
Obesity, Weight Loss, Lifestyle Interventions, and Autosomal Dominant Polycystic Kidney Disease


Abstract
1. Introduction
2. Epidemiological Data on BMI and Kidney Disease
2.1. General Population
2.2. Chronic Kidney Disease
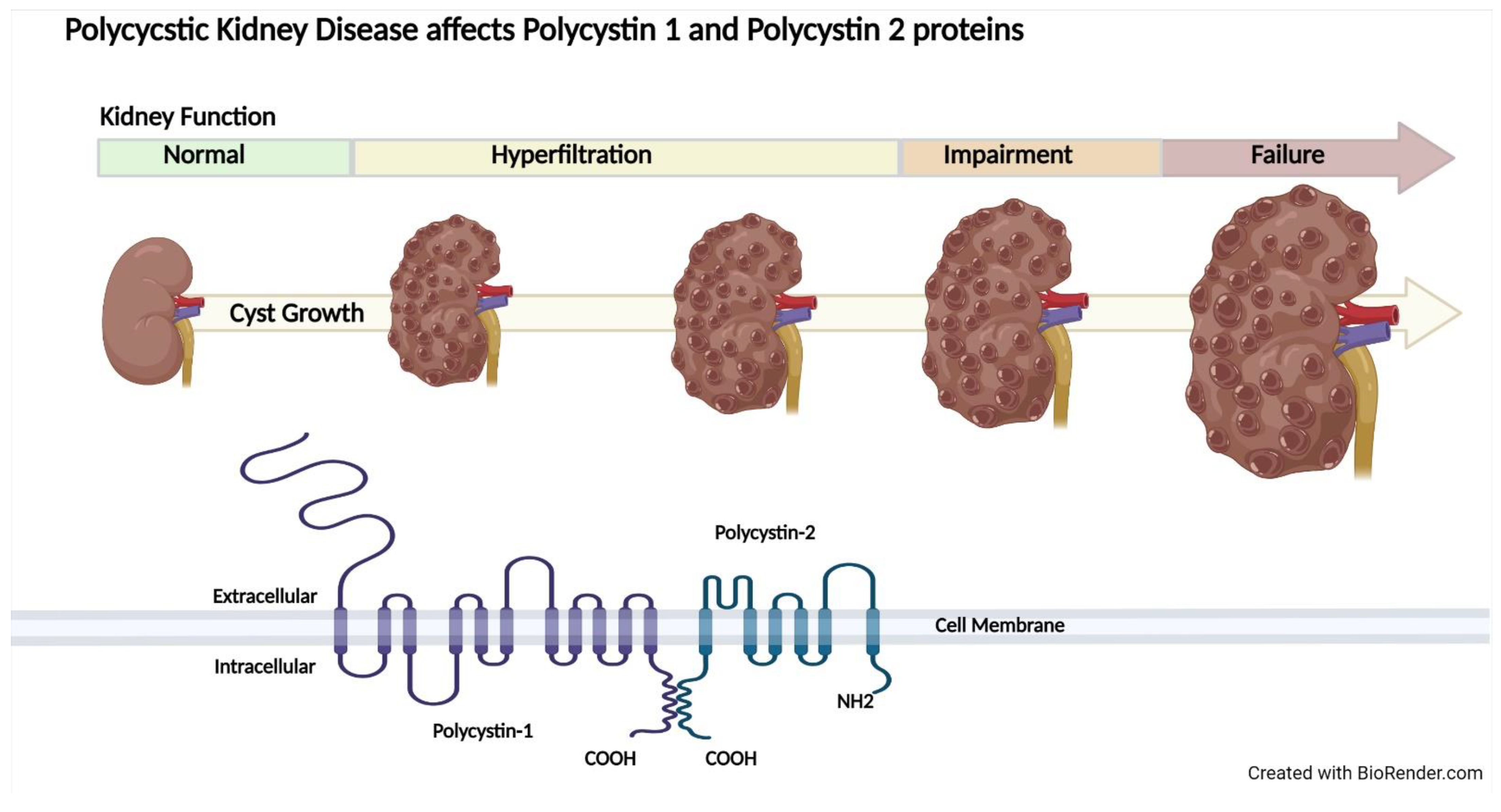
2.3. Autosomal Dominant Polycystic Kidney Disease
3. Potential Role of Adipose Tissue
3.1. Types and Distribution of Adipose Tissue
3.2. Harmful Effects of Adipose Tissue
4. Pathways Relevant to Obesity and ADPKD
5. Impaired Fatty Oxidation
6. Weight Loss and Kidney Function
6.1. General Population and Other Disease Profiles
6.2. Chronic Kidney Disease
6.3. Autosomal Dominant Polycystic Kidney Disease
7. Weight Loss Interventions and Kidney Function in ADPKD
7.1. Physical Activity Interventions
7.2. Dietary Interventions
8. Novel Future Direction and Clinical Implications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fruh, S.M. Obesity: Risk factors, complications, and strategies for sustainable long-term weight management. J. Am. Assoc. Nurse Pract. 2017, 29, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef]
- Stevens, G.A.; Singh, G.M.; Lu, Y.; Danaei, G.; Lin, J.K.; Finucane, M.M.; Bahalim, A.N.; McIntire, R.K.; Gutierrez, H.R.; Cowan, M.; et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul. Health Metr. 2012, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Baskin, M.L.; Ard, J.; Franklin, F.; Allison, D.B. Prevalence of obesity in the United States. Obes. Rev. 2005, 6, 5–7. [Google Scholar] [CrossRef]
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Curtin, L.R. Prevalence and Trends in Obesity Among US Adults, 1999–2008. J. Am. Med. Assoc. 2010, 303, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Valderas, J.M.; Starfield, B.; Sibbald, B.; Salisbury, C.; Roland, M. Defining comorbidity: Implications for understanding health and health services. Ann. Fam. Med. 2009, 7, 357–363. [Google Scholar] [CrossRef]
- Chalmers, L.; Kaskel, F.J.; Bamgbola, O. The Role of Obesity and Its Bioclinical Correlates in the Progression of Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2006, 13, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Brands, M.W.; Dixon, W.N.; Smith, M.J., Jr. Obesity-induced hypertension. Renal function and systemic hemodynamics. Hypertension 1993, 22, 292–299. [Google Scholar] [CrossRef]
- Hall, J.E.; Crook, E.D.; Jones, D.W.; Wofford, M.R.; Dubbert, P.M. Mechanisms of obesity-associated cardiovascular and renal disease. Am. J. Med. Sci. 2002, 324, 127–137. [Google Scholar] [CrossRef]
- Vaes, B.; Beke, E.; Truyers, C.; Elli, S.; Buntinx, F.; Verbakel, J.Y.; Goderis, G.; Van Pottelbergh, G. The correlation between blood pressure and kidney function decline in older people: A registry-based cohort study. British Med. J. Open. 2015, 5, e007571. [Google Scholar] [CrossRef]
- Hall, M.E.; do Carmo, J.M.; da Silva, A.A.; Juncos, L.A.; Wang, Z.; Hall, J.E. Obesity, hypertension, and chronic kidney disease. Int. J. Nephrol. Renov. Dis. 2014, 7, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.E.; Beske, S.D.; Ballard, T.P.; Davy, K.P. Sympathetic Neural Activation in Visceral Obesity. Circ. 2002, 106, 2533–2536. [Google Scholar] [CrossRef] [PubMed]
- Esler, M.; Rumantir, M.; Wiesner, G.; Kaye, D.; Hastings, J.; Lambert, G. Sympathetic nervous system and insulin resistance: From obesity to diabetes. Am. J. Hypertens. 2001, 14, 304S–309S. [Google Scholar] [CrossRef]
- Abate, N.I.; Mansour, Y.H.; Tuncel, M.; Arbique, D.; Chavoshan, B.; Kizilbash, A.; Howell-Stampley, T.; Vongpatanasin, W.; Victor, R.G. Overweight and sympathetic overactivity in black Americans. Hypertens. 2001, 38, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, G.L.; Sugerman, H.J.; Blocher, C.R.; Gehr, T.W.; Sica, D.A. Chronically increased intra-abdominal pressure produces systemic hypertension in dogs. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 819–824. [Google Scholar] [CrossRef]
