1. Introduction
The current COVID-19 pandemic caused by a novel coronavirus, SARS-CoV-2, started in December 2019 in Wuhan, China [
1,
2,
3,
4]. Understanding the interactions between human cells and SARS-CoV-2 is crucial for developing effective measures for medical intervention, such as vaccines and drugs. In general, the human immune system efficiently recognizes and eliminates foreign proteins but does not attack its own human proteins. This self/nonself discrimination is critical for immunological tolerance to avoid the development of autoimmune diseases, and it is antigen presentation by MHC (major histocompatibility complex) or HLA (human leukocyte antigen) class I and class II molecules that realizes this discrimination process [
5,
6,
7]. Pathogenic proteins are processed in professional antigen-presenting cells such as dendritic cells (DCs) and are presented as peptides on their surfaces. MHC class I cross-presentation of peptides derived from pathogens by DCs is important for the activation of CD8
+ cytotoxic T lymphocytes (CTLs) [
5,
6,
7]. Activation of CTLs is important for eliminating virus-infected cells because antibodies cannot recognize viral proteins inside cells. Therefore, an integrated strategy to overcome COVID-19 is not only to promote both efficient production of neutralizing antibodies and activation of CTLs against SARS-CoV-2 proteins but also to repress the production of self-targeted antibodies and CTLs to avoid potential side effects.
The potential risks of autoimmune diseases may be particularly relevant when an immunization must be administered repeatedly for years when a vaccine contains potential epitopes that are similar to those of humans. Additional concern on the potential autoimmune reaction to SARS-CoV-2 vaccines comes from the fact that various autoantibodies are produced in COVID-19 patients; clinical manifestations of COVID-19 resemble those of autoimmune diseases [
8,
9,
10]. Although its mechanism of SARS-CoV-2-induced autoantibody production is unclear at this moment, there is a possibility that the proteome of SARS-CoV-2 may contain many short amino acid sequences that have high similarities to those of humans. These short sequences would be overlooked by researchers because the BLAST (Basic Local Alignment Tool), the most popular alignment program, does not report such similarities among short sequences.
Peptides presented by MHC class II can be longer than 11 mer peptides, but those presented by MHC class I are constrained by the binding of their ends; 8–10 mer peptides are suitable for presentation [
11,
12]. Presented peptides are further constrained by interactions between amino acid (aa) residues within the peptides themselves, often at their middle positions [
13]. Because a presented peptide is held by an MHC class I molecule, only one side of the peptide is accessible from extracellular molecules. As a result, only a limited number of aa residues in the peptide may be recognized by T-cell receptors (TCRs). We speculate that a 5 aa stretch may be the minimum unit for recognition by TCRs. This may also be applicable to other systems. Molecular recognition by B-cell receptors (BCRs) and their corresponding antibodies is also mediated via short aa sequences, and an antibody often recognizes a few different short aa sequences that may form a 3-dimensional (3D) structure. In fact, several short epitopes that are recognized by neutralizing antibodies against SARS-CoV-2 spike (S) protein have already been identified [
14,
15,
16,
17,
18,
19,
20,
21,
22]. In addition to the DCs involved in antigen presentation, a subset of CD4
+ T cells, regulatory T (T
reg) cells, is responsible for the critical process of self/nonself discrimination [
23,
24].
For simplicity, we assume that the immune system preferentially (although not exclusively) uses short constituent sequences (SCSs) of 5 aa residues (5-aa SCSs, which may also be called pentats, pentapeptides, 5 mers, or other terms) as a recognition unit to perform these functions. This assumption may not be wholly accurate, because the presented peptides are longer than 5 aa, but we believe that 5 aa is an optimal SCS size because molecular recognition is often mediated via smaller SCSs. Molecular recognition by longer SCSs (6 aa or longer) may also be feasible, but longer SCSs can be realized as combinatorial use of two or more 5 aa SCSs. Although 3-aa and 4-aa SCSs are computationally more tractable, they may be too short to function as recognition units for epitopes, and their repertoire (203 for 3-aa SCS and 204 for 4-aa SCS) may be too small to fully describe sequence variations of larger datasets such as the human proteome. Thus, from the viewpoints of both immunology and computation, it is reasonable to start bioinformatics based on 5-aa SCS distributions (and then to extend the results to longer sequences as clusters (consecutive or overlapping sequences) of 5-aa SCSs) in a host-pathogen system.
