
SARS-CoV-2: An Update on the Biological Interplay with the Human Host





Abstract
:1. Introduction
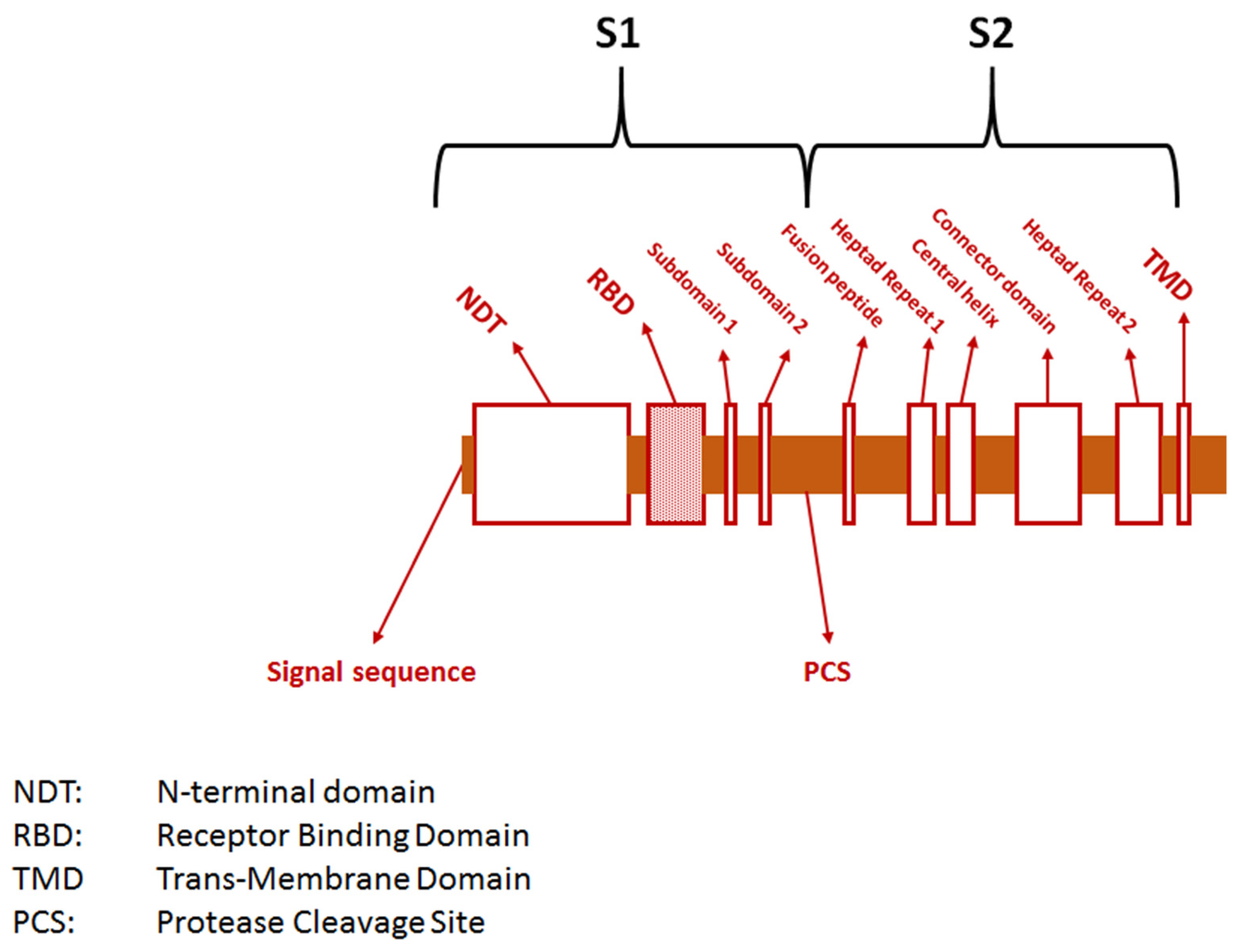
2. The Pathogen
3. Biological Characteristics of SARS-CoV-2 in Humans
3.1. Incubation
3.2. Viral Shedding
3.3. Infectivity
3.4. The Role of Super-Spreaders
4. SARS-CoV-2 Evolution
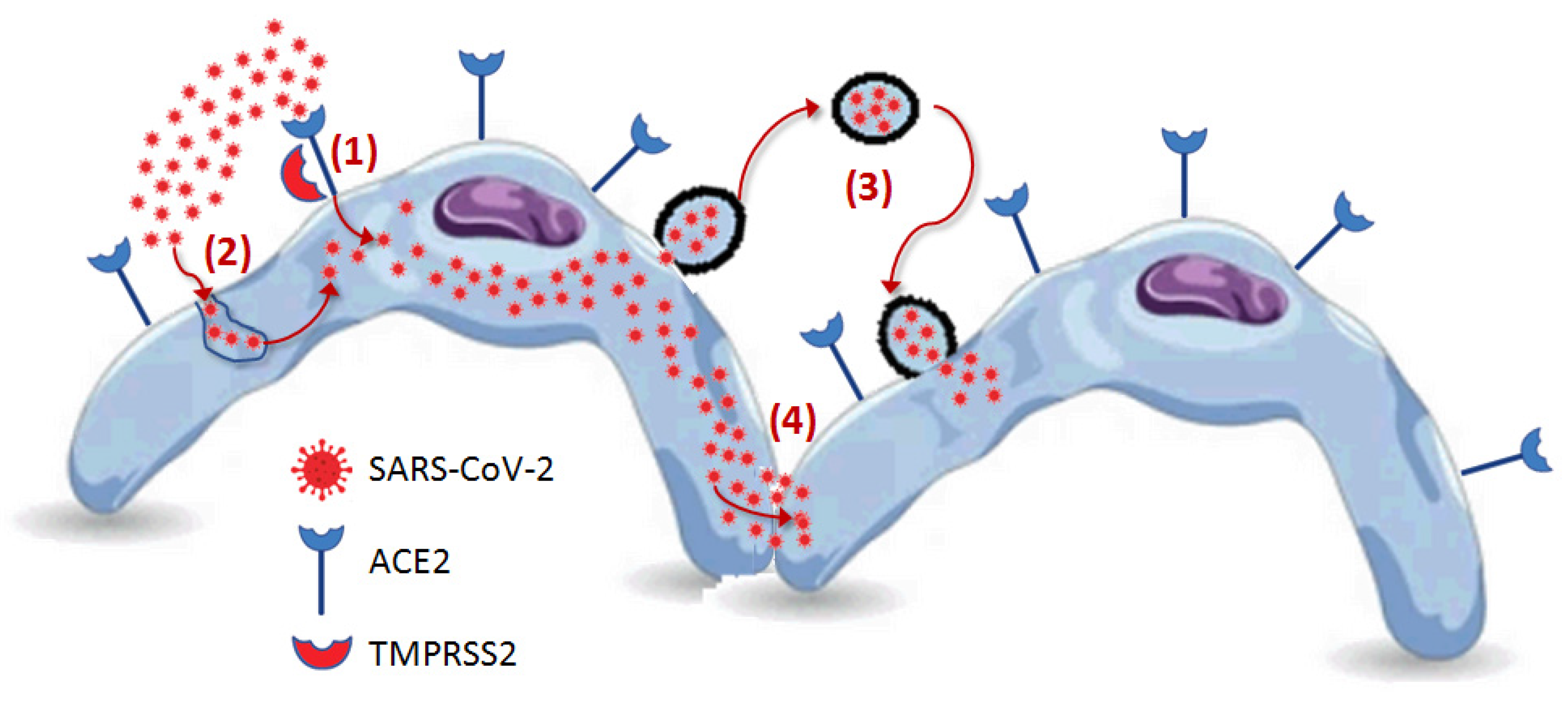
5. Human Host Cell Penetration
5.1. Receptor-Mediated Penetration
5.2. Cathepsin L-Mediated Endocytosis
5.3. SARS-CoV-2 Bearing Extracellular Particles
5.4. Cell-to-Cell Propagation
6. Intracellular Processing
7. The Human Host
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sampath, S.; Khedr, A.; Qamar, S.; Tekin, A.; Singh, R.; Green, R.; Kashyap, R. Pandemics Throughout the History. Cureus 2021, 13, e18136. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C.; Henry, B.M. Uncontrolled confounding in COVID-19 epidemiology. Diagnosis 2022, 10, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. COVID-19: WHO declares end of global health emergency. BMJ 2023, 381, 1041. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 10 September 2023).
