
Computational Study of the Therapeutic Potential of Novel Heterocyclic Derivatives against SARS-CoV-2

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
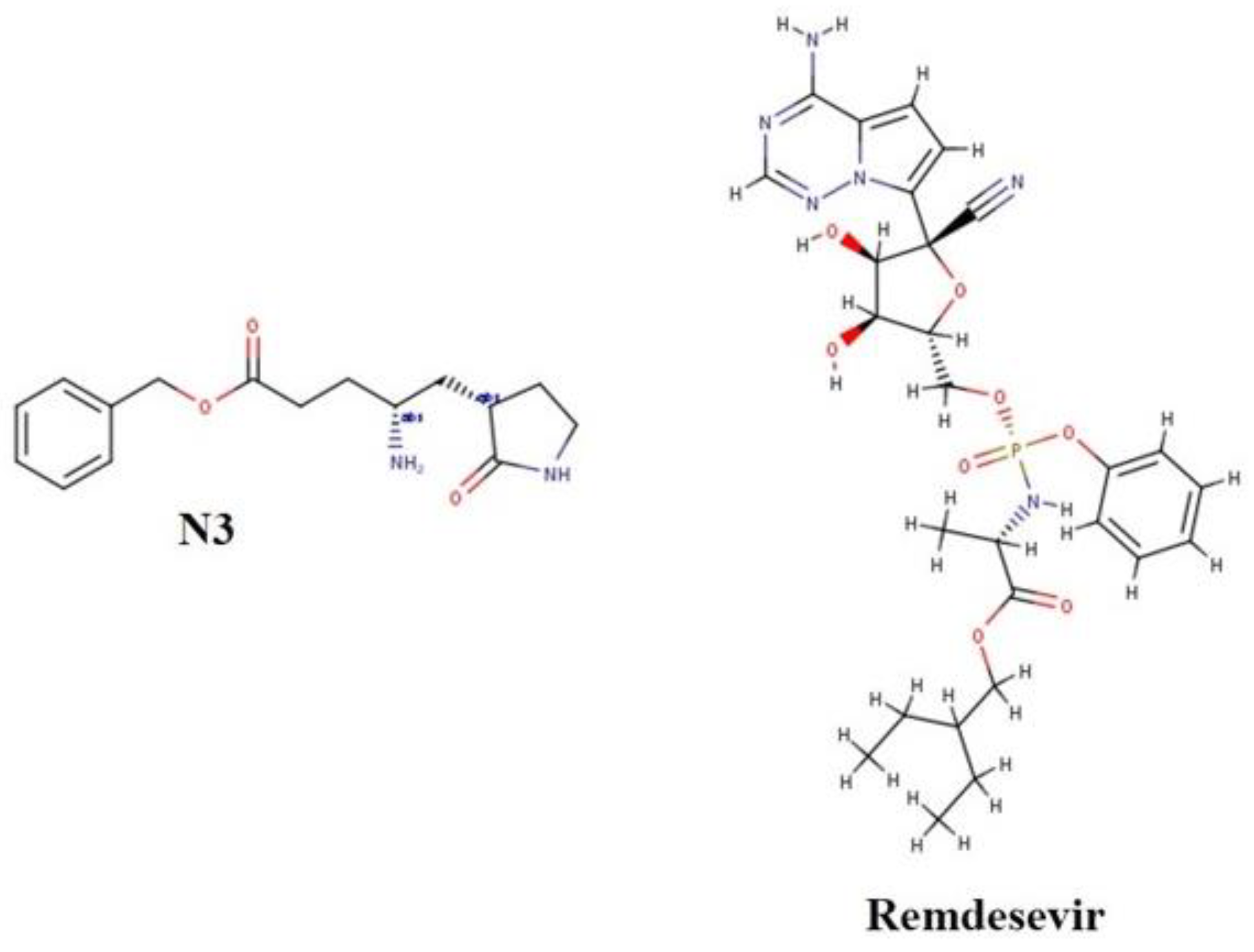
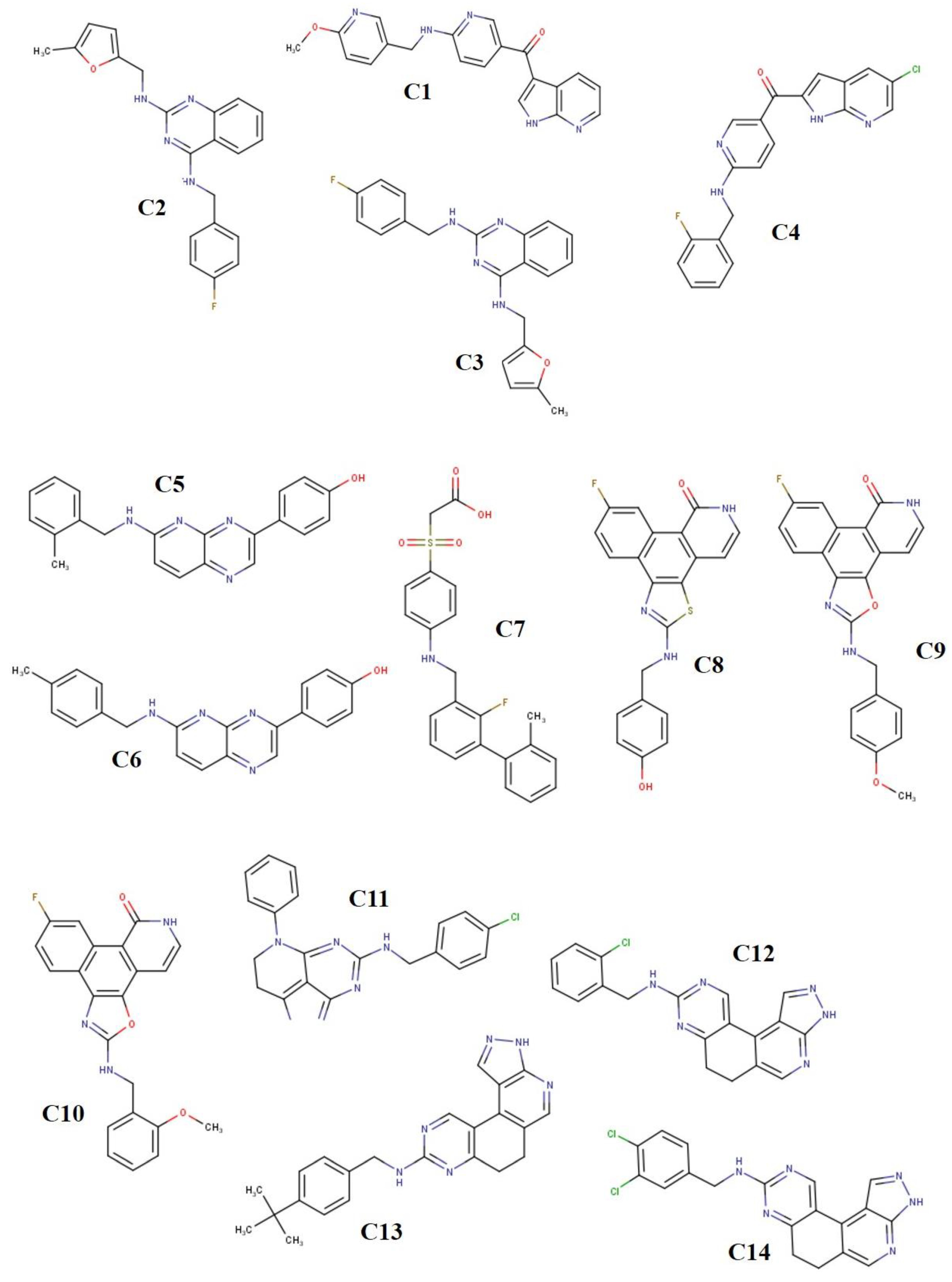
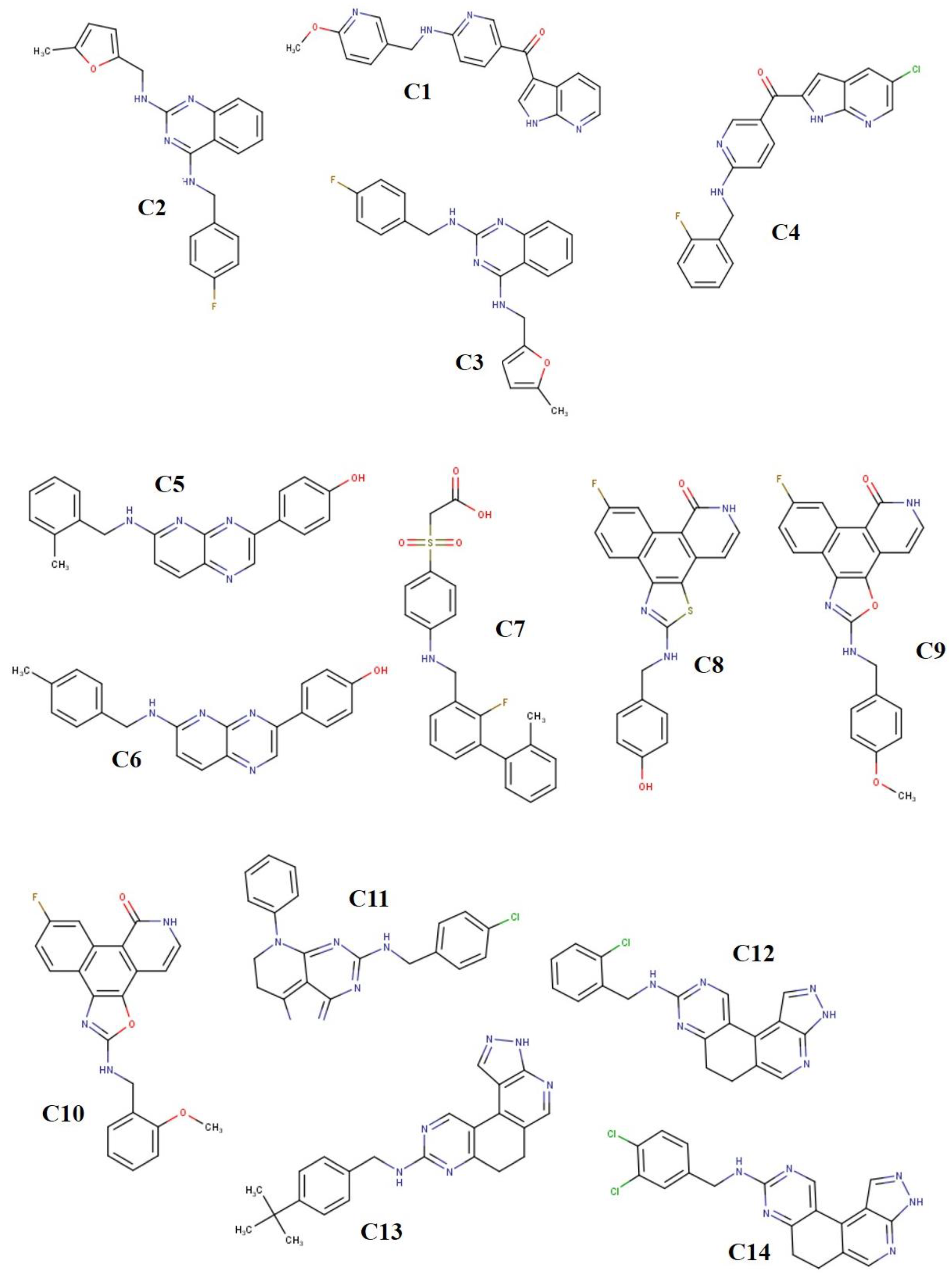
2.1. Ligand Derivatives
2.2. Ligand Preparation
2.3. Protein Preparation
2.4. Molecular Docking
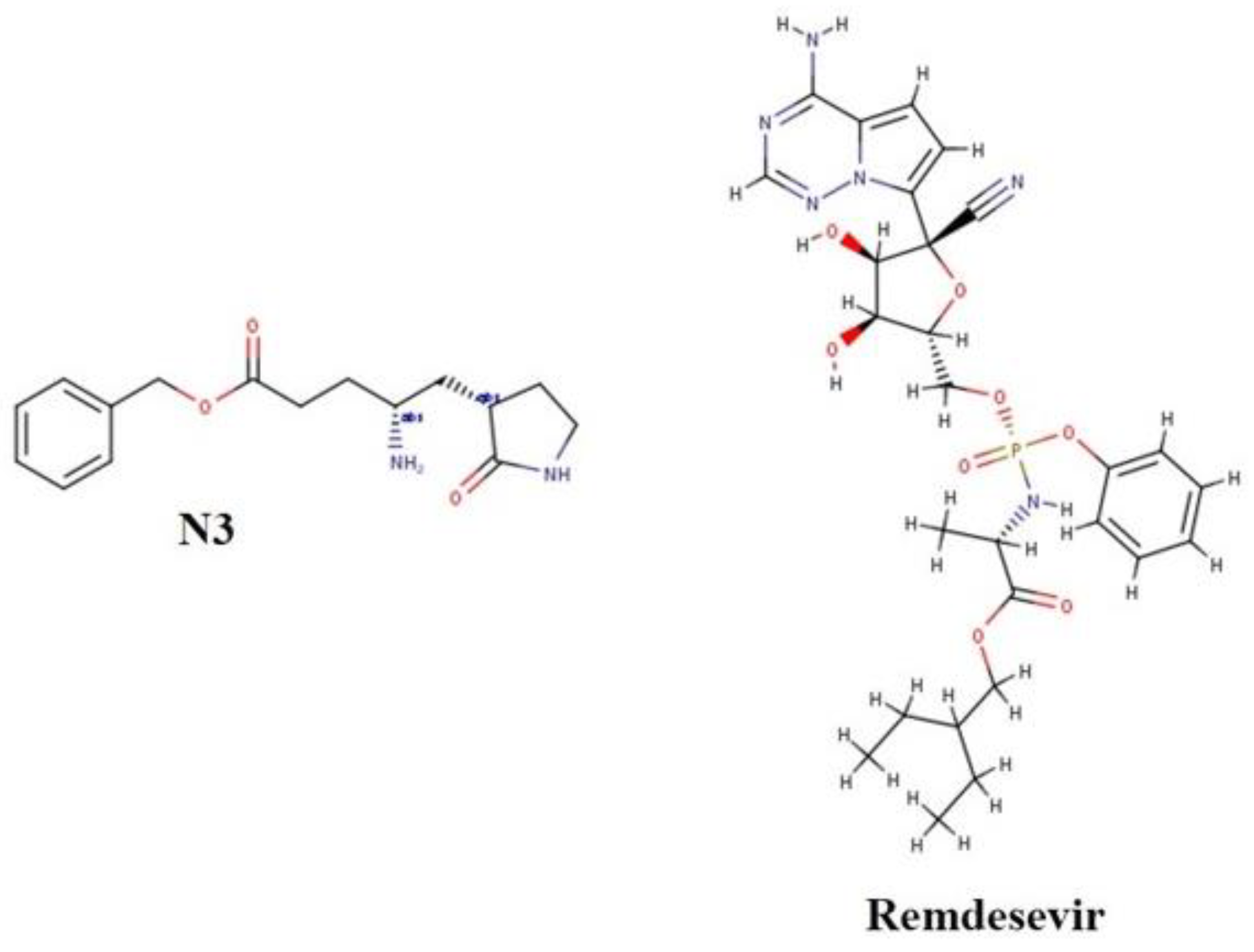
2.5. Validation of Docking Protocol
2.6. ADMET Predictions
3. Results
3.1. Binding Affinities and Stability of Test Compounds with SARS-CoV-2 Drug Targets
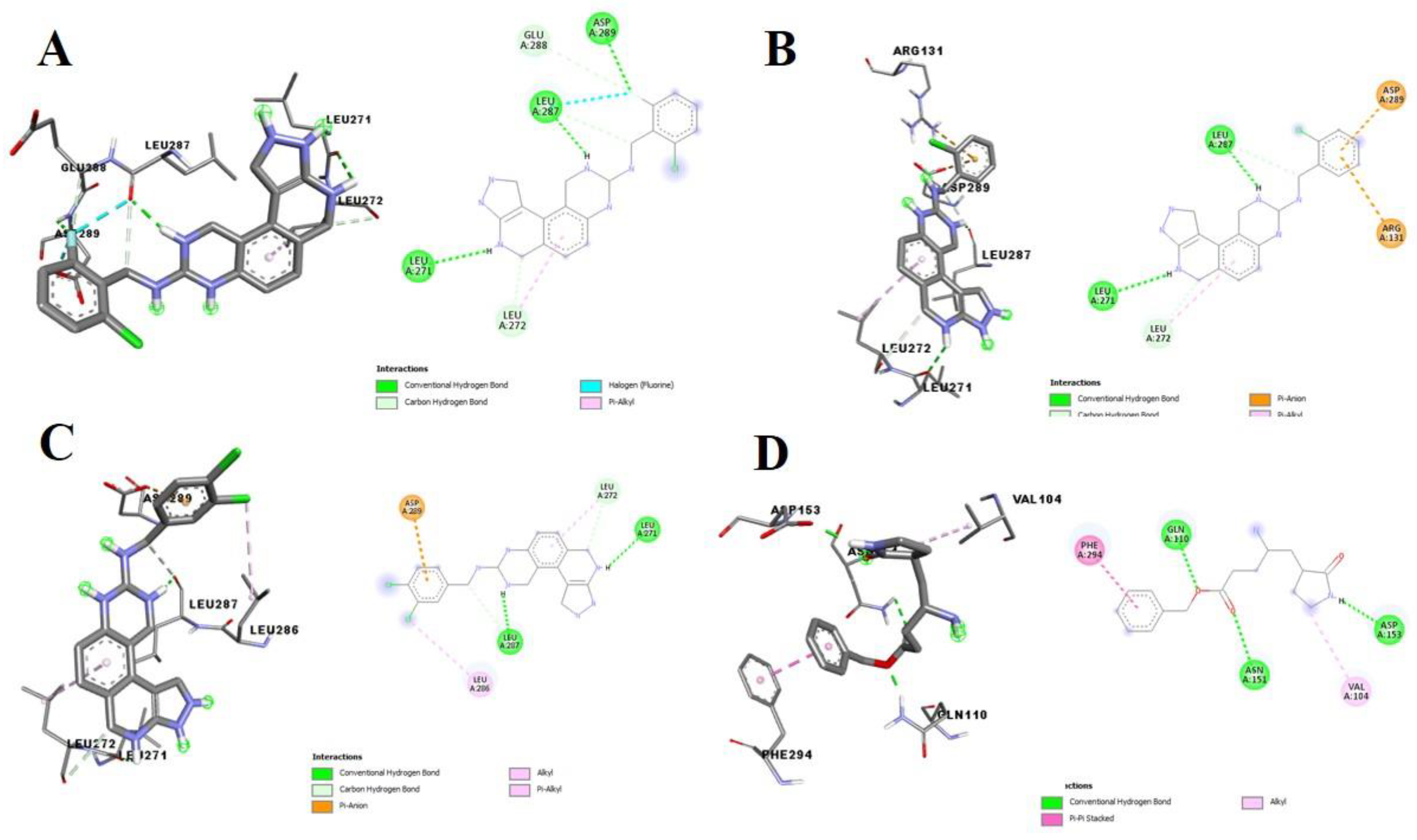
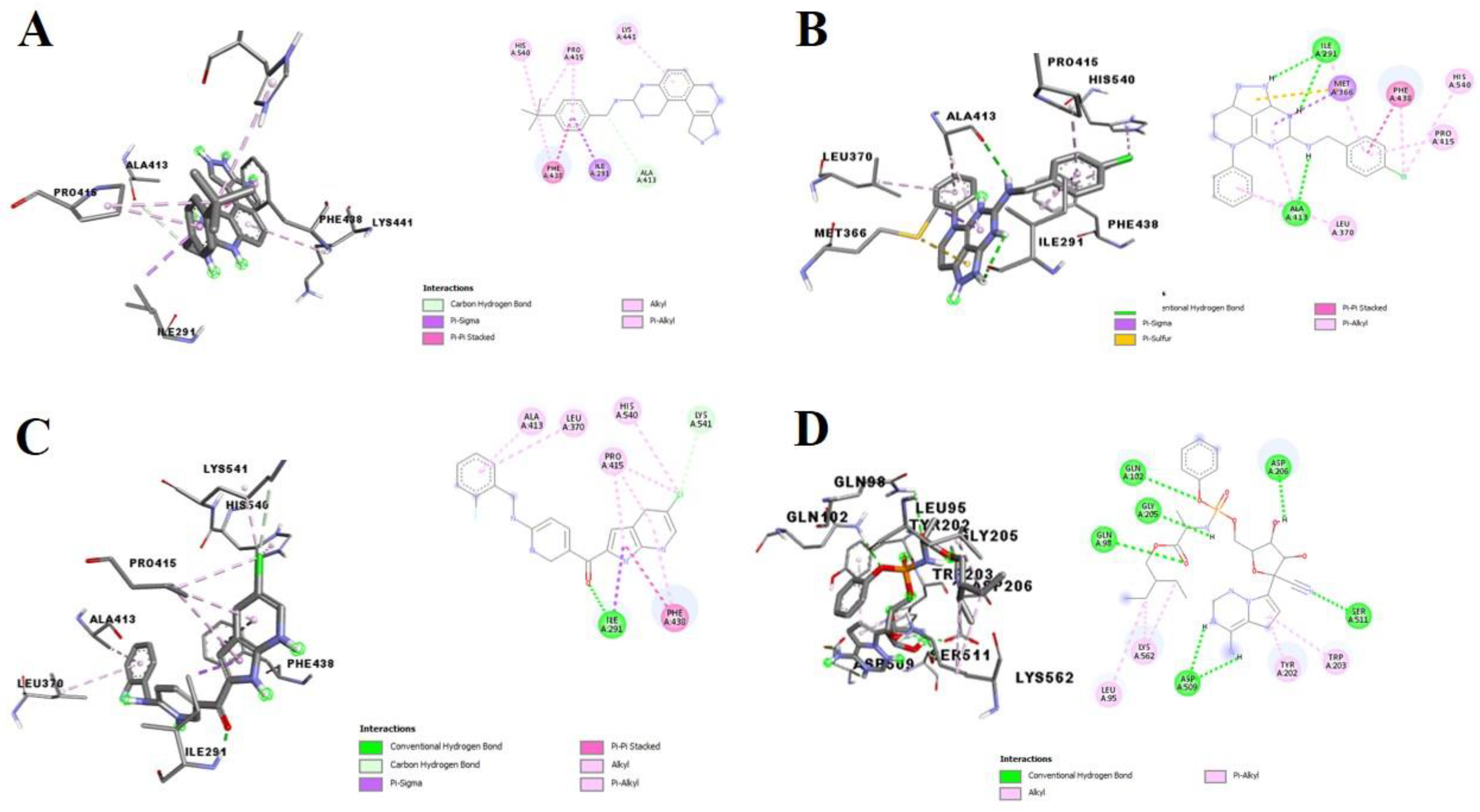
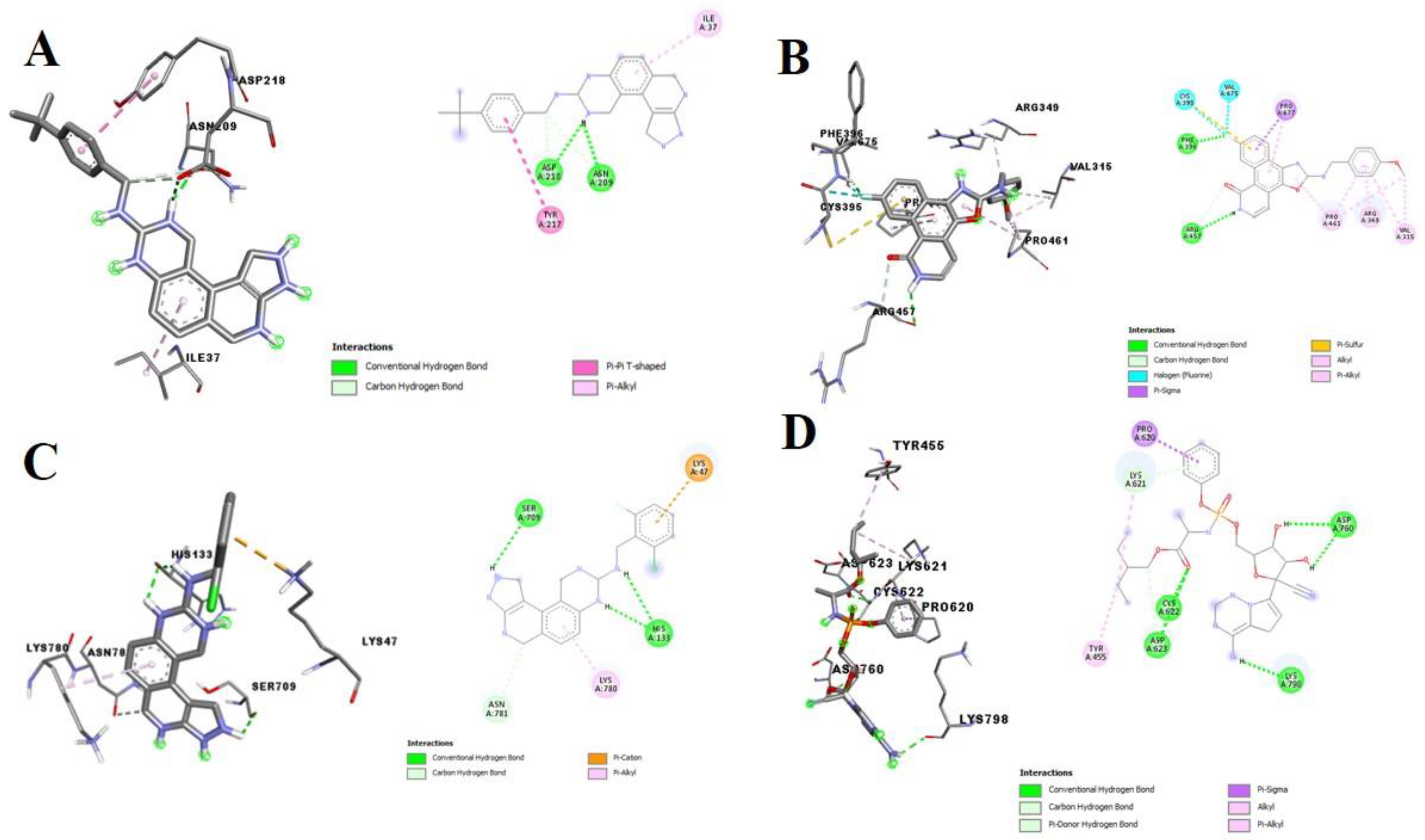
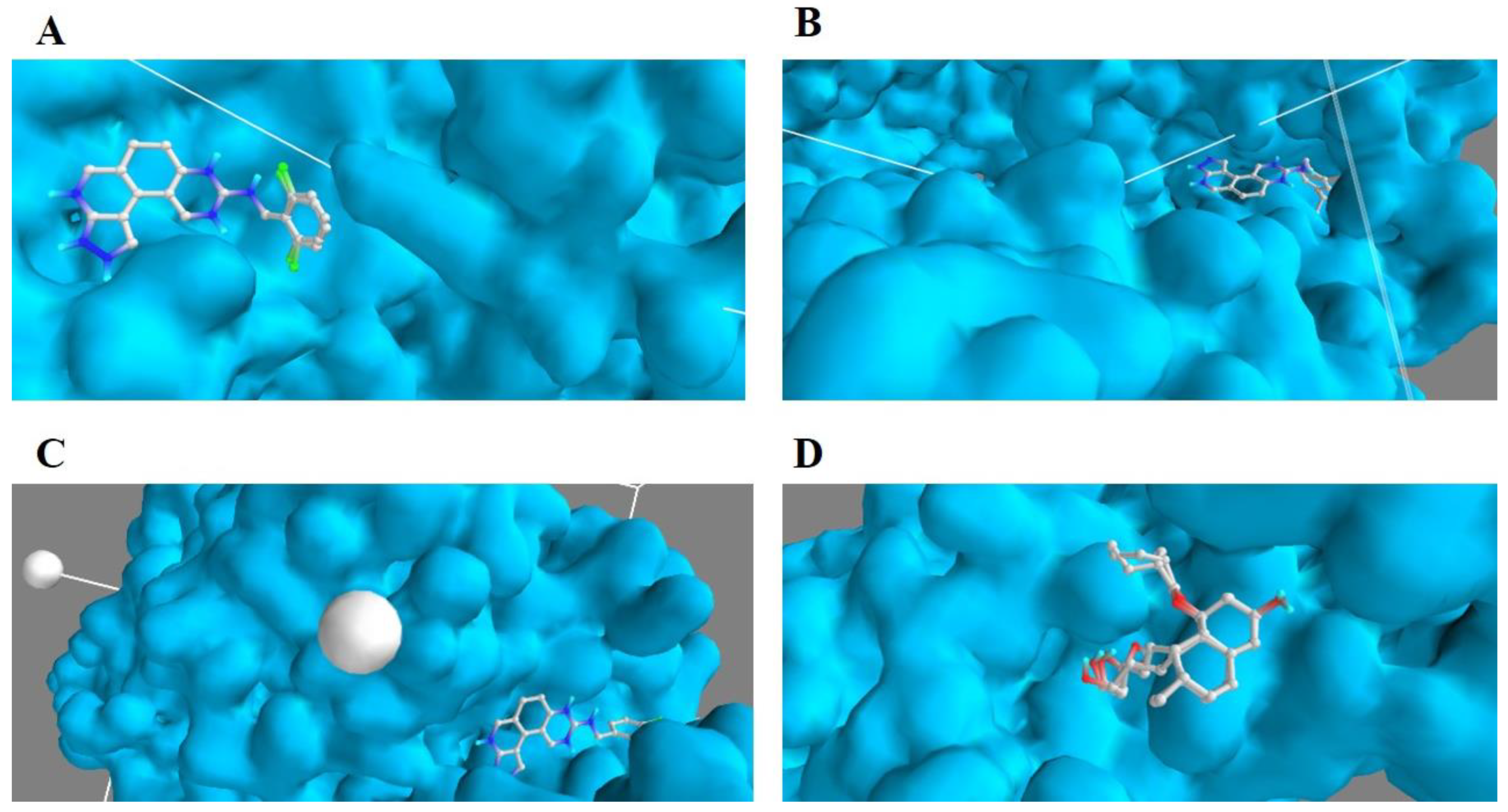
3.2. Molecular Docking Analysis of Selected Test Compounds
3.3. Validation of Docking Protocol
3.4. ADMET Profile
