleich, M.; Lang, F.; Barhanin, J.; Volkl, H.; Warth, R. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J. Am. Soc. Nephrol. 2001, 12, 2003–2011. [Google Scholar] [CrossRef]
- FARBER, S.J.; BERGER, E.Y.; EARLE, D.P. Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J. Clin. Investig. 1951, 30, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liao, Y.; Wang, J.; Wen, X.; Shu, L. Potential impacts of diabetes mellitus and anti-diabetes agents on expressions of sodium-glucose transporters (SGLTs) in mice. Endocrine 2021, 74, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Ward, M.S.; Fotheringham, A.K.; Zhuang, A.; Borg, D.J.; Flemming, N.B.; Harvie, B.M.; Kinneally, T.L.; Yeh, S.M.; McCarthy, D.A.; et al. Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci. Rep. 2016, 6, 26428. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol.-Ren. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Moreira, R.; Muscelli, E. Effect of Insulin on Proximal Tubules Handling of Glucose: A Systematic Review. J. Diabetes Res. 2020, 2020, 8492467. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol. 2012, 74, 351–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, K.N.; Lo, C.S.; Zhao, S.; Liao, M.C.; Pang, Y.; Chang, S.Y.; Peng, J.; Kretzler, M.; Filep, J.G.; Ingelfinger, J.R.; et al. Angiotensin II up-regulates sodium-glucose co-transporter 2 expression and SGLT2 inhibitor attenuates Ang II-induced hypertensive renal injury in mice. Clin. Sci. (Lond.) 2021, 135, 943–961. [Google Scholar] [CrossRef] [PubMed]
- Takesue, H.; Hirota, T.; Tachimura, M.; Tokashiki, A.; Ieiri, I. Nucleosome Positioning and Gene Regulation of the SGLT2 Gene in the Renal Proximal Tubular Epithelial Cells. Mol. Pharmacol. 2018, 94, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Yu, J.; Chen, Z.; Tang, Y.; Dong, R.; Yang, Y.; Luo, J.; Hu, S.; Tu, L.; Xu, X. Epoxyeicosatrienoic acids improve glucose homeostasis by preventing NF-κB-mediated transcription of SGLT2 in renal tubular epithelial cells. Mol. Cell. Endocrinol. 2021, 523, 111149. [Google Scholar] [CrossRef] [PubMed]
- Beloto-Silva, O.; Machado, U.F.; Oliveira-Souza, M. Glucose-induced regulation of NHEs activity and SGLTs expression involves the PKA signaling pathway. J. Membr. Biol. 2011, 239, 157–165. [Google Scholar] [CrossRef]
- Miyata, K.N.; Lo, C.S.; Zhao, S.; Zhao, X.P.; Chenier, I.; Yamashita, M.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Deletion of heterogeneous nuclear ribonucleoprotein F in renal tubules downregulates SGLT2 expression and attenuates hyperfiltration and kidney injury in a mouse model of diabetes. Diabetologia 2021, 64, 2589–2601. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.S.; Miyata, K.N.; Zhao, S.; Ghosh, A.; Chang, S.Y.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Tubular Deficiency of Heterogeneous Nuclear Ribonucleoprotein F Elevates Systolic Blood Pressure and Induces Glycosuria in Mice. Sci. Rep. 2019, 9, 15765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lo, C.S.; Miyata, K.N.; Ghosh, A.; Zhao, X.P.; Chenier, I.; Cailhier, J.F.; Ethier, J.; Lattouf, J.B.; Filep, J.G.; et al. Overexpression of Nrf2 in Renal Proximal Tubular Cells Stimulates Sodium-Glucose Cotransporter 2 Expression and Exacerbates Dysglycemia and Kidney Injury in Diabetic Mice. Diabetes 2021, 70, 1388–1403. [Google Scholar] [CrossRef] [PubMed]
- Gembardt, F.; Bartaun, C.; Jarzebska, N.; Mayoux, E.; Todorov, V.T.; Hohenstein, B.; Hugo, C. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am. J. Physiol.-Ren. Physiol. 2014, 307, F317–F325. [Google Scholar] [CrossRef] [Green Version]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Expression is increased in Human Diabetic Nephropathy: SGLT2 Inhibition Decreases Renal Lipid Accumulation, Inflammation and the Development of Nephropathy in Diabetic Mice. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [Green Version]