- Dwyer, T.M.; Banks, S.A.; Alonso-Galicia, M.; Cockrell, K.; Carroll, J.F.; Bigler, S.A.; Hall, J.E. Distribution of renal medullary hyaluronan in lean and obese rabbits. Kidney Int. 2000, 58, 721–729. [Google Scholar] [CrossRef]
- Hall, J.E.; Henegar, J.R.; Dwyer, T.M.; Liu, J.; da Silva, A.A.; Kuo, J.J.; Tallam, L. Is obesity a major cause of chronic kidney disease? Adv. Ren. Replace. Ther. 2004, 11, 41–54. [Google Scholar] [CrossRef]
- Fox, C.S.; Larson, M.G.; Leip, E.P.; Culleton, B.; Wilson, P.W.; Levy, D. Predictors of new-onset kidney disease in a community-based population. J. Amer. Med. Assoc. 2004, 291, 844–850. [Google Scholar] [CrossRef]
- Iseki, K.; Ikemiya, Y.; Kinjo, K.; Inoue, T.; Iseki, C.; Takishita, S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004, 65, 1870–1876. [Google Scholar] [CrossRef]
- Hsu, C.Y.; McCulloch, C.E.; Iribarren, C.; Darbinian, J.; Go, A.S. Body mass index and risk for end-stage renal disease. Ann. Intern. Med. 2006, 144, 21–28. [Google Scholar] [CrossRef]
- Tanner, R.M.; Brown, T.M.; Muntner, P. Epidemiology of Obesity, the Metabolic Syndrome, and Chronic Kidney Disease. Curr. Hypertens. Rep. 2012, 14, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.J.; Saranathan, A.; Luke, A.; Durazo-Arvizu, R.A.; Guichan, C.; Hou, S.; Cooper, R. Increasing body mass index and obesity in the incident ESRD population. J. Am. Soc. Nephrol. 2006, 17, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Chan, S.; Cameron, A.; Healy, H.G.; Venuthurupalli, S.K.; Tan, K.-S.; Hoy, W.E. BMI and its association with death and the initiation of renal replacement therapy (RRT) in a cohort of patients with chronic kidney disease (CKD). BMC Nephrol. 2019, 20, 329. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers. 2018, 4, 50. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abebe, K.Z.; Perrone, R.D.; Torres, V.E.; Braun, W.E.; Steinman, T.I.; Winklhofer, F.T.; Brosnahan, G.; Czarnecki, P.G.; Hogan, M.C.; et al. Blood Pressure in Early Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2014, 371, 2255–2266. [Google Scholar] [CrossRef]
- Nowak, K.L.; Steele, C.; Gitomer, B.; Wang, W.; Ouyang, J.; Chonchol, M.B. Overweight and Obesity and Progression of ADPKD. Clin. J. Am. Soc. Nephrol. 2021, 16, 908–915. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Song, Y.; Caballero, B.; Cheskin, L.J. Association between obesity and kidney disease: A systematic review and meta-analysis. Kidney Int. 2008, 73, 19–33. [Google Scholar] [CrossRef]
- Pinto-Sietsma, S.-J.; Navis, G.; Janssen, W.M.T.; de Zeeuw, D.; Gans, R.O.B.; de Jong, P.E. A central body fat distribution is related to renal function impairment, even in lean subjects. Am. J. Kidney Dis. 2003, 41, 733–741. [Google Scholar] [CrossRef]
- Chang, A.; Van Horn, L.; Jacobs, D.R., Jr.; Liu, K.; Muntner, P.; Newsome, B.; Shoham, D.A.; Durazo-Arvizu, R.; Bibbins-Domingo, K.; Reis, J.; et al. Lifestyle-related factors, obesity, and incident microalbuminuria: The CARDIA (Coronary Artery Risk Development in Young Adults) study. Am. J. Kidney Dis. 2013, 62, 267–275. [Google Scholar] [CrossRef]
- Thoenes, M.; Reil, J.C.; Khan, B.V.; Bramlage, P.; Volpe, M.; Kirch, W.; Böhm, M. Abdominal obesity is associated with microalbuminuria and an elevated cardiovascular risk profile in patients with hypertension. Vasc. Health Risk Manag. 2009, 5, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Hwang, S.J.; Massaro, J.M.; Hoffmann, U.; DeBoer, I.H.; Robins, S.J.; Vasan, R.S.; Fox, C.S. Association of subcutaneous and visceral adiposity with albuminuria: The Framingham Heart Study. Obesity 2011, 19, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Gelber, R.P.; Kurth, T.; Kausz, A.T.; Manson, J.E.; Buring, J.E.; Levey, A.S.; Gaziano, J.M. Association between body mass index and CKD in apparently healthy men. Am. J. Kidney Dis. 2005, 46, 871–880. [Google Scholar] [CrossRef]
- Kramer, H.; Luke, A.; Bidani, A.; Cao, G.; Cooper, R.; McGee, D. Obesity and prevalent and incident CKD: The Hypertension Detection and Follow-Up Program. Am. J. Kidney Dis. 2005, 46, 587–594. [Google Scholar] [CrossRef]