Self/nonself discrimination can be conceptually understood as a process for the immune system to scan all possible 5-aa SCSs in the human proteome to remember and tolerate these SCSs as “self” and then to recognize and eliminate 5-aa SCSs that are not remembered as “nonself”, as far as linear epitopes are concerned. This in vivo process can be performed in silico bioinformatically when all protein sequences of both host and pathogenic organisms are available. Under the above assumption, it is critical to note that a given foreign protein may contain both self and nonself 5-aa SCSs for the host. Therefore, we believe that SCS search studies can be applicable to immunological systems and may play an indispensable role in vaccine research.
The importance of 5-aa SCSs coincides with the usefulness of 5-aa SCSs in bioinformatics, as discussed below. In proteins, the frequencies of the 20 species of amino acids are not random; each amino acid has its own unique frequency [
25,
26,
27,
28]. Furthermore, the frequencies of SCS species are not random in proteins; each SCS has its own unique frequency that deviates from the probabilistically expected frequency [
27,
28,
29,
30,
31]. Among
n-aa SCSs, a set of 5-aa SCSs is practically most useful and structurally relevant, mainly for the following reasons. There are exactly 3.2 million (20
5) species of 5-aa SCSs, and this number is comparable to the number of 5 aa SCSs as components in many proteomes, such as the human proteome [
27,
28]. It is thus practically reasonable to characterize a collection of proteins or a proteome using 5 aa SCSs. Once the distribution patterns of 5 aa SCSs are obtained, longer sequences may be examined as adjacent clusters of 5 aa SCSs in proteins. Furthermore, 5-aa SCSs may be considered basic structural and functional units of protein sequences; 5 aa fragments may function as building blocks of protein structures [
32,
33]. Thus, in the present study, we have focused on 5-aa SCSs, which are simply described as SCSs unless noted otherwise.
The SCS concept may be useful for the elucidation of host-parasite interactions in general. For any parasite (including pathogen) to avoid being recognized and eliminated by the host immune system, their protein sequences might have been selected for similarity to the host’s repertoire of protein sequences. In this process, nonself sequences in a parasite proteome may evolve over time to resemble self sequences if such mutations are functionally allowed. In other words, nonself sequences that cannot be easily changed to self sequences (invariant nonself sequences) in a parasite proteome may be functionally important for that parasite. This sequence mimicry hypothesis in host-parasite interactions can be investigated by SCS analysis. Remarkably, it has been shown that the usage diversity (“vocabularies”) of 5-aa SCSs in proteomes is lower in parasitic organisms than in nonparasitic organisms with a similar phylogenetic status, suggesting that parasites might have reduced their vocabularies during evolution to escape immunological recognition by the host [
34]. The SCS approach is a kind of linguistic approach that analyzes frequencies of “words” in a large number of proteins [
35,
36], which is suitable to examine the sequence mimicry hypothesis.
To understand the self/nonself relationship between humans and SARS-CoV-2 and to find better epitopes for future vaccines, we first characterized the human proteome from the perspective of SCS distributions and then analyzed the SARS-CoV-2 proteome based on the SCS distributions in the human proteome. It is notable that the human proteome has been analyzed before, together with other mammalian proteomes, to search for human-specific proteins using the SCS methods [
37], demonstrating a promising way of analyzing protein aa sequences, but the present study incorporates the self/nonself concept; our SCS analyses here revealed self and nonself SCSs in the SARS-CoV-2 proteome. The use of such nonself sequences as vaccine targets may promote efficient production of neutralizing antibodies and activation of CTLs against SARS-CoV-2 but repress the production of self-targeted antibodies and CTL responses against noninfected cells. Thus, nonself sequences may serve as potential epitopes for highly efficient and safe vaccines that are suitable for long-term usage to resolve the current COVID-19 pandemic. Moreover, we analyzed self/nonself status changes associated with point mutations in the SARS-CoV-2 proteome. Since the exact time point of the host shift of this coronavirus from bats (or other organisms) to humans is known, the accumulation of mutations in SARS-CoV-2 may be an opportunity to observe a process of real-time evolution of the virus and to test the sequence mimicry hypothesis.
2. Materials and Methods
2.1. Genome and Proteome Sequences
The human reference genome sequence was obtained from NCBI (the National Center for Biotechnology Information, Bethesda, MD, USA) at
https://www.ncbi.nlm.nih.gov/datasets/genomes/?acc=GCF_000001405.39 (accessed on 14 November 2020). Protein sequence information in RefSeq: GCF_000001405.39 (NCBI Release 109.20210226 (GRCh38.p13)) in the file “protein.faa” was then examined using our computer programs. All analyses in this study were performed using this human reference proteome.