- Solini, A.; Rossi, C.; Mazzanti, C.M.; Proietti, A.; Koepsell, H.; Ferrannini, E. Sodium-glucose co-transporter (SGLT)2 and SGLT1 renal expression in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1289–1294. [Google Scholar] [CrossRef]
- Srinivasan Sridhar, V.; Ambinathan, J.P.N.; Kretzler, M.; Pyle, L.L.; Bjornstad, P.; Eddy, S.; Cherney, D.Z.; Reich, H.N.; European Renal cDNA Bank (ERCB); Nephrotic Syndrome Study Network (NEPTUNE). Renal SGLT mRNA expression in human health and disease: A study in two cohorts. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1224–F1230. [Google Scholar] [CrossRef]
- Zapata-Morales, J.R.; Galicia-Cruz, O.G.; Franco, M.; Martinez, Y.; Morales, F. Hypoxia-inducible factor-1α (HIF-1α) protein diminishes sodium glucose transport 1 (SGLT1) and SGLT2 protein expression in renal epithelial tubular cells (LLC-PK1) under hypoxia. J. Biol. Chem. 2014, 289, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Höcherl, K.; Schweda, F.; Kurtz, A.; Bucher, M. Regulation of renal sodium transporters during severe inflammation. J. Am. Soc. Nephrol. 2007, 18, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Onishi, A.; Fu, Y.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Patel, R.; Kim, Y.C.; Nespoux, J.; Freeman, B.; et al. Effect of renal tubule-specific knockdown of the Na+/H+ exchanger NHE3 in Akita diabetic mice. Am. J. Physiol.-Ren. Physiol. 2019, 317, F419–F434. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Lachin, J.M.; Inzucchi, S.E. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2016, 374, 1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von, E.M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Jongs, N.; Chertow, G.M.; Langkilde, A.M.; McMurray, J.J.V.; Correa-Rotter, R.; Rossing, P.; Sjostrom, C.D.; Stefansson, B.V.; Toto, R.D.; et al. Effect of dapagliflozin on the rate of decline in kidney function in patients with chronic kidney disease with and without type 2 diabetes: A prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 2021, 9, 743–754. [Google Scholar] [CrossRef]
- Rhee, J.J.; Jardine, M.J.; Chertow, G.M.; Mahaffey, K.W. Dedicated kidney disease-focused outcome trials with sodium-glucose cotransporter-2 inhibitors: Lessons from CREDENCE and expectations from DAPA-HF, DAPA-CKD, and EMPA-KIDNEY. Diabetes Obes. Metab. 2020, 22 (Suppl. 1), 46–54. [Google Scholar] [CrossRef]
- Lytvyn, Y.; Skrtic, M.; Yang, G.K.; Yip, P.M.; Perkins, B.A.; Cherney, D.Z. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. Am. J. Physiol.-Ren. Physiol. 2015, 308, F77–F83. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van, G.C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol.-Ren. Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.I.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suijk, D.; van Baar, M.; van Bommel, E.; Iqbal, Z.; Krebber, M.; Vallon, V.; Touw, D.; Hoorn, E.; Nieuwdorp, M.; Kramer, M.; et al. SGLT2 Inhibition and Uric Acid Excretion in Patients with Type 2 Diabetes and Normal Kidney Function. Clin. J. Am. Soc. Nephrol. 2022, 17, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Muhlbauer, B.; Osswald, H. Adenosine and kidney function. Physiol. Rev. 2006, 86, 901–940. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Osswald, H. Dipyridamole prevents diabetes-induced alterations of kidney function in rats. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994, 349, 217–222. [Google Scholar] [CrossRef]
- Vallon, V.; Richter, K.; Blantz, R.C.; Thomson, S.; Osswald, H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J. Am. Soc. Nephrol. 1999, 10, 2569–2576. [Google Scholar] [CrossRef]
- Thomson, S.C.; Rieg, T.; Miracle, C.; Mansoury, H.; Whaley, J.; Vallon, V.; Singh, P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2012, 302, R75–R83. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.C.; Vallon, V. Effects of SGLT2 inhibitor and dietary NaCl on glomerular hemodynamics assessed by micropuncture in diabetic rats. Am. J. Physiol.-Ren. Physiol. 2021, 320, F761–F771. [Google Scholar] [CrossRef]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol.-Ren. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [Green Version]