- Jones, J.M.; Manrique, I.M.; Stone, M.S.; Grebe, E.; Saa, P.; Germanio, C.D.; Spencer, B.R.; Notari, E.; Bravo, M.; Lanteri, M.C.; et al. Estimates of SARS-CoV-2 Seroprevalence and Incidence of Primary SARS-CoV-2 Infections Among Blood Donors, by COVID-19 Vaccination Status—United States, April 2021–September 2022. Morb. Mortal. Wkly. Rep. 2023, 72, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C.; Bovo, C.; Plebani, M. Current laboratory diagnostics of coronavirus disease 2019 (COVID-19). Acta Bio Medica Atenei Parm. 2020, 91, 137–145. [Google Scholar] [CrossRef]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef]
- Machitani, M.; Yasukawa, M.; Nakashima, J.; Furuichi, Y.; Masutomi, K. RNA-dependent RNA polymerase, RdRP, a promising therapeutic target for cancer and potentially COVID-19. Cancer Sci. 2020, 111, 3976–3984. [Google Scholar] [CrossRef]
- Sakkiah, S.; Guo, W.; Pan, B.; Ji, Z.; Yavas, G.; Azevedo, M.; Hawes, J.; Patterson, T.A.; Hong, H. Elucidating Interactions Between SARS-CoV-2 Trimeric Spike Protein and ACE2 Using Homology Modeling and Molecular Dynamics Simulations. Front. Chem. 2021, 8, 622632. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Lippi, G.; Henry, B.M.; Sanchis-Gomar, F. The real origin of SARS-CoV-2: Does it really matter? J. Lab. Precis. Med. 2021, 6, 9. [Google Scholar] [CrossRef]
- Wu, Y.; Kang, L.; Guo, Z.; Liu, J.; Liu, M.; Liang, W. Incubation Period of COVID-19 Caused by Unique SARS-CoV-2 Strains: A Systematic Review and Meta-analysis. JAMA Netw. Open 2022, 5, e2228008, Erratum in: JAMA Netw. Open 2022, 5, e2235424. [Google Scholar] [CrossRef]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; El-Shall, N.A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B.1.1.529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ Res. 2022, 209, 112816. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, M.; Saied, A.A.; Mitra, S.; Alhumaydhi, F.A.; Emran, T.B.; Wilairatana, P. Omicron variant (B.1.1.529) and its sublineages: What do we know so far amid the emergence of recombinant variants of SARS-CoV-2? Biomed. Pharmacother. 2022, 154, 113522. [Google Scholar] [CrossRef]
- Tan, K.S.; Ong, S.W.X.; Koh, M.H.; Tay, D.J.W.; Aw, D.Z.H.; Nah, Y.W.; Abdullah, M.R.B.; Coleman, K.K.; Milton, D.K. SARS-CoV-2 Omicron variant shedding during respiratory activities. Int. J. Infect. Dis. 2023, 131, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Zhang, X.; Chen, C.; Jiang, D.; Liu, X.; Zhou, Y.; Huang, C.; Zhou, Y.; Guan, Z.; Ding, C.; et al. Characteristics of Viral Shedding Time in SARS-CoV-2 Infections: A Systematic Review and Meta-Analysis. Front. Public Health 2021, 9, 652842. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.; Esmaeili-Wellman, S.; Schiffer, J.T. Heterogeneous SARS-CoV-2 kinetics due to variable timing and intensity of immune responses. medRxiv 2023, 2023, 23294350. [Google Scholar] [CrossRef]
- Marc, A.; Kerioui, M.; Blanquart, F.; Bertrand, J.; Mitjà, O.; Corbacho-Monné, M.; Marks, M.; Guedj, J. Quantifying the relationship between SARS-CoV-2 viral load and infectiousness. Elife 2021, 10, e69302. [Google Scholar] [CrossRef] [PubMed]