Similarly, the SARS-CoV-2 reference genome sequence and other related sequences were obtained at
https://www.ncbi.nlm.nih.gov/assembly/GCF_009858895.2/ and at
https://www.ncbi.nlm.nih.gov/assembly/organism/694009/latest/ (accessed on 15 November 2020), in which “protein.faa” files were examined. The SARS-CoV-2 proteome reference sequence used here was ASM985889v3 from NCBI, and 93 variant proteomes excluding the reference proteome were downloaded for variant analysis. Among them, proteomes that were not aligned exactly with the reference proteome due to insertion or deletion were excluded for simplicity; we focused on point mutations that changed amino acids and ignored insertions and deletions, resulting in 68 proteomes (including the single reference proteome) that were analyzed (
Supplementary Materials and Methods; Additional Data 3 at GitHub).
2.2. Frequency Calculations in the Human Proteome
Our strategy was first to focus on 5-aa SCSs comprehensively and to expand the output data to 6-aa and longer SCSs. There are 3.2 million species of 5-aa SCSs theoretically possible because a single position accommodates a single amino acid of a collection of 20 species. To calculate the number of 5-aa SCSs that occur in the protein aa files, the following operations were executed. First, a sequence of “occurrences” with 3.2 million in length was prepared. A bijective function (one-to-one correspondence), scscode: SCS→[0,3199999], was also prepared. Here, occurrence [i] stores the frequency of a 5 aa SCS having scscode i. The initial value of all occurrences (occurrence [0] to occurrence [3199999]) was zero. Second, 5-aa SCSs were counted from protein sequences of the protein aa files by sliding a 5 aa window of one aa residue at a time from the N-terminus to the C-terminus. A 5-aa SCS was converted to scscode, and 1 was added to the occurrence of the corresponding element. This operation was repeated until all 5 aa SCSs were treated. For example, we set scscode (‘AAAAA’) = 0, scscode(‘YYYYY’) = 3,199,999, and scscode (‘MKPAD’) = 1,668,802, and when these 5 aa SCSs were counted, their corresponding occurrence [0], occurrence [319999], and occurrence [1668802] acquired +1 values.
Based on the SCS frequency information obtained above, various basic statistical values were calculated. First, the total number of 5-aa SCSs was obtained by adding all values of occurrence [0] to occurrence [3199999]. Second, the number of 5-aa SCS species that occur in the human proteome was obtained by counting the number of frequency sequence “occurrences” that were not equal to 0. Third, the most frequent and least frequent 5-aa SCSs were obtained by sorting the occurrence values in ascending or descending order. These calculations were performed by Source Codes for Human SCS Analysis available online at
https://adslab-uryukyu.github.io/scs-sars-cov-2/ (accessed on 4 October 2021). Calculations were performed not only for SCS frequencies but also for other tasks such as exact SCSs for a particular rank range and availability scores [
27,
28]. SCS length can be adjusted as 3 aa, 4 aa, or 5 aa residues.
2.3. Self and Nonself Assignments for the SARS-CoV-2 Proteome
Each 5 aa SCS in the SARS-CoV-2 proteome was assigned 0 or 1 at the first position of its amino acid in a protein sequence. To do so, numbers were assigned to proteins in the order of their appearance in the protein aa file. Within a protein, numbers were assigned to aa positions from the N-terminus to the C-terminus. Each aa position was specified in this way. For example, the third amino acid in the sixth protein was signified as 6-3. Using this positional number system, a consecutive 5 aa sequence was extracted; positional numbers were considered not for numbers of aa positions but for numbers of 5 aa SCS positions.
Then, SCS was converted to decimal numbers (n). When occurrence [n] = 0, the corresponding SCS does not exist in the human proteome. That is, this SCS is a zero-count SCS (ZCS), and it is a nonself SCS. In that case, “1” was assigned at the first position of the 5-aa SCS together with its positional number in a csv file. When a 5 aa SCS in question was not a ZCS, it is a self SCS. In that case, “0” was assigned, implying “invisibility” from the host immune system. For example, when SCS = ASDRG, it is a ZCS, and thus, the location number of A such as 2-16 and its identity of 1 were recorded as “2-16, 1” in a csv file.