- Kidokoro, K.; Cherney, D.Z.I.; Bozovic, A.; Nagasu, H.; Satoh, M.; Kanda, E.; Sasaki, T.; Kashihara, N. Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using In Vivo Imaging. Circulation 2019, 140, 303–315. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef] [Green Version]
- van Bommel, E.J.M.; Muskiet, M.H.A.; van Baar, M.J.B.; Tonneijck, L.; Smits, M.M.; Emanuel, A.L.; Bozovic, A.; Danser, A.H.J.; Geurts, F.; Hoorn, E.J.; et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 2020, 97, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.; Vijapurkar, U.; Meininger, G. Renal effects of canagliflozin in type 2 diabetes mellitus. Curr. Med. Res. Opin. 2015, 31, 2219–2231. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.; Desai, M.; Jardine, M.; Balis, D.; Meininger, G.; Perkovic, V. Canagliflozin Slows Progression of Renal Function Decline Independently of Glycemic Effects. J. Am. Soc. Nephrol. 2018, 28, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohan, D.E.; Fioretto, P.; Johnsson, K.; Parikh, S.; Ptaszynska, A.; Ying, L. The effect of dapagliflozin on renal function in patients with type 2 diabetes. J. Nephrol. 2016, 29, 391–400. [Google Scholar] [CrossRef]
- Yale, J.F.; Bakris, G.; Cariou, B.; Yue, D.; David-Neto, E.; Xi, L.; Figueroa, K.; Wajs, E.; Usiskin, K.; Meininger, G. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 2013, 15, 463–473. [Google Scholar] [CrossRef]
- Barnett, A.H.; Mithal, A.; Manassie, J.; Jones, R.; Rattunde, H.; Woerle, H.J.; Broedl, U.C. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 369–384. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Petrykiv, S.; Laverman, G.D.; Cherney, D.Z.; Gansevoort, R.T.; Heerspink, H.J.L. Effects of the SGLT-2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes. Metab. 2018, 20, 1988–1993. [Google Scholar] [CrossRef] [Green Version]
- Satirapoj, B.; Korkiatpitak, P.; Supasyndh, O. Effect of sodium-glucose cotransporter 2 inhibitor on proximal tubular function and injury in patients with type 2 diabetes: A randomized controlled trial. Clin. Kidney J. 2019, 12, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Thorpe, K.E. Acute kidney injury with sodium-glucose co-transporter-2 inhibitors: A meta-analysis of cardiovascular outcome trials. Diabetes Obes. Metab. 2019, 21, 1996–2000. [Google Scholar] [CrossRef]
- Vallon, V.; Komers, R. Pathophysiology of the diabetic kidney. Compr. Physiol. 2011, 1, 1175–1232. [Google Scholar] [PubMed]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Maxwell, P. HIF-1: An oxygen response system with special relevance to the kidney. J. Am. Soc. Nephrol. 2003, 14, 2712–2722. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, Y.; Tanaka, T.; Nangaku, M. Mechanisms of metabolic memory and renal hypoxia as a therapeutic target in diabetic kidney disease. J. Diabetes Investig. 2017, 8, 261–271. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.T.; Vallon, V.; Edwards, A. Modeling oxygen consumption in the proximal tubule: Effects of NHE and SGLT2 inhibition. Am. J. Physiol.-Ren. Physiol. 2015, 308, F1343–F1357. [Google Scholar] [CrossRef] [Green Version]
- Layton, A.T.; Vallon, V.; Edwards, A. Predicted Consequences of Diabetes and SGLT Inhibition on Transport and Oxygen Consumption along a Rat Nephron. Am. J. Physiol.-Ren. Physiol. 2016, 310, F1269–F1283. [Google Scholar] [CrossRef] [Green Version]
- Neill, O.; Fasching, A.; Pihl, L.; Patinha, D.; Franzen, S.; Palm, F. Acute SGLT inhibition normalizes oxygen tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol.-Ren. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef] [Green Version]