- SeyedAlinaghi, S.; Karimi, A.; Mojdeganlou, H.; Pashaei, Z.; Mirzapour, P.; Shamsabadi, A.; Barzegary, A.; Afroughi, F.; Dehghani, S.; Janfaza, N.; et al. Minimum infective dose of severe acute respiratory syndrome coronavirus 2 based on the current evidence: A systematic review. SAGE Open Med. 2022, 10, 20503121221115053. [Google Scholar] [CrossRef]
- Hamner, L.; Dubbel, P.; Capron, I.; Ross, A.; Jordan, A.; Lee, J.; Lynn, J.; Ball, A.; Narwal, S.; Russell, S.; et al. High SARS-CoV-2 Attack Rate Following Exposure at a Choir Practice—Skagit County, Washington, March 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 606–610. [Google Scholar] [CrossRef]
- Park, M.; Pawliuk, C.; Nguyen, T.; Griffitt, A.; Dix-Cooper, L.; Fourik, N.; Dawes, M. Determining the communicable period of SARS-CoV-2: A rapid review of the literature, March to September 2020. Eurosurveillance 2021, 26, 2001506. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, L.J.; Khan, M.; Yoo, S.J.; Zapiec, B.; Van Gerven, L.; Van Slambrouck, J.; Vanstapel, A.; Van Raemdonck, D.; Vos, R.; Wauters, E.; et al. Persistence of SARS-CoV-2 RNA in lung tissue after mild COVID-19. Lancet Respir. Med. 2021, 9, e78–e79. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Ishikane, M.; Ujiie, M.; Iwamoto, N.; Okumura, N.; Sato, T.; Nagashima, M.; Moriya, A.; Suzuki, M.; Hojo, M.; et al. Duration of Infectious Virus Shedding by SARS-CoV-2 Omicron Variant-Infected Vaccinees. Emerg. Infect. Dis. 2022, 28, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Alimohamadi, Y.; Taghdir, M.; Sepandi, M. Estimate of the Basic Reproduction Number for COVID-19: A Systematic Review and Meta-analysis. J. Prev. Med. Public Health 2020, 53, 151–157. [Google Scholar] [CrossRef]
- Nishiura, H.; Ito, K.; Anzai, A.; Kobayashi, T.; Piantham, C.; Rodríguez-Morales, A.J. Relative Reproduction Number of SARS-CoV-2 Omicron (B.1.1.529) Compared with Delta Variant in South Africa. J. Clin. Med. 2021, 11, 30. [Google Scholar] [CrossRef]
- Omer, S.B.; Yildirim, I.; Forman, H.P. Herd Immunity and Implications for SARS-CoV-2 Control. JAMA 2020, 324, 2095–2096. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Hosseini, M.; Poon, L.L.M.; Chin, A.W.H.; Ducker, W.A. Effect of Surface Porosity on SARS-CoV-2 Fomite Infectivity. ACS Omega 2022, 7, 18238–18246. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Henry, B.M.; Lippi, G. Regional Association between Mean Air Temperature and Case Numbers of Multiple SARS-CoV-2 Lineages throughout the Pandemic. Viruses 2022, 14, 1913. [Google Scholar] [CrossRef]
- Greenhalgh, T.; Jimenez, J.L.; Prather, K.A.; Tufekci, Z.; Fisman, D.; Schooley, R. Ten scientific reasons in support of airborne transmission of SARS-CoV-2. Lancet 2021, 397, 1603–1605, Erratum in: Lancet 2021, 397, 1808. [Google Scholar] [CrossRef] [PubMed]