Above, 5 aa SCSs were converted to decimal numbers in accordance with the letter-to-number correspondence as follows: A = 0, C = 1, D = 2, E = 3, F = 4, G = 5, H = 6, I = 7, K = 8, L = 9, M = 10, N = 11, P = 12, Q = 13, R = 14, S = 15, T = 16, V = 17, W = 18, Y = 19. Converted
n-th letter’s value was set as “SCS_n”. Then, SCS_1, SCS_2, SCS_3, and SCS_4 were multiplied by 20
4, 20
3, 20
2, and 20, respectively. The decimally converted 5 aa SCS was thus expressed as SCS_1 + SCS_2 + SCS_3 + SCS_4 + SCS_5. For example, using M = 10, K = 8, P = 12, A = 0, and D = 2, MKPAD can be converted as follows: 10 × 20
4 + 8 × 20
3 + 12 × 20
2 + 0 × 20 + 2 = 1,600,000 + 64,000 + 4800 + 0 + 2 = 1,668,802. These calculations were performed by Source Codes for SARS-CoV-2 SCS Analysis available online at
https://adslab-uryukyu.github.io/scs-sars-cov-2/ (accessed on 4 October 2021).
2.4. Identification of Nonself SCSs in a 3D Model of the Spike Protein
Nonself SCSs identified as potential candidates for vaccine targets were further examined in their locations in a 3D model of the spike protein. To do so, 3D structural data of the spike protein (PDB ID: 6VYB) [
38] in the Protein Data Bank (PDB) managed by the Research Collaboratory for Structural Bioinformatics (RCSB) were accessed [
39]. This structure has been determined by cryo-EM at a 3.20-Å resolution [
38]. The structure was viewed and the nonself SCSs were highlighted by Mol *, a built-in viewer of the RCSB-PDB [
40].
2.5. Self/Nonself Status Change Frequencies
We first focused on the 68 variant proteomes from NCBI (see
Section 2.1). The numbers of mutations in the 68 SARS-CoV-2 proteomes in reference to the reference proteome (ASM985889v3) were counted manually based on the self (0) or nonself (1) assignments in Microsoft Excel. This is to exclude artifactual mutations due to sequencing or translational errors resulting in frameshifts and unknown amino acid residue X. To examine frequency differences between self-to-nonself and nonself-to-self status changes associated with mutations, the χ
2 test was performed using JSTAT 16.1 (Yokohama, Japan).
We referred to the literature for information regarding cellular infection, spike protein structure, and current vaccines [
41,
42,
43,
44,
45]. Mutational data of spike protein were collected from the literature [
46,
47,
48,
49] (
Supplementary Materials and Methods; Additional Data 4 at GitHub) and were input in csv files, and calculations for self/nonself assignments were performed as above. Status changes were then examined by visual inspections, and the status change frequencies were subjected to the χ
2 test as above.
3. Results
3.1. SCS Distributions in the Human Proteome
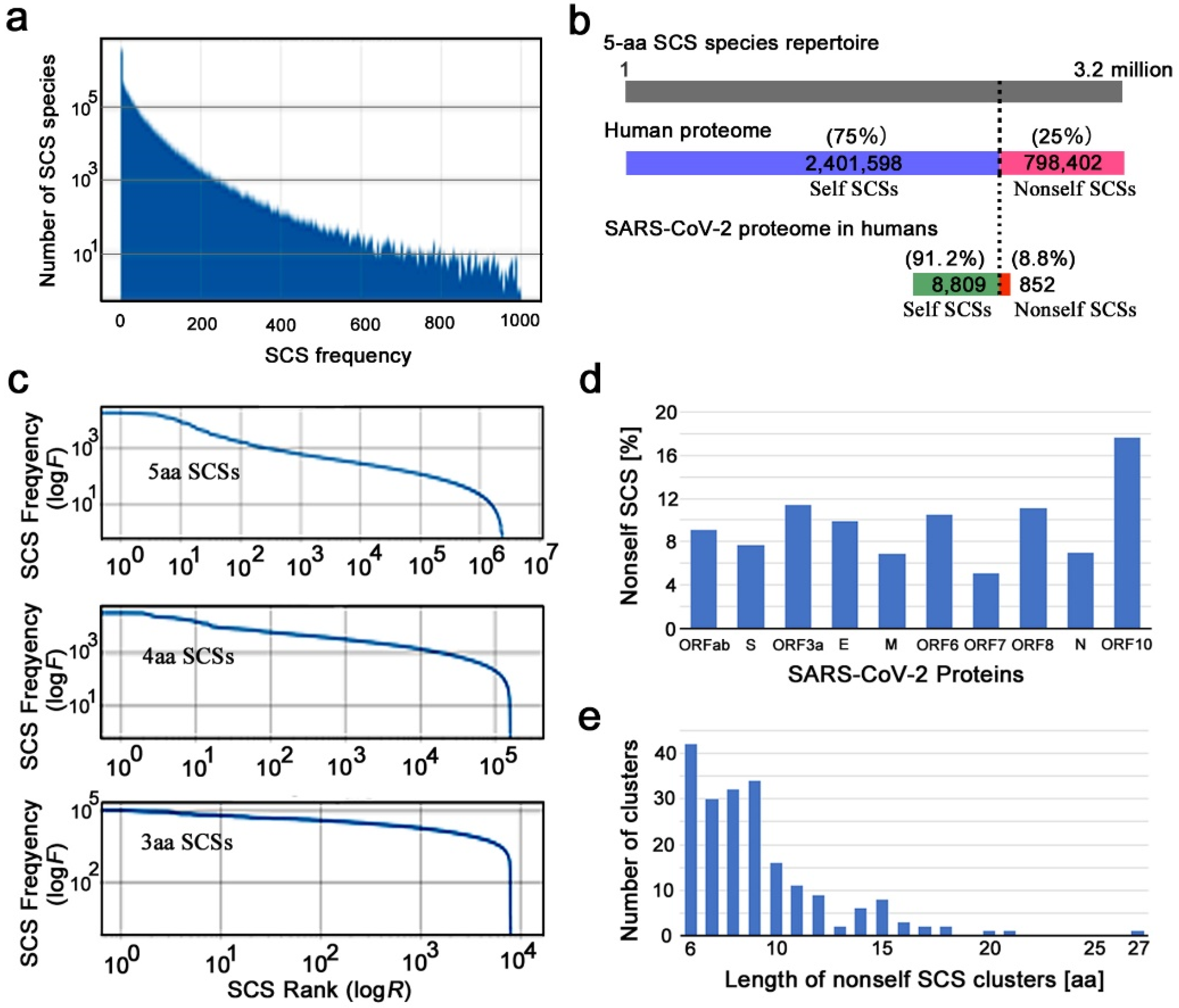