- de Boer, I.H.; Katz, R.; Fried, L.F.; Ix, J.H.; Luchsinger, J.; Sarnak, M.J.; Shlipak, M.G.; Siscovick, D.S.; Kestenbaum, B. Obesity and change in estimated GFR among older adults. Am. J. Kidney Dis. 2009, 54, 1043–1051. [Google Scholar] [CrossRef]
- Lu, J.L.; Molnar, M.Z.; Naseer, A.; Mikkelsen, M.K.; Kalantar-Zadeh, K.; Kovesdy, C.P. Association of age and BMI with kidney function and mortality: A cohort study. Lancet Diabetes Endocrinol. 2015, 3, 704–714. [Google Scholar] [CrossRef]
- Vivante, A.; Golan, E.; Tzur, D.; Leiba, A.; Tirosh, A.; Skorecki, K.; Calderon-Margalit, R. Body mass index in 1.2 million adolescents and risk for end-stage renal disease. Arch. Intern. Med. 2012, 172, 1644–1650. [Google Scholar] [CrossRef]
- Bray, G.A. Overweight is risking fate. Definition, classification, prevalence, and risks. Ann. N. Y. Acad. Sci. 1987, 499, 14–28. [Google Scholar] [CrossRef]
- Chang, T.-J.; Zheng, C.-M.; Wu, M.-Y.; Chen, T.-T.; Wu, Y.-C.; Wu, Y.-L.; Lin, H.-T.; Zheng, J.-Q.; Chu, N.-F.; Lin, Y.-M.; et al. Relationship between body mass index and renal function deterioration among the Taiwanese chronic kidney disease population. Sci. Rep. 2018, 8, 6908. [Google Scholar] [CrossRef]
- Herrington, W.G.; Smith, M.; Bankhead, C.; Matsushita, K.; Stevens, S.; Holt, T.; Hobbs, F.D.R.; Coresh, J.; Woodward, M. Body-mass index and risk of advanced chronic kidney disease: Prospective analyses from a primary care cohort of 1.4 million adults in England. PLoS ONE 2017, 12, e0173515. [Google Scholar] [CrossRef]
- Iseki, K. Body mass index and the risk of chronic renal failure: The Asian experience. Contrib. Nephrol. 2006, 151, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Leng, C.; Chia, K.S.; Koh, D.; Tai, E.S.; Saw, S.M.; Lim, S.C.; Wong, T.Y. Association between body mass index and chronic kidney disease in men and women: Population-based study of Malay adults in Singapore. Nephrol. Dial. Transplant. 2008, 23, 1910–1918. [Google Scholar] [CrossRef]
- Horber, F.F.; Gruber, B.; Thomi, F.; Jensen, E.X.; Jaeger, P. Effect of sex and age on bone mass, body composition and fuel metabolism in humans. Nutrition 1997, 13, 524–534. [Google Scholar] [CrossRef]
- Kuk, J.L.; Lee, S.; Heymsfield, S.B.; Ross, R. Waist circumference and abdominal adipose tissue distribution: Influence of age and sex. Am. J. Clin. Nutr. 2005, 81, 1330–1334. [Google Scholar] [CrossRef]
- Othman, M.; Kawar, B.; El Nahas, A.M. Influence of obesity on progression of non-diabetic chronic kidney disease: A retrospective cohort study. Nephron. Clin. Pract. 2009, 113, c16–c23. [Google Scholar] [CrossRef] [PubMed]
- MacLaughlin, H.L.; Pike, M.; Selby, N.M.; Siew, E.; Chinchilli, V.M.; Guide, A.; Stewart, T.G.; Himmelfarb, J.; Go, A.S.; Parikh, C.R.; et al. Body mass index and chronic kidney disease outcomes after acute kidney injury: A prospective matched cohort study. BMC Nephrol. 2021, 22, 200. [Google Scholar] [CrossRef]
- Khedr, A.; Khedr, E.; House, A.A. Body mass index and the risk of progression of chronic kidney disease. J. Ren. Nutr. 2011, 21, 455–461. [Google Scholar] [CrossRef]
- Mohsen, A.; Brown, R.; Hoefield, R.; Kalra, P.A.; O’Donoghue, D.; Middleton, R.; New, D. Body mass index has no effect on rate of progression of chronic kidney disease in subjects with type 2 diabetes mellitus. J. Nephrol. 2012, 25, 384–393. [Google Scholar] [CrossRef]
- Brown, R.N.K.L.; Mohsen, A.; Green, D.; Hoefield, R.A.; Summers, L.K.M.; Middleton, R.J.; O’Donoghue, D.J.; Kalra, P.A.; New, D.I. Body mass index has no effect on rate of progression of chronic kidney disease in non-diabetic subjects. Nephrol. Dial. Transplant. 2012, 27, 2776–2780. [Google Scholar] [CrossRef]