- Laursen, J.C.; Sondergaard-Heinrich, N.; de Melo, J.M.L.; Haddock, B.; Rasmussen, I.K.B.; Safavimanesh, F.; Hansen, C.S.; Storling, J.; Larsson, H.B.W.; Groop, P.H.; et al. Acute effects of dapagliflozin on renal oxygenation and perfusion in type 1 diabetes with albuminuria: A randomised, double-blind, placebo-controlled crossover trial. EClinicalMedicine 2021, 37, 100895. [Google Scholar] [CrossRef] [PubMed]
- Bessho, R.; Takiyama, Y.; Takiyama, T.; Kitsunai, H.; Takeda, Y.; Sakagami, H.; Ota, T. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci. Rep. 2019, 9, 14754. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef]
- Pruijm, M.; Milani, B.; Pivin, E.; Podhajska, A.; Vogt, B.; Stuber, M.; Burnier, M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.T.; Vallon, V. SGLT2 inhibition in a kidney with reduced nephron number: Modeling and analysis of solute transport and metabolism. Am. J. Physiol.-Ren. Physiol. 2018, 314, F969–F984. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, T.D.; Campos, L.C.; Carraro-Lacroix, L.; Girardi, A.C.; Malnic, G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J. Am. Soc. Nephrol. 2014, 25, 2028–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobulescu, I.A.; Moe, O.W. Luminal Na(+)/H(+) exchange in the proximal tubule. Pflug. Arch. 2009, 458, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Watanabe, Y.; Fukuda, K.; Watanabe, M.; Onishi, A.; Ohara, K.; Imai, T.; Koepsell, H.; Muto, S.; Vallon, V.; et al. Unmasking a sustained negative effect of SGLT2 inhibition on body fluid volume in the rat. Am. J. Physiol.-Ren. Physiol. 2018, 315, F653–F664. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na(+)/H(+) exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol.-Ren. Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Borges-Júnior, F.A.; Silva Dos Santos, D.; Benetti, A.; Polidoro, J.Z.; Wisnivesky, A.C.T.; Crajoinas, R.O.; Antônio, E.L.; Jensen, L.; Caramelli, B.; Malnic, G.; et al. Empagliflozin Inhibits Proximal Tubule NHE3 Activity, Preserves GFR, and Restores Euvolemia in Nondiabetic Rats with Induced Heart Failure. J. Am. Soc. Nephrol. 2021, 32, 1616–1629. [Google Scholar] [CrossRef]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 2011, 286, 13869–13878. [Google Scholar] [CrossRef] [Green Version]
- Mazer, C.D.; Hare, G.M.T.; Connelly, P.W.; Gilbert, R.E.; Shehata, N.; Quan, A.; Teoh, H.; Leiter, L.A.; Zinman, B.; Juni, P.; et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores and Red Blood Cell Morphology in Patients with Type 2 Diabetes and Coronary Artery Disease. Circulation 2019, 141, 704–707. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Hejna, J.; Green, K.; Batra, M.; Makdissi, A.; Chaudhuri, A.; Dandona, P. Dapagliflozin Suppresses Hepcidin And Increases Erythropoiesis. J. Clin. Endocrinol. Metab. 2020, 105, e1056–e1063. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018, 41, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Neal, B.; Perkovic, V.; de Zeeuw, D.; Neuen, B.L.; Arnott, C.; Simpson, R.; Oh, R.; Mahaffey, K.W.; Heerspink, H.J.L. Mediators of the effects of canagliflozin on kidney protection in patients with type 2 diabetes. Kidney Int. 2020, 98, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Woodward, M.; Perkovic, V.; Figtree, G.A.; Heerspink, H.J.L.; Mahaffey, K.W.; de Zeeuw, D.; Vercruysse, F.; Shaw, W.; Matthews, D.R.; et al. Mediators of the Effects of Canagliflozin on Heart Failure in Patients With Type 2 Diabetes. JACC Heart Fail. 2020, 8, 57–66. [Google Scholar] [CrossRef]
- Mulder, S.; Heerspink, H.J.L.; Darshi, M.; Kim, J.J.; Laverman, G.D.; Sharma, K.; Pena, M.J. Effects of dapagliflozin on urinary metabolites in people with type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 2422–2428. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Sugiura, Y.; Saito, H.; Sugahara, M.; Higashijima, Y.; Yamaguchi, J.; Inagi, R.; Suematsu, M.; Nangaku, M.; Tanaka, T. Sodium-glucose cotransporter 2 inhibition normalizes glucose metabolism and suppresses oxidative stress in the kidneys of diabetic mice. Kidney Int. 2018, 94, 912–925. [Google Scholar] [CrossRef]