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ∆G | Change in free energy |
| ACE-2 | Angiotensin-converting Enzyme-2 |
| ADMET | Absorption, Distribution, Metabolism, Excretion, Toxicity |
| AMES | Ames Mutagenicity |
| ARG | Arginine |
| ASN | Asparagine |
| ATP | Adenosine Triphosphate |
| CEST | Central European Summer Time |
| COVID | Coronavirus Disease |
| CYP | Cytochrome P450 |
| CYS | Cysteine |
| DILI | Drug-Induced Liver Injury |
| EGFR | Estimated Glomerular Filtration Rate |
| FDA | Food and Drug Administration |
| GLN | Glutamine |
| GLU | Glutamic acid |
| H-bond | Hydrogen bond |
| HCoV-229E | Human Corona Virus from specimen coded 229E |
| HCov-HKU1 | Human Coronavirus from Hong Kong University 1 |
| HCov-NL63 | Human Coronavirus NetherLand63 |
| HCoV-OC43 | Human Coronavirus from Organ Culture 43 |
| hERG | human ether-a-go-go related gene potassium channel |
| H-HT | Human Hepatotoxicity |
| HIS | Histidine |
| ILE | Isoleucine |
| kcal/mol | Kilocalorie per mole |
| LD50 | Lethal Dose 50 |
| LEU | Leucine |
| Log Kp | Logarithmic skin permeation coefficient |
| Log P | Lipophilicity |
| LYS | Lysine |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MET | Methionine |
| SDF | Structural Data File |
| Mpro | Main protease |
| MR | Magnetic Resonance |
| N3 | N-[(5-Methyl-3-isoxazolyl)carbonyl]-L-alanyl-L-valyl-N-[(1S,2E)-4-oxo-1-[[(3S)-2-oxo-3-pyrrolidinyl]methyl]-4-(phenylmethoxy)-2-buten-1-yl]-L-leucinamide |
| NCBI | National Center for Biotechnology Information |
| PDB | Protein Data Bank |
| PDBQT | Protein Data Bank, Partial Charge(Q) and Atom Type(T) |
| Pgp | Protein Gene Product |
| PHE | Phenylalanine |
| PRO | Proline |
| RBD | receptor-binding domain |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| RdRp | RNA-dependent RNA polymerase |
| RNA | Ribonucleic acid |
| SARS-CoV | Severe Acute Respiratory Syndrome Coronavirus |
| SER | Serine |
| Spro | Spike protein |
| TPSA | Topological Polar Surface Area |
| TYR | Tyrosine |
| VAL | Valine |
References
- Amanat, F.; Krammer, F. SARS-CoV-2 vaccines: A status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar]
- World Health Organization—WHO. WHO Coronavirus (COVID-19) Dashboard. 2021. Available online: https://covid19.who.int/ (accessed on 11 October 2021).
- Bager, P.; Ramussen, M.W.; Albertsen, M.; Krause, T.G. Hospitalisation associated with SARS-CoV-2 delta variant in Denmark. Lancet 2021, 21, 1351. [Google Scholar] [CrossRef]
- Khan, A. Has the Delta Variant Changed the Symptoms of COVID-19? Aljazeera. 2021. Available online: https://www.aljazeera.com/features/2021/9/22/has-the-delta-variant-changed-the-symptoms-of-covid-19 (accessed on 23 September 2021).