- Lowrie, E.G.; Lew, N.L. Death risk in hemodialysis patients: The predictive value of commonly measured variables and an evaluation of death rate differences between facilities. Am. J. Kidney Dis. 1990, 15, 458–482. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Block, G.; Humphreys, M.H.; Kopple, J.D. Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int. 2003, 63, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Block, G.; Horwich, T.; Fonarow, G.C. Reverse epidemiology of conventional cardiovascular risk factors in patients with chronic heart failure. J. Am. Coll. Cardiol. 2004, 43, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Leavey, S.F.; McCullough, K.; Hecking, E.; Goodkin, D.; Port, F.K.; Young, E.W. Body mass index and mortality in ‘healthier’ as compared with ‘sicker’ haemodialysis patients: Results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol. Dial. Transplant. 2001, 16, 2386–2394. [Google Scholar] [CrossRef]
- Molnar, M.Z.; Streja, E.; Kovesdy, C.P.; Bunnapradist, S.; Sampaio, M.S.; Jing, J.; Krishnan, M.; Nissenson, A.R.; Danovitch, G.M.; Kalantar-Zadeh, K. Associations of body mass index and weight loss with mortality in transplant-waitlisted maintenance hemodialysis patients. Am. J. Transplant. 2011, 11, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Horwich, T.B.; Fonarow, G.C.; Hamilton, M.A.; MacLellan, W.R.; Woo, M.A.; Tillisch, J.H. The relationship between obesity and mortality in patients with heart failure. J. Am. Coll. Cardiol. 2001, 38, 789–795. [Google Scholar] [CrossRef]
- Fung, F.; Sherrard, D.J.; Gillen, D.L.; Wong, C.; Kestenbaum, B.; Seliger, S.; Ball, A.; Stehman-Breen, C. Increased risk for cardiovascular mortality among malnourished end-stage renal disease patients. Am. J. Kidney Dis. 2002, 40, 307–314. [Google Scholar] [CrossRef]
- Lee, M.J.; Park, J.T.; Park, K.S.; Kwon, Y.E.; Han, S.H.; Kang, S.-W.; Choi, K.H.; Oh, K.-H.; Park, S.K.; Chae, D.W.; et al. Normal body mass index with central obesity has increased risk of coronary artery calcification in Korean patients with chronic kidney disease. Kidney Int. 2016, 90, 1368–1376. [Google Scholar] [CrossRef]
- Grantham, J.J.; Torres, V.E.; Chapman, A.B.; Guay-Woodford, L.M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Baumgarten, D.A.; Kenney, P.J.; Harris, P.C.; et al. Volume Progression in Polycystic Kidney Disease. N. Engl. J. Med. 2006, 354, 2122–2130. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Rangel, L.J.; Bergstralh, E.J.; Osborn, S.L.; Harmon, A.J.; Sundsbak, J.L.; Bae, K.T.; Chapman, A.B.; Grantham, J.J.; Mrug, M.; et al. Imaging Classification of Autosomal Dominant Polycystic Kidney Disease: A Simple Model for Selecting Patients for Clinical Trials. J. Am. Soc. Nephrol. 2015, 26, 160–172. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Bae, K.T.; Abebe, K.Z.; Bost, J.E.; Miskulin, D.C.; Steinman, T.I.; Braun, W.E.; Winklhofer, F.T.; et al. Analysis of baseline parameters in the HALT polycystic kidney disease trials. Kidney Int. 2012, 81, 577–585. [Google Scholar] [CrossRef]
- Nowak, K.L.; You, Z.; Gitomer, B.; Brosnahan, G.; Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Steinman, T.I.; Abebe, K.Z.; Rahbari-Oskoui, F.F.; et al. Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Freise, J.; Tavakol, M.; Gao, Y.; Klein, O.; Lee, B.K.; Freise, C.; Park, M. The Effect of Enlarged Kidneys on Calculated Body Mass Index Categorization in Transplant Recipients With ADPKD. Kidney Int. Rep. 2019, 4, 606–609. [Google Scholar] [CrossRef]
- Barberio, A.M.; Alareeki, A.; Viner, B.; Pader, J.; Vena, J.E.; Arora, P.; Friedenreich, C.M.; Brenner, D.R. Central body fatness is a stronger predictor of cancer risk than overall body size. Nat. Commun. 2019, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, F.X. The epidemiology of central fat distribution in relation to disease. Nutr. Rev. 2004, 62, S120–S126. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Hopp, K. Metabolic Reprogramming in Autosomal Dominant Polycystic Kidney Disease: Evidence and Therapeutic Potential. Clin. J. Am. Soc. Nephrol. 2020, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Shuster, A.; Patlas, M.; Pinthus, J.H.; Mourtzakis, M. The clinical importance of visceral adiposity: A critical review of methods for visceral adipose tissue analysis. Br. J. Radiol. 2012, 85, 1–10. [Google Scholar] [CrossRef]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef]