- Lau, M.S.Y.; Grenfell, B.; Thomas, M.; Bryan, M.; Nelson, K.; Lopman, B. Characterizing superspreading events and age-specific infectiousness of SARS-CoV-2 transmission in Georgia, USA. Proc. Natl. Acad. Sci. USA 2020, 117, 22430–22435. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Singanayagam, A.; Goonawardane, N.; Moshe, M.; Sweeney, F.P.; Sukhova, K.; Killingley, B.; Kalinova, M.; Mann, A.J.; Catchpole, A.P.; et al. Viral emissions into the air and environment after SARS-CoV-2 human challenge: A phase 1, open label, first-in-human study. Lancet Microbe 2023, 4, e579–e590, Erratum in: Lancet Microbe 2023, 4, e576. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The coronavirus is mutating—Does it matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. New Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Losos, J.B. Convergence, adaptation, and constraint. Evolution 2011, 65, 1827–1840. [Google Scholar] [CrossRef]
- Boyle, L.; Hletko, S.; Huang, J.; Lee, J.; Pallod, G.; Tung, H.R.; Durrett, R. Selective sweeps in SARS-CoV-2 variant competition. Proc. Natl. Acad. Sci. USA 2022, 119, e2213879119. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Abe, H.; Yasuda, J. Comparison of genome replication fidelity between SARS-CoV-2 and influenza A virus in cell culture. Sci. Rep. 2023, 13, 13105. [Google Scholar] [CrossRef]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol. Evol. 2022, 39, msac013. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients with Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 713–720, Erratum in: Clin. Infect. Dis. 2021, 73, 2374. [Google Scholar] [CrossRef]
- Li, J.; Du, P.; Yang, L.; Zhang, J.; Song, C.; Chen, D.; Song, Y.; Ding, N.; Hua, M.; Han, K.; et al. Two-step fitness selection for intra-host variations in SARS-CoV-2. Cell Rep. 2022, 38, 110205. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.C. Intra-Host Diversity of SARS-CoV-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.K.; Mishra, G.P.; Uppili, B.; Walia, S.; Fatihi, S.; Abbas, T.; Banu, S.; Ghosh, A.; Kanampalliwar, A.; Jha, A.; et al. Spatio-temporal dynamics of intra-host variability in SARS-CoV-2 genomes. Nucleic Acids Res. 2022, 50, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Laskar, R.; Ali, S. Differential mutation profile of SARS-CoV-2 proteins across deceased and asymptomatic patients. Chem. Biol. Interact. 2021, 347, 109598. [Google Scholar] [CrossRef]
- Al-Khatib, H.A.; Smatti, M.K.; Ali, F.H.; Zedan, H.T.; Thomas, S.; Ahmed, M.N.; El-Kahlout, R.A.; Al Bader, M.A.; Elgakhlab, D.; Coyle, P.V.; et al. Comparative analysis of within-host diversity among vaccinated COVID-19 patients infected with different SARS-CoV-2 variants. iScience 2022, 25, 105438. [Google Scholar] [CrossRef]
- He, Y.; Ma, W.; Dang, S.; Chen, L.; Zhang, R.; Mei, S.; Wei, X.; Lv, Q.; Peng, B.; Chen, J.; et al. Possible recombination between two variants of concern in a COVID-19 patient. Emerg. Microbes Infect. 2022, 11, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, R.K.; Kandi, V.; Tuli, H.S.; Chakraborty, C.; Dhama, K. The recombinant variants of SARS-CoV-2: Concerns continues amid COVID-19 pandemic. J. Med. Virol. 2022, 94, 3506–3508. [Google Scholar] [CrossRef]
- Scarpa, F.; Ciccozzi, M. On the SARS-CoV-2 BA.2.86 lineage: A mutation point of view. J. Med. Virol. 2023, 95, e29079. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Mykytyn, A.Z.; Fouchier, R.A.; Haagmans, B.L. Antigenic evolution of SARS coronavirus 2. Curr. Opin. Virol. 2023, 62, 101349. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi, C.; Lippi, G. Timeline analysis of clinical severity of COVID-19 in the general population. Eur. J. Intern. Med. 2023, 110, 97–98. [Google Scholar] [CrossRef]