- Johnson, T.O.; Adegboyega, A.E.; Iwaloye, O.; Eseola, O.A.; Plass, W.; Afolabi, B.; Rotimi, D.; Ahmed, E.I.; Albrakati, A.; Batiha, G.E.; et al. Computational study of the therapeutic potentials of a new series of imidazole derivatives against SARS-CoV-2. J. Pharmacol. Sci. 2021, 147, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, anti-viral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2021, 39, 3409–3418. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shabat, S.; Yarmolinsky, L.; Porat, D.; Dahan, A. Anti-viral effect of phytochemicals from medicinal plants: Applications and drug delivery strategies. Drug Del. Trans. Res. 2020, 10, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.O.; Kim, Y.; Lovell, S.; Rathnayake, A.D.; Groutas, W.C. Anti-viral drug discovery: Norovirus proteases and development of inhibitors. Viruses 2019, 11, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elekofehinti, O.O.; Iwaloye, O.; Famusiwa, C.D.; Akinseye, O.; Rocha, J.B. Identification of main protease of coronavirus SARS-CoV-2 (Mpro) inhibitors from melissa officinalis. Curr. Drug Discov. Technol. 2020, 17, e17092020186048. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668. [Google Scholar] [CrossRef]
- Chukwudozie, O.S.; Duru, V.C.; Ndiribe, C.C.; Aborode, A.T.; Oyebanji, V.O.; Emikpe, B.O. The Relevance of Bioinformatics Applications in the Discovery of Vaccine Candidates and Potential Drugs for COVID-19 Treatment. Bioinform. Biol. Insights 2021, 15, 11779322211002168. [Google Scholar] [CrossRef]
- Sumon, T.A.; Hussain, M.; Hasan, M.; Hasan, M.; Jang, W.J.; Bhuiya, E.H.; Chowdhury, A.A.M.; Sharifuzzaman, S.M.; Brown, C.L.; Kwon, H.J.; et al. A revisit to the research updates of drugs, vaccines, and bioinformatics approaches in combating COVID-19 pandemic. Front. Mol. Biosci. 2021, 7, 493. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, G.; Kumar, A.; Rajkhowa, S.; Tiwari, V.K. Synthesis of biologically relevant heterocyclic skeletons under solvent-free condition. In Green Synthetic Approaches for Biologically Relevant Heterocycles; Elsevier: Amsterdam, The Netherlands, 2021; pp. 421–459. [Google Scholar]
- Babalola, B.A.; Adebami, G.E.; Akinsuyi, S.E. Mechanistic basis for Cancer Immune Evasion and role of immune checkpoint blockades in Immuno-Oncology. Glob. J. Cancer Ther. 2021, 7, 35–42. [Google Scholar] [CrossRef]
- Kumura, K.; Wakiyama, Y.; Ueda, K.; Umemura, E.; Watanabe, T.; Kumura, M.; Yoshida, T.; Ajito, K. Synthesis and antibacterial activity of novel lincomycin derivatives. II. Exploring (7 S)-7-(5-aryl-1,3,4-thiadiazol-2-yl-thio)-7-deoxylincomycin derivatives. J. Antibiot. Res. 2017, 70, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, K.; Sun, Y.; Sun, L.; Wei, Z.; Guo, H.; Wu, J.; Liu, M. Design, Synthesis, and in vitro Antibacterial Activity of Fluoroquinolone Derivatives Containing a Chiral 3-(Alkoxyimino)-2-(aminomethyl) azetidine Moiety. Chem. Med. Chem. 2012, 7, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G. Azetidines of pharmacological interest. Arch. Pharm. 2021, 354, e2100062. [Google Scholar] [CrossRef] [PubMed]
- Tarr, J.C.; Wood, M.R.; Noetzel, M.J.; Melancon, B.J.; Lamsal, A.; Luscombe, V.B.; Rodriguez, A.L.; Byers, F.W.; Chang, S.; Cho, H.P.; et al. Challenges in the development of an M4 PAM preclinical candidate: The discovery, SAR, and biological characterization of a series of azetidine-derived tertiary amides. Bioorg. Med. Chem. Lett. 2017, 27, 5179–5184. [Google Scholar] [CrossRef] [PubMed]
- Maetani, M.; Kato, N.; Jabor, V.A.; Calil, F.A.; Nonato, M.C.; Scherer, C.A.; Schreiber, S.L. Discovery of antimalarial Azetidine-2-carbonitriles that inhibit P. falciparum dihydroorotate dehydrogenase. ACS Med. Chem. Lett. 2017, 8, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Maetani, M.; Zoller, J.; Melillo, B.; Verho, O.; Kato, N.; Pu, J.; Comer, E.; Schreiber, S.L. Synthesis of a bicyclic azetidine with in vivo antimalarial activity enabled by stereospecific, directed C (sp3)–H arylation. J. Am. Chem. Soc. 2017, 139, 11300–11306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, A.; Lofberg, C.; Antonsson, M.; von Unge, S.; Hayes, M.A.; Judkins, R.; Ploj, K.; Benthem, L.; Linden, D.; Brodin, P.; et al. Discovery of (3-(4-(2-oxa-6-azaspiro [3.3] heptan-6-ylmethyl) phenoxy) azetidin-1-yl)(5-(4-methoxyphenyl)-1, 3, 4-oxadiazol-2-yl) methanone (AZD1979), a melanin concentrating hormone receptor 1 (MCHr1) antagonist with favorable physicochemical properties. J. Med. Chem. 2016, 59, 2497–2511. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, V.K.; Janero, D.R.; Makriyannis, A. Pharmacotherapeutic targeting of the endocannabinoid signaling system: Drugs for obesity and the metabolic syndrome. Physiol. Behav. 2008, 93, 671–686. [Google Scholar]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A.; et al. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [PubMed]