- Young, J.A.; Hwang, S.-J.; Sarnak, M.J.; Hoffmann, U.; Massaro, J.M.; Levy, D.; Benjamin, E.J.; Larson, M.G.; Vasan, R.S.; O’Donnell, C.J.; et al. Association of visceral and subcutaneous adiposity with kidney function. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 1786–1791. [Google Scholar] [CrossRef]
- Reddy, P.; Lent-Schochet, D.; Ramakrishnan, N.; McLaughlin, M.; Jialal, I. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hao, J.; Tarrago, M.G.; Warner, G.M.; Giorgadze, N.; Wei, Q.; Huang, Y.; He, K.; Chen, C.; Peclat, T.R.; et al. FBF1 deficiency promotes beiging and healthy expansion of white adipose tissue. Cell Rep. 2021, 36, 109481. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Farias, M.; Fos-Domenech, J.; Serra, D.; Herrero, L.; Sánchez-Infantes, D. White adipose tissue dysfunction in obesity and aging. Biochem. Pharmacol. 2021, 192, 114723. [Google Scholar] [CrossRef]
- Okhunov, Z.; Mues, A.C.; Kline, M.; Haramis, G.; Xu, B.; Mirabile, G.; Vira, M.; Landman, J. Evaluation of perirenal fat as a predictor of cT 1a renal cortical neoplasm histopathology and surgical outcomes. J. Endourol. 2012, 26, 911–916. [Google Scholar] [CrossRef]
- Foster, M.C.; Hwang, S.J.; Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Fox, C.S. Fatty kidney, hypertension, and chronic kidney disease: The Framingham Heart Study. Hypertens 2011, 58, 784–790. [Google Scholar] [CrossRef]
- Wei, G.; Sun, H.; Dong, K.; Hu, L.; Wang, Q.; Zhuang, Q.; Zhu, Y.; Zhang, X.; Shao, Y.; Tang, H.; et al. The thermogenic activity of adjacent adipocytes fuels the progression of ccRCC and compromises anti-tumor therapeutic efficacy. Cell Metab. 2021, 33, 2021–2039.e8. [Google Scholar] [CrossRef]
- Ahima, R.S.; Lazar, M.A. Adipokines and the peripheral and neural control of energy balance. Mol. Endocrinol. 2008, 22, 1023–1031. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- de Souza Batista, C.M.; Yang, R.Z.; Lee, M.J.; Glynn, N.M.; Yu, D.Z.; Pray, J.; Ndubuizu, K.; Patil, S.; Schwartz, A.; Kligman, M.; et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes 2007, 56, 1655–1661. [Google Scholar] [CrossRef]
- Samaras, K.; Botelho, N.K.; Chisholm, D.J.; Lord, R.V. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity 2010, 18, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Degawa-Yamauchi, M.; Bovenkerk, J.E.; Juliar, B.E.; Watson, W.; Kerr, K.; Jones, R.; Zhu, Q.; Considine, R.V. Serum resistin (FIZZ3) protein is increased in obese humans. J. Clin. Endocrinol. Metab. 2003, 88, 5452–5455. [Google Scholar] [CrossRef] [PubMed]
- McTernan, P.G.; McTernan, C.L.; Chetty, R.; Jenner, K.; Fisher, F.M.; Lauer, M.N.; Crocker, J.; Barnett, A.H.; Kumar, S. Increased resistin gene and protein expression in human abdominal adipose tissue. J. Clin. Endocrinol. Metab. 2002, 87, 2407–2410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yeung, D.C.; Karpisek, M.; Stejskal, D.; Zhou, Z.G.; Liu, F.; Wong, R.L.; Chow, W.S.; Tso, A.W.; Lam, K.S.; et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008, 57, 1246–1253. [Google Scholar] [CrossRef]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Christiansen, T.; Richelsen, B.; Bruun, J.M. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int. J. Obes. 2005, 29, 146–150. [Google Scholar] [CrossRef]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef]
- Sag, S.; Yildiz, A.; Gullulu, S.; Gungoren, F.; Ozdemir, B.; Cegilli, E.; Oruc, A.; Ersoy, A.; Gullulu, M. Early atherosclerosis in normotensive patients with autosomal dominant polycystic kidney disease: The relation between epicardial adipose tissue thickness and carotid intima-media thickness. Springerplus. 2016, 5, 211. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Calabro, P.; Yeh, E.T. Intra-abdominal adiposity, inflammation, and cardiovascular risk: New insight into global cardiometabolic risk. Curr. Hypertens. Rep. 2008, 10, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Lee, Y.S.; Kim, K.H.; Lee, J.H.; Lee, H.K.; Jang, S.