- Litvinov, D.; Selvarajan, K.; Garelnabi, M.; Brophy, L.; Parthasarathy, S. Anti-atherosclerotic actions of azelaic acid, an end product of linoleic acid peroxidation, in mice. Atherosclerosis 2010, 209, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Thach, T.T.; Wu, C.; Hwang, K.Y.; Lee, S.J. Azelaic Acid Induces Mitochondrial Biogenesis in Skeletal Muscle by Activation of Olfactory Receptor 544. Front. Physiol. 2020, 11, 329. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Hwang, S.H.; Jia, Y.; Choi, J.; Kim, Y.J.; Choi, D.; Pathiraja, D.; Choi, I.G.; Koo, S.H.; Lee, S.J. Olfactory receptor 544 reduces adiposity by steering fuel preference toward fats. J. Clin. Investig. 2017, 127, 4118–4123. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Wu, J.; Sun, Z.; Yang, S.; Fu, J.; Fan, Y.; Wang, N.; Hu, J.; Ma, L.; Peng, C.; Wang, Z.; et al. Profiling of kidney transcriptome at the single-cell level reveals a distinct response of proximal tubular cells to SGLT2 inhibitor and angiotensin receptor blocker treatment in diabetic mice. Mol. Ther. 2021, 30, 1741–1753. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Gödel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mór, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwagata, S.; Kume, S.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Nakazawa, J.; Sugaya, T.; Koya, D.; Haneda, M.; Maegawa, H.; et al. MicroRNA148b-3p inhibits mTORC1-dependent apoptosis in diabetes by repressing TNFR2 in proximal tubular cells. Kidney Int. 2016, 90, 1211–1225. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Hinden, L.; Riahi, Y.; Israeli, T.; Tirosh, B.; Cerasi, E.; Mizrachi, E.B.; Tam, J.; Mosenzon, O.; Leibowitz, G. Proximal Tubule mTORC1 Is a Central Player in the Pathophysiology of Diabetic Nephropathy and Its Correction by SGLT2 Inhibitors. Cell Rep. 2020, 32, 107954. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A.; Shah, S.V. Autophagy Function and Regulation in Kidney Disease. Biomolecules 2020, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kitada, M.; Ogura, Y.; Liu, H.; Koya, D. Dapagliflozin Restores Impaired Autophagy and Suppresses Inflammation in High Glucose-Treated HK-2 Cells. Cells 2021, 10, 1457. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol.-Ren. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Satriano, J.; Mansoury, H.; Deng, A.; Sharma, K.; Vallon, V.; Blantz, R.C.; Thomson, S.C. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am. J. Physiol.-Cell Physiol. 2010, 299, C374–C380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R.; Ferrario, F.; Villaggio, B.; Gianiorio, F.; Tosetti, F.; Weiss, U.; et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol.-Ren. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, K.; Nakano, D.; Ohsaki, H.; Hitomi, H.; Minamino, T.; Yatabe, J.; Felder, R.A.; Mori, H.; Masaki, T.; Kobori, H.; et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J. Diabetes Its Complicat. 2014, 28, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Wu, X. Endoplasmic Reticulum Stress in Diabetic Nephrology: Regulation, Pathological Role, and Therapeutic Potential. Oxidative Med. Cell. Longev. 2021, 2021, 7277966. [Google Scholar] [CrossRef]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Rastaldi, M.P.; Ikehata, M.; Neusser, M.A.; Kretzler, M.; Cohen, C.D.; Schlöndorff, D. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J. Am. Soc. Nephrol. 2008, 19, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Jaikumkao, K.; Pongchaidecha, A.; Chueakula, N.; Thongnak, L.O.; Wanchai, K.; Chatsudthipong, V.; Chattipakorn, N.; Lungkaphin, A. Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes. Metab. 2018, 20, 2617–2626. [Google Scholar] [CrossRef]