- Metkar, S.D.; Bhatia, M.S.; Desai, U.V. Synthesis and biological evaluation of novel azetidine derivatives as dopamine antagonist. Med. Chem. Res. 2013, 22, 5982–5989. [Google Scholar] [CrossRef]
- Hart, T.; Macias, A.T.; Benwell, K.; Brooks, T.; D’Alessandro, J.; Dokurno, P.; Francis, G.; Gibbons, B.; Haymes, T.; Kennett, G.; et al. Fatty acid amide hydrolase inhibitors. Surprising selectivity of chiral azetidine ureas. Bioorg. Med. Chem. Lett. 2009, 19, 4241–4244. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Malhotra, N.; Yogavel, M.; Harlos, K.; Melilo, B.; Comer, E.; Gonse, A.; Parvez, S.; Mitasev, B.; Fang, F.G.; et al. Structural basis of malaria parasite phenylalanine tRNA-synthetase inhibition by bicyclic azetidines. Nat. Commun. 2021, 12, 343. [Google Scholar] [CrossRef]
- Yue, P.; Lopez-Tapia, F.; Zhu, Y.; Brotherton-Pleiss, C.; Fu, W.; Alonso-Valenteen, F.; Mikhael, S.; Medina-Kauwe, L.; Tius, M.; Turkson, J. High-affinity azetidine-based small-molecules as a new class of direct inhibitors of STAT3 activity and breast cancer phenotype (abstract). In Proceedings of the American Association for Cancer Research Annual Meeting, Philadelphia, PA, USA, 10–15 April and 17–21 May 2021; Volume 81. [Google Scholar]
- Kim, S.H.; Semenya, D.; Castagnolo, D. Antimicrobial drugs bearing guanidine moieties: A review. Eur. J. Med. Chem. 2021, 216, 113293. [Google Scholar] [CrossRef]
- Shen, Y.; You, Q.; Wu, Y.; Wu, J. Inhibition of PAD4-mediated NET formation by cl-amidine prevents diabetes development in nonobese diabetic mice. Eur. J. Pharmacol. 2021, 174623. [Google Scholar] [CrossRef] [PubMed]
- Čikoš, A.; Dragojević, S.; Kubiček, A. Degradation products of azetidine core G334089–Isolation, structure elucidation and pathway. J. Pharm. Biomed. Anal. 2021, 203, 114232. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; Yagen, B.; Mechoulam, R.; Becker, Y. Structure-activity relationships of pyrrole amidine antiviral antibiotics. 2. Preparation of mono-and tripyrrole derivatives of congocidine. J. Med. Chem. 1980, 23, 1144–1148. [Google Scholar] [CrossRef]
- Naesens, L.; Guddat, L.W.; Keough, D.T.; van Kuilenburg, A.B.; Meijer, J.; Voorde, J.V.; Balzarini, J. Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T-705 (favipiravir). Mol. Pharmacol. 2013, 84, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, G.C.; Martins, L.M.; Bregadiolli, B.A.; Moreno, V.F.; da Silva-Filho, L.C.; da Silva, B.H.S.T. Heterocyclic compounds as anti-viral drugs: Synthesis, structure–activity relationship and traditional applications. J. Heterocycl. Chem. 2021. [Google Scholar] [CrossRef]
- Głowacka, I.E.; Grabkowska-Drużyc, M.; Andrei, G.; Schols, D.; Snoeck, R.; Witek, K.; Podlewska, S.; Handzlik, J.; Piotrowska, D.G. Novel N-Substituted 3-Aryl-4-(diethoxyphosphoryl)azetidin-2-ones as Antibiotic Enhancers and Antiviral Agents in Search for a Successful Treatment of Complex Infections. Int. J. Mol. Sci. 2021, 22, 8032. [Google Scholar] [CrossRef]
- Hasan, M.J.; Rabbani, R.; Anam, A.M.; Huq, S.M.R.; Polash, M.M.I.; Nessa, S.S.T.; Bachar, S.C. Impact of high dose of baricitinib in severe COVID-19 pneumonia: A prospective cohort study in Bangladesh. BMC Infect Dis. 2021, 21, 427. [Google Scholar] [CrossRef] [PubMed]
- Boghuma, K.T.; Monica, M.F.; Ashish, M.; Randi, C.S.; Abeer, M.; Sushma, K.C.; Jesse, O.S.; Kathryn, D.S.; Bonnie, C.; Alex, E.; et al. Use of Baricitinib in Patients with Moderate to Severe Coronavirus Disease 2019. Arch. Clin. Infect. Dis. 2021, 72, 1247–1250. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminfor 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, G.O.; Afees, O.J.; Nwamaka, N.C.; Simon, N.; Oluwaseun, A.A.; Soyinka, T.; Oluwaseun, A.S.; Bankole, S. Selection of Luteolin as a potential antagonist from molecular docking analysis of EGFR mutant. Bioinformation 2018, 14, 241–247. [Google Scholar] [CrossRef]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.P.; Cao, D.S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef]