-H.; Kim, S.-E.; Lee, G.Y.; Lee, J.-W.; Jung, S.-A.; et al. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol. Endocrinol. 2010, 24, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Zhao, J.; Lawless, M.W. Stop feeding cancer: Pro-inflammatory role of visceral adiposity in liver cancer. Cytokine 2013, 64, 626–637. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Li, B.; Leung, J.C.K.; Chan, L.Y.Y.; Yiu, W.H.; Tang, S.C.W. A global perspective on the crosstalk between saturated fatty acids and Toll-like receptor 4 in the etiology of inflammation and insulin resistance. Prog. Lipid Res. 2020, 77, 101020. [Google Scholar] [CrossRef]
- Menezes, L.F.; Lin, C.C.; Zhou, F.; Germino, G.G. Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef]
- Kim, K.; Trott, J.F.; Gao, G.; Chapman, A.; Weiss, R.H. Plasma metabolites and lipids associate with kidney function and kidney volume in hypertensive ADPKD patients early in the disease course. BMC Nephrol. 2019, 20, 66. [Google Scholar] [CrossRef]
- Klawitter, J.; Klawitter, J.; McFann, K.; Pennington, A.T.; Abebe, K.Z.; Brosnahan, G.; Cadnapaphornchai, M.A.; Chonchol, M.; Gitomer, B.; Christians, U.; et al. Bioactive lipid mediators in polycystic kidney disease. J. Lipid. Res. 2014, 55, 1139–1149. [Google Scholar] [CrossRef]
- Ma, C.; Avenell, A.; Bolland, M.; Hudson, J.; Stewart, F.; Robertson, C.; Sharma, P.; Fraser, C.; MacLennan, G. Effects of weight loss interventions for adults who are obese on mortality, cardiovascular disease, and cancer: Systematic review and meta-analysis. Br. Med. J. 2017, 359, j4849. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.J.; Boucher, J.L.; Rutten-Ramos, S.; VanWormer, J.J. Lifestyle Weight-Loss Intervention Outcomes in Overweight and Obese Adults with Type 2 Diabetes: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. J. Acad. Nutr. Diet. 2015, 115, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Kanda, E.; Muneyuki, T.; Suwa, K.; Nakajima, K. Effects of Weight Loss Speed on Kidney Function Differ Depending on Body Mass Index in Nondiabetic Healthy People: A Prospective Cohort. PLoS ONE 2015, 10, e0143434. [Google Scholar] [CrossRef]
- Afshinnia, F.; Wilt, T.J.; Duval, S.; Esmaeili, A.; Ibrahim, H.N. Weight loss and proteinuria: Systematic review of clinical trials and comparative cohorts. Nephrol. Dial. Transplant. 2010, 25, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Morales, E.; Praga, M. The effect of weight loss in obesity and chronic kidney disease. Curr. Hypertens. Rep. 2012, 14, 170–176. [Google Scholar] [CrossRef]
- Bolignano, D.; Zoccali, C. Effects of weight loss on renal function in obese CKD patients: A systematic review. Nephrol. Dial. Transplant. 2013, 28 (Suppl. 4), iv82–iv98. [Google Scholar] [CrossRef]
- Vasquez, B.; Flock, E.V.; Savage, P.J.; Nagulesparan, M.; Bennion, L.J.; Baird, H.R.; Bennett, P.H. Sustained reduction of proteinuria in type 2 (non-insulin-dependent) diabetes following diet-induced reduction of hyperglycaemia. Diabetologia 1984, 26, 127–133. [Google Scholar] [CrossRef]
- Solerte, S.B.; Fioravanti, M.; Schifino, N.; Ferrari, E. Effects of diet-therapy on urinary protein excretion albuminuria and renal haemodynamic function in obese diabetic patients with overt nephropathy. Int. J. Obes. 1989, 13, 203–211. [Google Scholar]
- Saiki, A.; Nagayama, D.; Ohhira, M.; Endoh, K.; Ohtsuka, M.; Koide, N.; Oyama, T.; Miyashita, Y.; Shirai, K. Effect of weight loss using formula diet on renal function in obese patients with diabetic nephropathy. Int. J. Obes. 2005, 29, 1115–1120. [Google Scholar] [CrossRef]
- Gilardini, L.; Zulian, A.; Girola, A.; Redaelli, G.; Conti, A.; Invitti, C. Predictors of the early impairment of renal disease in human obesity. Int. J. Obes. 2010, 34, 287–294. [Google Scholar] [CrossRef]
- Shen, W.-W.; Chen, H.-M.; Chen, H.; Xu, F.; Li, L.-S.; Liu, Z.-H. Obesity-related glomerulopathy: Body mass index and proteinuria. Clin. J. Am. Soc. Nephrol. 2010, 5, 1401–1409. [Google Scholar] [CrossRef]