- Shibusawa, R.; Yamada, E.; Okada, S.; Nakajima, Y.; Bastie, C.C.; Maeshima, A.; Kaira, K.; Yamada, M. Dapagliflozin rescues endoplasmic reticulum stress-mediated cell death. Sci. Rep. 2019, 9, 9887. [Google Scholar] [CrossRef]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef]
- Coca, S.G.; Nadkarni, G.N.; Huang, Y.; Moledina, D.G.; Rao, V.; Zhang, J.; Ferket, B.; Crowley, S.T.; Fried, L.F.; Parikh, C.R. Plasma Biomarkers and Kidney Function Decline in Early and Established Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2786–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, N.; Skupien, J.; Smiles, A.M.; Yamanouchi, M.; Niewczas, M.A.; Galecki, A.T.; Duffin, K.L.; Breyer, M.D.; Pullen, N.; Bonventre, J.V.; et al. Markers of early progressive renal decline in type 2 diabetes suggest different implications for etiological studies and prognostic tests development. Kidney Int. 2018, 93, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Hinokio, Y.; Suzuki, S.; Hirai, M.; Suzuki, C.; Suzuki, M.; Toyota, T. Urinary excretion of 8-oxo-7, 8-dihydro-2′-deoxyguanosine as a predictor of the development of diabetic nephropathy. Diabetologia 2002, 45, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Wu, Y.; Tian, M.; Sjöström, C.D.; Johansson, U.; Peng, X.R.; Smith, D.M.; Huang, Y. Dapagliflozin slows the progression of the renal and liver fibrosis associated with type 2 diabetes. Am. J. Physiol.-Endocrinol. Metab. 2017, 313, E563–E576. [Google Scholar] [CrossRef]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Murakoshi, M.; Ichikawa, S.; Koshida, T.; Adachi, E.; Suzuki, C.; Ueda, S.; Gohda, T.; Suzuki, Y. The sodium-glucose cotransporter 2 inhibitor tofogliflozin prevents diabetic kidney disease progression in type 2 diabetic mice. FEBS Open Bio. 2020, 10, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Parvez, F.; Rahman, M.M.; Khan, F.; Subhan, N.; Alam, M.A. Canagliflozin ameliorates renal oxidative stress and inflammation by stimulating AMPK-Akt-eNOS pathway in the isoprenaline-induced oxidative stress model. Sci. Rep. 2020, 10, 14659. [Google Scholar] [CrossRef]
- Yao, D.; Wang, S.; Wang, M.; Lu, W. Renoprotection of dapagliflozin in human renal proximal tubular cells via the inhibition of the high mobility group box 1-receptor for advanced glycation end products-nuclear factor-κB signaling pathway. Mol. Med. Rep. 2018, 18, 3625–3630. [Google Scholar] [CrossRef] [Green Version]
- Zaibi, N.; Li, P.; Xu, S.Z. Protective effects of dapagliflozin against oxidative stress-induced cell injury in human proximal tubular cells. PLoS ONE 2021, 16, e0247234. [Google Scholar] [CrossRef]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Bajaj, M.; Yang, H.C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’Antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Müller, G.A.; Colasanti, G.; D’Amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.H.; Kim, J.S.; Chang, J.W.; Kim, S.B.; Park, J.S.; Lee, S.K. AMP-activated protein kinase inhibits TGF-β-, angiotensin II-, aldosterone-, high glucose-, and albumin-induced epithelial-mesenchymal transition. Am. J. Physiol.-Ren. Physiol. 2013, 304, F686–F697. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhao, Y.; Wang, Q.; Hillebrands, J.L.; van den Born, J.; Ji, L.; An, T.; Qin, G. Dapagliflozin Attenuates Renal Tubulointerstitial Fibrosis Associated With Type 1 Diabetes by Regulating STAT1/TGFβ1 Signaling. Front. Endocrinol. (Lausanne) 2019, 10, 441. [Google Scholar] [CrossRef] [Green Version]