- Singh, S.; Khera, D.; Chugh, A.; Khera, P.S.; Chugh, V.K. Efficacy and safety of remdesivir in COVID-19 caused by SARS-CoV-2: A systematic review and meta-analysis. BMJ Open 2021, 11, e048416. [Google Scholar] [CrossRef]
- Young, B.; Tan, T.T.; Leo, Y.S. The place for remdesivir in COVID-19 treatment. Lancet Infect. Dis. 2021, 21, 20–21. [Google Scholar] [CrossRef]
- Liu, L.; Hu, Y.; Shen, Y.F.; Wang, G.X.; Zhu, B. Evaluation on anti-viral activity of coumarin derivatives against spring viraemia of carp virus in epithelioma papulosum cyprini cells. Antivir. Res. 2017, 144, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.K.; Ghosh, S.; Naesens, L.; Bhat, H.R.; Singh, U.P. Facile synthesis, antimicrobial and anti-viral evaluation of novel substituted phenyl 1, 3-thiazolidin-4-one sulfonyl derivatives. Bioorg. Chem. 2021, 114, 105153. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, A.; Ahmad, M.; Aslam, S.; Naqvi, S.A.R.; Saif, M.J. Recent advances in anti-viral benzimidazole derivatives: A mini review. Pharmaceut. Chem. J. 2019, 53, 179–187. [Google Scholar] [CrossRef]
- Singh, V.P. Aromatic interactions in biological systems. Sci. Technol. Jpn. 2015, 3, 42–48. [Google Scholar]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Pant, S.; Zhang, Q.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Unusual zwitterionic catalytic site of SARS–CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. [Google Scholar] [CrossRef]
- Novak, J.; Rimac, H.; Kandagalla, S.; Pathak, P.; Grishina, M.; Potemkin, V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Pagadala, N.S.; Landi, A.; Maturu, P.; Tuszynski, J. In silico identification of RBD subdomain of spike protein from Pro322-Thr581 for applications in vaccine development against SARS-CoV2. J. Mol. Struct. 2021, 1240, 130534. [Google Scholar] [CrossRef]
- López-Cortés, G.I.; Palacios-Pérez, M.; Zamudio, G.S.; Velediaz, H.F.; Ortega, E.; Jose, M.V. Neutral evolution test of the spike protein of SARS-CoV-2 and its implications in the binding to ACE2. Sci. Rep. 2021, 11, 18847. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.P. Conformational Analysis between 6M71 (SARS COV2 RNA-Dependent RNA Polymerase) and CHEMBL3120791 using GROMACS Molecular Dynamic Simulation. 2021. Available online: https://chemrxiv.org/engage/api-gateway/chemrxiv/assets/orp/resource/item/60e1686a9bb5dd588390ddef/original/conformational-analysis-between-6m71-sars-cov2-rna-dependent-rna-polymerase-and-chembl3120791-using-gromacs-molecular-dynamic-simulation.pdf (accessed on 17 August 2021).
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-score—A comprehensive scoring function for evaluation of chemical drug-likeness. MedChemComm 2018, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Ursu, O.; Rayan, A.; Goldblum, A.; Oprea, T.I. Understanding drug-likeness. WIREs Comput. Mol. Sci. 2011, 1, 760–781. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computa-tional approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecularproperties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; al Habet, S.; Baweja, R.K.; Burckart, G.J.; et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
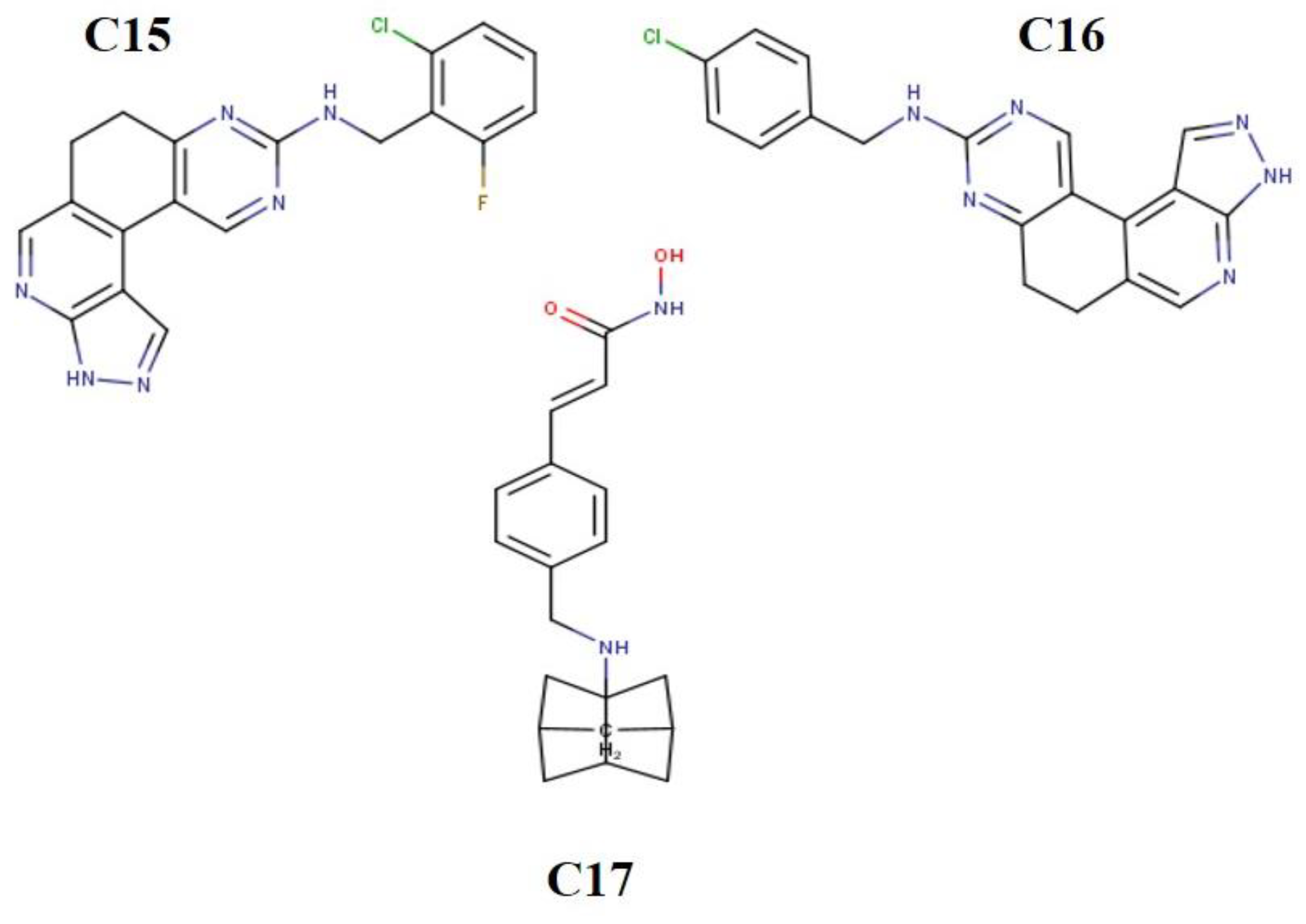{kind=link}
{kind=link}
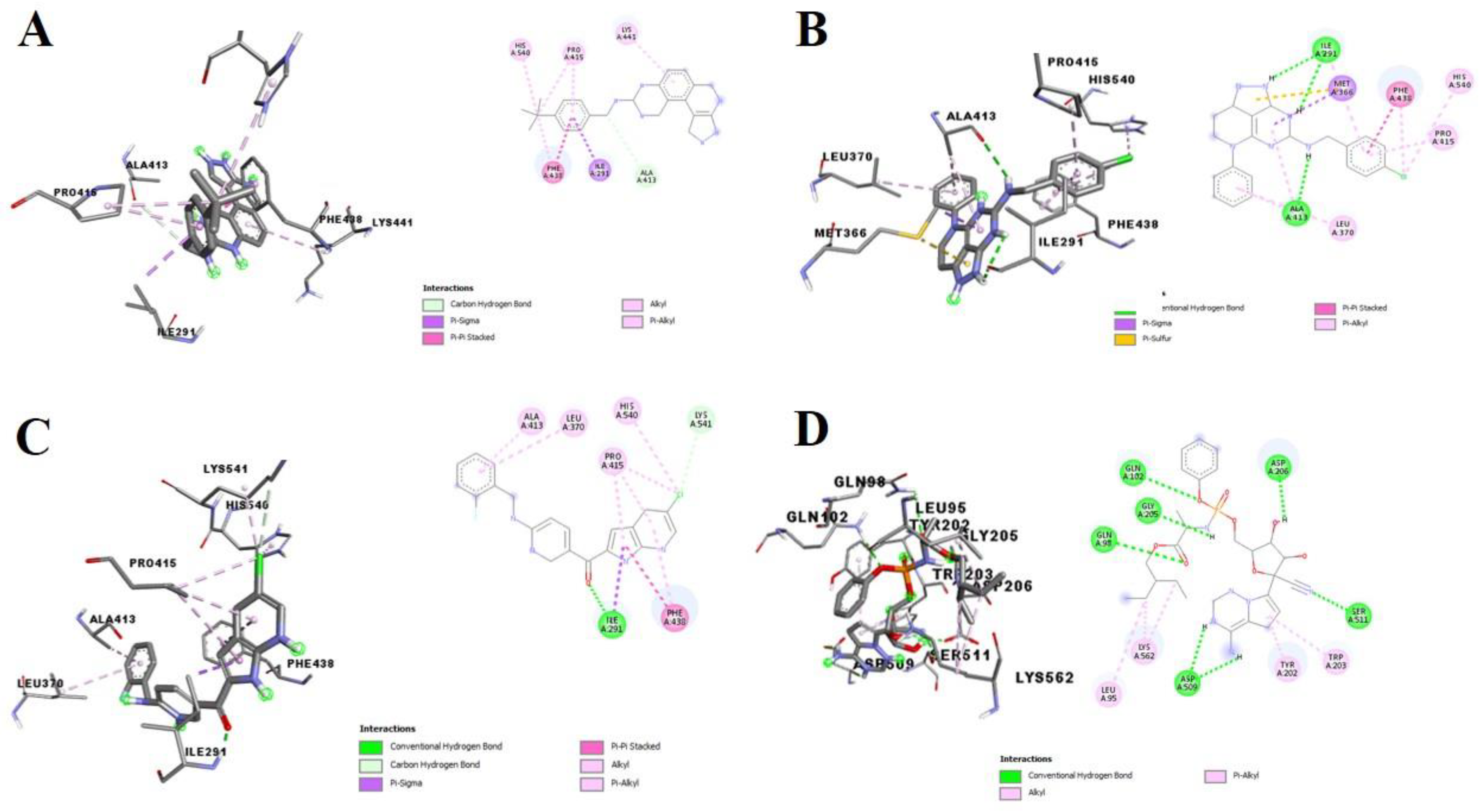{kind=link}
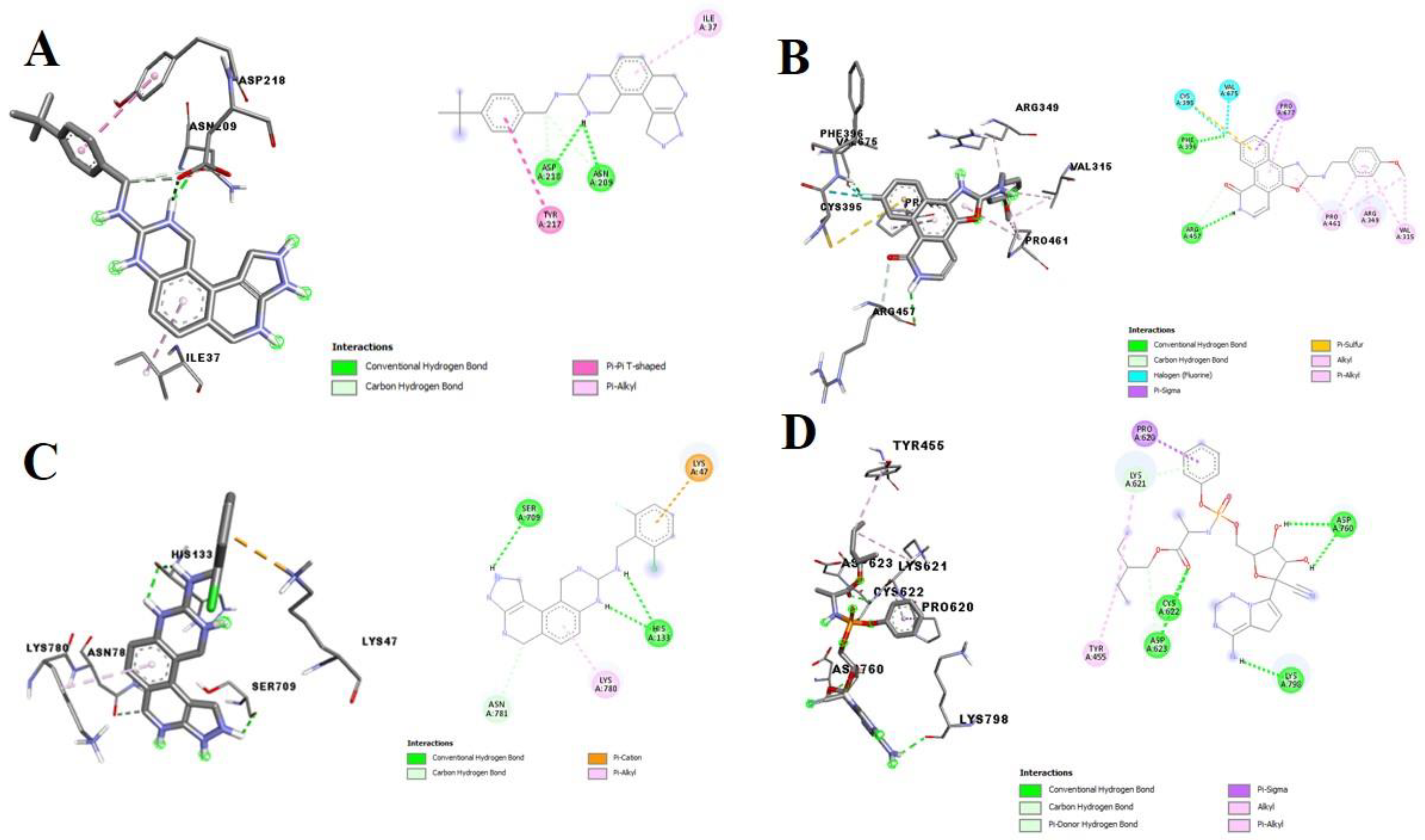{kind=link}
{kind=link}
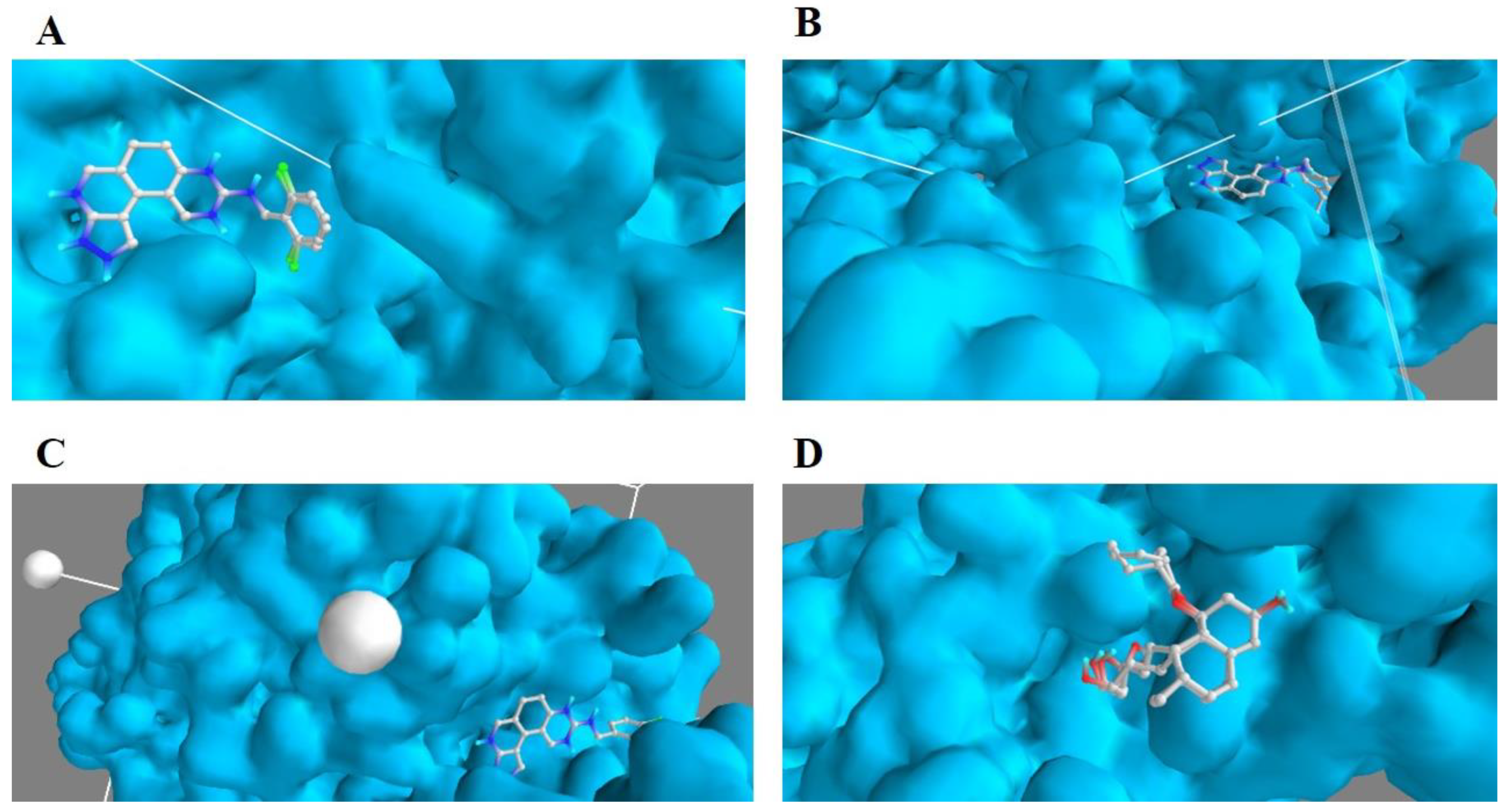{kind=link}
| Compounds | ∆G Energy (Kcal/mol) | ||
|---|---|---|---|
| Mpro (6LU7) | Spro (6LZG) | RdRp (6M71) | |
| Standard ligands | |||
| Remdesivir | −7.1 | −6.8 | −7.6 |
| N3 | −7.3 | −6.3 | −5.6 |
| Derivatives | |||
| C1 | −7.3 | −9.7 | −8.1 |
| C2 | −7.3 | −10.1 | −8.9 |
| C3 | −8.1 | −9.3 | −8.4 |
| C4 | −7.9 | −10.2 | −8.5 |
| C5 | −7.5 | −9.8 | −8.7 |
| C6 | −8.2 | −9.2 | −8.3 |
| C7 | −7.6 | −9.8 | −7.5 |
| C8 | −7.9 | −9.5 | −8.9 |
| C9 | −7.6 | −9.4 | −9.0 |
| C10 | −8.0 | −8.8 | −8.6 |
| C11 | −7.8 | −10.3 | −8.5 |
| C12 | −8.7 | −9.4 | −8.9 |
| C13 | −8.3 | −10.6 | −9.5 |