- Ezequiel, D.G.; Costa, M.B.; Chaoubah, A.; de Paula, R.B. Weight loss improves renal hemodynamics in patients with metabolic syndrome. J. Bras. Nefrol. 2012, 34, 36–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conley, M.M.; McFarlane, C.M.; Johnson, D.W.; Kelly, J.T.; Campbell, K.L.; MacLaughlin, H.L. Interventions for weight loss in people with chronic kidney disease who are overweight or obese. Cochrane Database Syst. Rev. 2021, 2021, CD013119. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Weimbs, T.; Shillingford, J.M.; Torres, J.; Kruger, S.L.; Bourgeois, B.C. Emerging targeted strategies for the treatment of autosomal dominant polycystic kidney disease. Clin. Kidney. J. 2018, 11, i27–i38. [Google Scholar] [CrossRef]
- Moore, S.C.; Patel, A.V.; Matthews, C.E.; de Gonzalez, A.B.; Park, Y.; Katki, H.A.; Linet, M.S.; Weiderpass, E.; Visvanathan, K.; Helzlsouer, K.J.; et al. Leisure time physical activity of moderate to vigorous intensity and mortality: A large pooled cohort analysis. PLoS Med. 2012, 9, e1001335. [Google Scholar] [CrossRef]
- Zelle, D.M.; Klaassen, G.; van Adrichem, E.; Bakker, S.J.; Corpeleijn, E.; Navis, G. Physical inactivity: A risk factor and target for intervention in renal care. Nat. Rev. Nephrol. 2017, 13, 152–168. [Google Scholar] [CrossRef]
- Beddhu, S.; Baird, B.C.; Zitterkoph, J.; Neilson, J.; Greene, T. Physical activity and mortality in chronic kidney disease (NHANES III). Clin. J. Am. Soc. Nephrol. 2009, 4, 1901–1906. [Google Scholar] [CrossRef]
- Martins, P.; Marques, E.A.; Leal, D.V.; Ferreira, A.; Wilund, K.R.; Viana, J.L. Association between physical activity and mortality in end-stage kidney disease: A systematic review of observational studies. BMC Nephrol. 2021, 22, 227. [Google Scholar] [CrossRef]
- American College of Sports Medicine. ACSM Guidelines for Exercise Testing and Prescription, 11th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2020. [Google Scholar]
- Chebib, F.T.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am. J. Kidney Dis. 2016, 67, 792–810. [Google Scholar] [CrossRef]
- Pickel, L.; Iliuta, I.A.; Scholey, J.; Pei, Y.; Sung, H.K. Dietary Interventions in Autosomal Dominant Polycystic Kidney Disease. Adv. Nutr. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Catenacci, V.A.; Dwivedi, N.; Kline, T.L.; Wang, W.; You, Z.; Nguyen, D.T.; Bing, K.; Poudyal, B.; Johnson, G.C.; et al. Weight loss and cystic disease progression in autosomal dominant polycystic kidney disease. iScience 2022, 25, 103697. [Google Scholar] [CrossRef]
- Testa, F.; Marchiò, M.; Belli, M.; Giovanella, S.; Ligabue, G.; Cappelli, G.; Biagini, G.; Magistroni, R. A pilot study to evaluate tolerability and safety of a modified Atkins diet in ADPKD patients. PharmaNutrition 2019, 9, 100154. [Google Scholar] [CrossRef]
- Testa, F.; Marchiò, M.; D’Amico, R.; Giovanella, S.; Ligabue, G.; Fontana, F.; Alfano, G.; Cappelli, G.; Biagini, G.; Magistroni, R. GREASE II. A phase II randomized, 12-month, parallel-group, superiority study to evaluate the efficacy of a Modified Atkins Diet in Autosomal Dominant Polycystic Kidney Disease patients. PharmaNutrition. 2020, 13, 100206. [Google Scholar] [CrossRef]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Renal. Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef]
- Torres, J.A.; Kruger, S.L.; Broderick, C.; Amarlkhagva, T.; Agrawal, S.; Dodam, J.R.; Mrug, M.; Lyons, L.A.; Weimbs, T. Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab. 2019, 30, 1007–1023.e5. [Google Scholar] [CrossRef]
- Strubl, S.; Oehm, S.; Torres, J.A.; Grundmann, F.; Haratani, J.; Decker, M.; Vuong, S.; Kaur Bhandal, A.; Methot, N.; Haynie-Cion, R.; et al. Ketogenic dietary interventions in autosomal dominant polycystic kidney disease—A retrospective case series study: First insights into feasibility, safety and effects. Clin. Kidney J. 2021, 1–14, in press. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | Study Design | Length (n) | Baseline BMI (kg/m2) or Inclusion | Weight Loss | Kidney Outcomes |