- Das, N.A.; Carpenter, A.J.; Belenchia, A.; Aroor, A.R.; Noda, M.; Siebenlist, U.; Chandrasekar, B.; DeMarco, V.G. Empagliflozin reduces high glucose-induced oxidative stress and miR-21-dependent TRAF3IP2 induction and RECK suppression, and inhibits human renal proximal tubular epithelial cell migration and epithelial-to-mesenchymal transition. Cell. Signal. 2020, 68, 109506. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Takagi, S.; Nitta, K.; Kitada, M.; Srivastava, S.P.; Takagaki, Y.; Kanasaki, K.; Koya, D. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight 2020, 5, e129034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.R.; DaCosta, C.M.; Gay, J.; Ding, Z.M.; Smith, M.; Greer, J.; Doree, D.; Jeter-Jones, S.; Mseeh, F.; Rodriguez, L.A.; et al. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am. J. Physiol.-Endocrinol. Metab. 2013, 304, E117–E130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Chan, O.; Ding, Y.; Zhu, W.; Mastaitis, J.; Sherwin, R. Reduction in SGLT1 mRNA Expression in the Ventromedial Hypothalamus Improves the Counterregulatory Responses to Hypoglycemia in Recurrently Hypoglycemic and Diabetic Rats. Diabetes 2015, 64, 3564–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danne, T.; Cariou, B.; Banks, P.; Brandle, M.; Brath, H.; Franek, E.; Kushner, J.A.; Lapuerta, P.; McGuire, D.K.; Peters, A.L.; et al. HbA1c and Hypoglycemia Reductions at 24 and 52 Weeks With Sotagliflozin in Combination With Insulin in Adults With Type 1 Diabetes: The European inTandem2 Study. Diabetes Care 2018, 41, 1981–1990. [Google Scholar] [CrossRef] [Green Version]
- Zambrowicz, B.; Freiman, J.; Brown, P.M.; Frazier, K.S.; Turnage, A.; Bronner, J.; Ruff, D.; Shadoan, M.; Banks, P.; Mseeh, F.; et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin. Pharmacol. Ther. 2012, 92, 158–169. [Google Scholar] [CrossRef]
- Dobbins, R.L.; Greenway, F.L.; Chen, L.; Liu, Y.; Breed, S.L.; Andrews, S.M.; Wald, J.A.; Walker, A.; Smith, C.D. Selective sodium-dependent glucose transporter 1 inhibitors block glucose absorption and impair glucose-dependent insulinotropic peptide release. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G946–G954. [Google Scholar] [CrossRef]
- Gorboulev, V.; Schurmann, A.; Vallon, V.; Kipp, H.; Jaschke, A.; Klessen, D.; Friedrich, A.; Scherneck, S.; Rieg, T.; Cunard, R.; et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 2012, 61, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Moriya, R.; Shirakura, T.; Ito, J.; Mashiko, S.; Seo, T. Activation of sodium-glucose cotransporter 1 ameliorates hyperglycemia by mediating incretin secretion in mice. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E1358–E1365. [Google Scholar] [CrossRef] [Green Version]
- Alicic, R.Z.; Cox, E.J.; Neumiller, J.J.; Tuttle, K.R. Incretin drugs in diabetic kidney disease: Biological mechanisms and clinical evidence. Nat. Rev. Nephrol. 2021, 17, 227–244. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Welch, W.J.; Murad, F.; Gross, S.S.; Taylor, G.; Levi, R.; Schmidt, H.H. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc. Natl. Acad. Sci. USA 1992, 89, 11993–11997. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Thomson, S. Inhibition of local nitric oxide synthase increases homeostatic efficiency of tubuloglomerular feedback. Am. J. Physiol. 1995, 269, F892–F899. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Carretero, O.A.; Ren, Y.; Garvin, J.L. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int. 2005, 67, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Traynor, T.; Barajas, L.; Huang, Y.G.; Briggs, J.P.; Schnermann, J. Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J. Am. Soc. Nephrol. 2001, 12, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Komers, R.; Lindsley, J.N.; Oyama, T.T.; Allison, K.M.; Anderson, S. Role of neuronal nitric oxide synthase (NOS1) in the pathogenesis of renal hemodynamic changes in diabetes. Am. J. Physiol.-Ren. Physiol. 2000, 279, F573–F583. [Google Scholar] [CrossRef]
- Tolins, J.P.; Shultz, P.J.; Raij, L.; Brown, D.M.; Mauer, S.M. Abnormal renal hemodynamic response to reduced renal perfusion pressure in diabetic rats: Role of NO. Am. J. Physiol. 1993, 265, F886–F895. [Google Scholar] [CrossRef]