- Perez-Guzman, P.N.; Knock, E.; Imai, N.; Rawson, T.; Elmaci, Y.; Alcada, J.; Whittles, L.K.; Thekke Kanapram, D.; Sonabend, R.; Gaythorpe, K.A.M.; et al. Epidemiological drivers of transmissibility and severity of SARS-CoV-2 in England. Nat. Commun. 2023, 14, 4279. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Merle, U.; Schirmacher, P.; Longerich, T. Lethality of SARS-CoV-2 infection-a comparative autopsy study focusing on COVID-19 development and virus variants. Histopathology 2023, 83, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Nocini, R.; Henry, B.M.; Mattiuzzi, C.; Lippi, G. Improving Nasal Protection for Preventing SARS-CoV-2 Infection. Biomedicines 2022, 10, 2966. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Lavie, C.J.; Henry, B.M.; Sanchis-Gomar, F. Do genetic polymorphisms in angiotensin converting enzyme 2 (ACE2) gene play a role in coronavirus disease 2019 (COVID-19)? Clin. Chem. Lab. Med. 2020, 58, 1415–1422. [Google Scholar] [CrossRef]
- Kishimoto, M.; Uemura, K.; Sanaki, T.; Sato, A.; Hall, W.W.; Kariwa, H.; Orba, Y.; Sawa, H.; Sasaki, M. TMPRSS11D and TMPRSS13 Activate the SARS-CoV-2 Spike Protein. Viruses 2021, 13, 384. [Google Scholar] [CrossRef]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.J.; Soundararajan, V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife 2020, 9, e58603. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Evans, J.P.; Liu, S.L. Role of host factors in SARS-CoV-2 entry. J. Biol. Chem. 2021, 297, 100847. [Google Scholar] [CrossRef]
- Lim, S.; Zhang, M.; Chang, T.L. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses 2022, 14, 2535. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Soriaga, L.B.; Montiel-Ruiz, M.; Benigni, F.; Noack, J.; Park, Y.J.; Bianchi, S.; Walls, A.C.; Bowen, J.E.; Zhou, J.; et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 2021, 598, 342–347. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh RMJr Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P.Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Kakizaki, M.; Shiwa-Sudo, N.; Okura, T.; Tahara, M.; Fukushi, S.; Maeda, K.; Kawase, M.; Asanuma, H.; Tomita, Y.; et al. Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat. Commun. 2022, 13, 6100. [Google Scholar] [CrossRef] [PubMed]
- Kongsomros, S.; Pongsakul, N.; Panachan, J.; Khowawisetsut, L.; Somkird, J.; Sangma, C.; Kanjanapruthipong, T.; Wongtrakoongate, P.; Chairoungdua, A.; Pattanapanyasat, K.; et al. Comparison of viral inactivation methods on the characteristics of extracellular vesicles from SARS-CoV-2 infected human lung epithelial cells. J. Extracell. Vesicles 2022, 11, e12291. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Pan, X.; Luo, R.H.; Shen, X.; Li, S.; Wang, Y.; Zuo, X.; Wu, Y.; Guo, Y.; Xiao, G.; et al. Extracellular vesicles mediate antibody-resistant transmission of SARS-CoV-2. Cell Discov. 2023, 9, 2. [Google Scholar] [CrossRef]