|---|---|---|---|---|---|
| Caloric Restriction (34% reduction in caloric intake, NCT03342742) Status: Complete [124] | Randomized, double blind, parallel assignment, two experimental arms | 12-months (n = 15) | 34.6 ± 5.1 | 3-months: −7.1 ± 4.2% 12-months: −9.1 ± 6.0% | Annual % change in htTKV was highly correlated with % change in weight (r = 0.68, p = 0.001) and change in BMI at 12-months (r = 0.63, p < 0.01) |
| Intermittent Fasting (20% reduction on three non-consecutive days per week, NCT03342742) Status: Complete [124] | Randomized, double blind, parallel assignment two experimental arms | 12-months (n = 13) | 34.8 ± 5.1 | 3-months: −5.5 ± 3.3% 12-months: −4.9 ± 5.6% | |
| Ketogenic Diet (lipids 65%, proteins 30%, and carbohydrate 5% total caloric intake, modified Atkins diet) Status: Complete [125] | Single-arm interventional pilot | 3-months (n = 3) | 25.3 ± 1.4 | Hypocaloric ketogenic diet invoked weight loss 1–4.2 kg in those who were overweight | eGFR did not change |
| Caloric Restriction (30% reduction in caloric intake and increased physical activity, NCT04907799) Status: On-going | Randomized, double-blind, parallel assignment one experimental arm, one control arm | 24-months (n = 126) | 25–45 | Secondary outcome change in abdominal adiposity | Primary outcome change in htTKV |
| Time-Restricted Eating (food intake restricted to an 8-h window, NCT04534985). Status: On-going | Randomized, double-blind, parallel assignment one experimental arm, one active comparator | 12-months (n = 30) | 25–45 | Secondary outcomes change in body weight, abdominal adiposity, and body composition | Secondary outcome change in htTKV |
| Ketogenic Diet (High fat, moderate protein, very low carbohydrate <20 g per day, NCT04680780) Status: On-going | Randomized, parallel assignment, two experimental arms, and one control arm | 3-months (n = 21) | 18.6–34.9 | Secondary outcome change in BMI | Secondary outcome change in TKV |
| Water Fasting (water fasting on 3 consecutive days within the first 14 days of each month, NCT04680780) Status: On-going | Randomized, parallel assignment, two experimental arms, and one control arm | 3-months (n = 21) | 18.6–34.9 | Secondary outcome change in BMI | Secondary outcome change in TKV |
| Acute fasting for 72 h or intake of a ketogenic diet for 14 days (NCT04472624) Status: Complete (results not posted) | Non-randomized (participant selected experimental arm), parallel assignment | 72 h (fasting) or 14 days (ketogenic diet) (n = 10) | 18–35 | Secondary outcome absolute and relative change in weight | Primary outcome relative difference in TKV immediately before and after the ketonic state [Time Frame: Visit 2: 2–4 Weeks after enrolment; Visit 3: 3–21 days after Visit 2] |
| Ketogenic Diet [4–6% carbohydrates, 25–30% proteins, and 60–70% lipids; modified Atkins diet], or a balanced normocaloric diet [55–60% carbohydrates, 10–15% proteins, 25–30% lipids]. Status: On-going [126] | Randomized, parallel group, two experimental arms | 12 months (n = 90) | >20 | Caloric intake will be adjusted for participants to remain weight stable | Primary outcome change in TKV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steele, C.; Nowak, K. Obesity, Weight Loss, Lifestyle Interventions, and Autosomal Dominant Polycystic Kidney Disease. Kidney Dial. 2022, 2, 106-122. https://doi.org/10.3390/kidneydial2010013
Steele C, Nowak K. Obesity, Weight Loss, Lifestyle Interventions, and Autosomal Dominant Polycystic Kidney Disease. Kidney and Dialysis. 2022; 2(1):106-122. https://doi.org/10.3390/kidneydial2010013
Chicago/Turabian StyleSteele, Cortney, and Kristen Nowak. 2022. "Obesity, Weight Loss, Lifestyle Interventions, and Autosomal Dominant Polycystic Kidney Disease" Kidney and Dialysis 2, no. 1: 106-122. https://doi.org/10.3390/kidneydial2010013
APA StyleSteele, C., & Nowak, K. (2022). Obesity, Weight Loss, Lifestyle Interventions, and Autosomal Dominant Polycystic Kidney Disease. Kidney and Dialysis, 2(1), 106-122. https://doi.org/10.3390/kidneydial2010013