- Thomson, S.C.; Deng, A.; Komine, N.; Hammes, J.S.; Blantz, R.C.; Gabbai, F.B. Early diabetes as a model for testing the regulation of juxtaglomerular NOS I. Am. J. Physiol.-Ren. Physiol. 2004, 287, F732–F738. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Cui, Y.; Jiang, S.; Wei, J.; Chan, J.; Thalakola, A.; Le, T.; Xu, L.; Zhao, L.; et al. Knockout of Macula Densa Neuronal Nitric Oxide Synthase Increases Blood Pressure in db/db Mice. Hypertension 2021, 78, 1760–1770. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, J.; Cui, Y.; Jiang, S.; Wei, J.; Kim, Y.C.; Chan, J.; Thalakola, A.; Le, T.; Xu, L.; et al. Role of the macula densa sodium glucose cotransporter type 1-neuronal nitric oxide synthase-tubuloglomerular feedback pathway in diabetic hyperfiltration. Kidney Int. 2022, 101, 541–550. [Google Scholar] [CrossRef]
- Guyton, A.C. Blood pressure control—Special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef]
- Nespoux, J.; Patel, R.; Hudkins, K.L.; Huang, W.; Freeman, B.; Kim, Y.; Koepsell, H.; Alpers, C.E.; Vallon, V. Gene deletion of the Na-glucose cotransporter SGLT1 ameliorates kidney recovery in a murine model of acute kidney injury induced by ischemia-reperfusion. Am. J. Physiol.-Ren. Physiol. 2019, 316, F1201–F1210. [Google Scholar] [CrossRef]
- Li, Z.; Agrawal, V.; Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Sincoular, A.; Jakubiak, M.; Music, M.L.; Kutschke, W.J.; Huang, X.N.; et al. Cardiac Sodium-Glucose Co-Transporter 1 (SGLT1) is a Novel Mediator of Ischemia/Reperfusion Injury. Cardiovasc. Res. 2019, 115, 1646–1658. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Sands, A.T.; Zambrowicz, B.P.; Rosenstock, J.; Lapuerta, P.; Bode, B.W.; Garg, S.K.; Buse, J.B.; Banks, P.; Heptulla, R.; Rendell, M.; et al. Sotagliflozin, a Dual SGLT1 and SGLT2 Inhibitor, as Adjunct Therapy to Insulin in Type 1 Diabetes. Diabetes Care 2015, 38, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.K.; Henry, R.R.; Banks, P.; Buse, J.B.; Davies, M.J.; Fulcher, G.R.; Pozzilli, P.; Gesty-Palmer, D.; Lapuerta, P.; Simo, R.; et al. Effects of Sotagliflozin Added to Insulin in Patients with Type 1 Diabetes. N. Engl. J. Med. 2017, 377, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mathieu, C.; Phillip, M.; Hansen, L.; Griffen, S.C.; Tschope, D.; Thoren, F.; Xu, J.; Langkilde, A.M.; DEPICT-1 Investigators. Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes (DEPICT-1): 24 week results from a multicentre, double-blind, phase 3, randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 864–876. [Google Scholar] [CrossRef]
- Henry, R.R.; Thakkar, P.; Tong, C.; Polidori, D.; Alba, M. Efficacy and Safety of Canagliflozin, a Sodium-Glucose Cotransporter 2 Inhibitor, as Add-on to Insulin in Patients with Type 1 Diabetes. Diabetes Care 2015, 38, 2258–2265. [Google Scholar] [CrossRef] [Green Version]
- Fattah, H.; Vallon, V. The Potential Role of SGLT2 Inhibitors in the Treatment of Type 1 Diabetes Mellitus. Drugs 2018, 78, 717–726. [Google Scholar] [CrossRef]
- Pieber, T.R.; Famulla, S.; Eilbracht, J.; Cescutti, J.; Soleymanlou, N.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Kaspers, S. Empagliflozin as adjunct to insulin in patients with type 1 diabetes: A 4-week, randomized, placebo-controlled trial (EASE-1). Diabetes Obes. Metab. 2015, 17, 928–935. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oe, Y.; Vallon, V. The Pathophysiological Basis of Diabetic Kidney Protection by Inhibition of SGLT2 and SGLT1. Kidney Dial. 2022, 2, 349-368. https://doi.org/10.3390/kidneydial2020032
Oe Y, Vallon V. The Pathophysiological Basis of Diabetic Kidney Protection by Inhibition of SGLT2 and SGLT1. Kidney and Dialysis. 2022; 2(2):349-368. https://doi.org/10.3390/kidneydial2020032
Chicago/Turabian StyleOe, Yuji, and Volker Vallon. 2022. "The Pathophysiological Basis of Diabetic Kidney Protection by Inhibition of SGLT2 and SGLT1" Kidney and Dialysis 2, no. 2: 349-368. https://doi.org/10.3390/kidneydial2020032
APA StyleOe, Y., & Vallon, V. (2022). The Pathophysiological Basis of Diabetic Kidney Protection by Inhibition of SGLT2 and SGLT1. Kidney and Dialysis, 2(2), 349-368. https://doi.org/10.3390/kidneydial2020032