| C14 | −8.6 | −9.1 | −8.7 |
| C15 | −8.8 | −9.6 | −8.9 |
| C16 | −8.4 | −9.8 | −8.8 |
| C17 | −7.1 | −9.9 | −7.6 |
| Parameters | C4 | C9 | C11 | C12 | C13 | C14 | C15 | N3 | Remdesivir |
|---|---|---|---|---|---|---|---|---|---|
| Molecular weight (g/mol) | 380.8 | 389.38 | 376.84 | 362.82 | 384.48 | 397.26 | 380.81 | 290.36 | 602.58 |
| Consensus Log P | 3.98 | 4.1 | 3.63 | 3.27 | 3.93 | 3.78 | 3.55 | 1.48 | 1.56 |
| Log Sw (Silicos-IT) | 5.03 | 5 | 3.72 | 4.22 | 4.92 | 4.85 | 4.63 | 2.35 | −0.05 |
| Solubility class | Moderately soluble | Moderately soluble | Moderately soluble | Moderately soluble | Moderately soluble | Moderately soluble | Moderately soluble | soluble | Moderately soluble |
| #Heavy atoms | 27 | 29 | 27 | 26 | 29 | 27 | 27 | 21 | 42 |
| #Aromatic heavy atoms | 21 | 23 | 21 | 21 | 21 | 21 | 21 | 6 | 15 |
| Fraction Csp3 | 0.05 | 0.09 | 0.15 | 0.16 | 0.3 | 0.16 | 0.16 | 0.5 | 0.48 |
| #Rotatable bonds | 5 | 4 | 4 | 3 | 4 | 3 | 3 | 8 | 14 |
| #H-bond acceptors | 4 | 5 | 3 | 4 | 4 | 4 | 5 | 4 | 12 |
| #H-bond donors | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 |
| MR | 102.52 | 109.88 | 110.72 | 101.54 | 115.8 | 106.55 | 101.49 | 83.47 | 150.43 |
| TPSA (Å2) | 70.67 | 80.15 | 69.73 | 79.38 | 79.38 | 79.38 | 79.38 | 81.42 | 213.36 |
| Lipinski violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Ghose violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Veber violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Egan violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Muegge violations | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.17 |
| Synthetic availability | 2.75 | 3.07 | 3.13 | 3.04 | 3.45 | 3.07 | 3.11 | 2.9 | 6.33 |
| Parameters | C4 | C9 | C11 | C12 | C13 | C14 | C15 | N3 | Remdesivir |
|---|---|---|---|---|---|---|---|---|---|
| GI Absorption | High | High | High | High | High | High | High | High | Low |
| Blood-brain permeant | Yes | No | Yes | No | No | Yes | No | No | No |
| Pgp substrate | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| CYP1A2 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No |
| CYP2C9 inhibitor | Yes | Yes | Yes | No | Yes | Yes | Yes | No | No |
| CYP2D6 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | No |
| CYP3A4 inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Skin permeant Log Kp (cm/s) | −5.46 | −5.62 | −5.27 | −6.04 | −5.42 | −5.27 | −6.07 | −7.50 | −8.62 |
| Parameters | C4 | C9 | C11 | C12 | C13 | C14 | C15 | N3 | Remdesivir |
|---|---|---|---|---|---|---|---|---|---|
| hERG-Blockers | - | - | - | - | - | - | - | - | - |
| H-HT (Human Hepatotoxicity) | - | - | - | - | - | - | - | - | - |
| AMES (Ames Mutagenicity) | - | - | - | - | - | - | - | - | - |
| LD50 (LD50 of acute toxicity) | 2.503-log mol/kg (1195.937 mg/kg) | 2.616-log mol/kg (942.712 mg/kg) | 2.569-log mol/kg (1016.646 mg/kg) | 2.579-log mol/kg (956.524 mg/kg) | 2.704-log mol/kg (760.119 mg/kg) | 2.651-log mol/kg (887.329 mg/kg) | 2.598-log mol/kg (960.977 mg/kg) | 2.452-log mol/kg (1025.513 mg/kg) | 2.989-log mol/kg (618.042 mg/kg) |
| DILI (Drug Induced Liver Injury) | - | - | - | - | - | - | - | - | - |
| FDAMDD (Maximum Recommended Daily Dose) | - | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babalola, B.A.; Adetobi, T.E.; Akinsuyi, O.S.; Adebisi, O.A.; Folajimi, E.O. Computational Study of the Therapeutic Potential of Novel Heterocyclic Derivatives against SARS-CoV-2. COVID 2021, 1, 757-774. https://doi.org/10.3390/covid1040061
Babalola BA, Adetobi TE, Akinsuyi OS, Adebisi OA, Folajimi EO. Computational Study of the Therapeutic Potential of Novel Heterocyclic Derivatives against SARS-CoV-2. COVID. 2021; 1(4):757-774. https://doi.org/10.3390/covid1040061
Chicago/Turabian StyleBabalola, Benjamin Ayodipupo, Tosin Emmanuel Adetobi, Oluwamayowa Samuel Akinsuyi, Otunba Ahmed Adebisi, and Elizabeth Oreoluwa Folajimi. 2021. "Computational Study of the Therapeutic Potential of Novel Heterocyclic Derivatives against SARS-CoV-2" COVID 1, no. 4: 757-774. https://doi.org/10.3390/covid1040061
APA StyleBabalola, B. A., Adetobi, T. E., Akinsuyi, O. S., Adebisi, O. A., & Folajimi, E. O. (2021). Computational Study of the Therapeutic Potential of Novel Heterocyclic Derivatives against SARS-CoV-2. COVID, 1(4), 757-774. https://doi.org/10.3390/covid1040061