- Ning, B.; Huang, Z.; Youngquist, B.M.; Scott, J.W.; Niu, A.; Bojanowski, C.M.; Zwezdaryk, K.J.; Saba, N.S.; Fan, J.; Yin, X.M.; et al. Liposome-mediated detection of SARS-CoV-2 RNA-positive extracellular vesicles in plasma. Nat. Nanotechnol. 2021, 16, 1039–1044. [Google Scholar] [CrossRef]
- Zeng, C.; Evans, J.P.; King, T.; Zheng, Y.M.; Oltz, E.M.; Whelan, S.P.J.; Saif, L.J.; Peeples, M.E.; Liu, S.L. SARS-CoV-2 spreads through cell-to-cell transmission. Proc. Natl. Acad. Sci. USA 2022, 119, e2111400119. [Google Scholar] [CrossRef]
- Li, X.; Yuan, H.; Li, X.; Wang, H. Spike protein mediated membrane fusion during SARS-CoV-2 infection. J. Med. Virol. 2023, 95, e28212. [Google Scholar] [CrossRef]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 2021, 81, 2656–2668.e8. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R., 3rd; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Cobat, A.; Casanova, J.L. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef]
- Matuozzo, D.; Talouarn, E.; Marchal, A.; Zhang, P.; Manry, J.; Seeleuthner, Y.; Zhang, Y.; Bolze, A.; Chaldebas, M.; Milisavljevic, B.; et al. Rare predicted loss-of-function variants of type I IFN immunity genes are associated with life-threatening COVID-19. Genome Med. 2023, 15, 22. [Google Scholar] [CrossRef]
- Biancolella, M.; Colona, V.L.; Luzzatto, L.; Watt, J.L.; Mattiuz, G.; Conticello, S.G.; Kaminski, N.; Mehrian-Shai, R.; Ko, A.I.; Gonsalves, G.S.; et al. COVID-19 annual update: A narrative review. Hum. Genom. 2023, 17, 68. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. A second update on mapping the human genetic architecture of COVID-19. Nature 2023, 621, E7–E26. [Google Scholar] [CrossRef]
- Lenharo, M. WHO declares end to COVID-19’s emergency phase. Nature, 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M. COVID-19: The global health emergency is over for the WHO, but not yet for laboratory medicine. J. Lab. Precis. Med. 2023, 8, 17. [Google Scholar] [CrossRef]
- Callaway, E. COVID’s future: Mini-waves rather than seasonal surges. Nature 2023, 617, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Ruaño, G.; Ha, T. Living with respiratory viruses: The next saga in human/viral coexistence? Bioessays 2021, 43, e2000321. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Variant | Spike Protein Mutations |
|---|---|
| Ancestral | - |
| Alpha | ~8 |
| Beta | ~9 |
| Gamma | ~10 |
| Delta | ~10 |
| Omicron | >35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippi, G.; Sanchis-Gomar, F.; Mattiuzzi, C.; Henry, B.M. SARS-CoV-2: An Update on the Biological Interplay with the Human Host. COVID 2023, 3, 1586-1600. https://doi.org/10.3390/covid3100108
Lippi G, Sanchis-Gomar F, Mattiuzzi C, Henry BM. SARS-CoV-2: An Update on the Biological Interplay with the Human Host. COVID. 2023; 3(10):1586-1600. https://doi.org/10.3390/covid3100108
Chicago/Turabian StyleLippi, Giuseppe, Fabian Sanchis-Gomar, Camilla Mattiuzzi, and Brandon M. Henry. 2023. "SARS-CoV-2: An Update on the Biological Interplay with the Human Host" COVID 3, no. 10: 1586-1600. https://doi.org/10.3390/covid3100108
APA StyleLippi, G., Sanchis-Gomar, F., Mattiuzzi, C., & Henry, B. M. (2023). SARS-CoV-2: An Update on the Biological Interplay with the Human Host. COVID, 3(10), 1586-1600. https://doi.org/10.3390/covid3100108