We first characterized the human reference proteome in terms of SCS distributions. The number of 5-aa SCSs in the human reference proteome was 75,727,600 when U (selenocysteine) and X (unknown) residues were included. When nonstandard SCSs containing U or X were excluded, the human reference proteome contained 75,727,187 SCSs. This number is 24 times larger than the number of theoretically possible 5-aa SCS species, which is exactly 205 = 3,200,000. In other words, if these SCS species are present at equal frequency in the human proteome, a given SCS species will be found approximately 24 times in the human proteome. This number suggests that 5-aa SCS usage is reasonable to analyze SCS distributions in the proteome. In contrast, if 6-aa SCSs are used, a given SCS is present only 1.2 times on average, making it more difficult to analyze and interpret SCS distributions rationally despite a need for higher computational power.
The actual human proteome does not satisfy the above assumption of equal frequency of SCS species. Each SCS species had its own unique frequency (maximum = 18,073, minimum = 0, mean = 14.7) in the human proteome; the number of SCS species found with a particular frequency decreased rapidly as the SCS frequency increased (
Figure 1a). The highest number of SCS species was found at zero; that is, these SCS species do not exist at all in the human proteome. Among the 3.2 million SCS species, 2,401,598 (75.05%) were present in the human proteome, whereas 798,402 (24.95%) were not present at all (
Figure 1b). The former SCSs are considered self SCSs because the human self is defined as the collection of these existing SCSs. The latter SCSs are zero-count SCSs (ZCSs) or nonself SCSs.
A rank–frequency plot of SCS species (5-aa, 4-aa, and 3-aa SCSs) showed a nearly linear distribution over several orders of magnitude on the
x-axis (SCS rank) and a few orders of magnitude on the
y-axis (SCS frequency), although the linear distribution was disturbed sharply at the largest rank ranges (
Figure 1c). This result suggests that the SCS distribution in the human proteome has a scale-free nature that follows Zipf’s law (a special case of power law), confirming the results of previous studies [
35,
36]. We focused on 5-aa SCSs and did not pursue shorter ones (4-aa and 3-aa SCSs) in this study. Shorter ones are more tractable computationally but less feasible biologically and immunologically (see
Section 1).
3.2. Self and Nonself SCS Mapping in the SARS-CoV-2 Proteome
We next characterized the SARS-CoV-2 reference proteome based on self and nonself SCS information obtained from the human reference proteome above. The SARS-CoV-2 proteome contains 10 open reading frames that were defined by conceptual translation in the original file: ORF1ab, surface glycoprotein (spike, S), ORF3a, envelope protein (E), membrane glycoprotein (M), ORF6, ORF7a, ORF8, nucleocapsid phosphoprotein (N), and ORF10. We assigned self (0 for invisibility from the host immune system) or nonself (1 for visibility) identity to all SCSs in these proteins (Additional Data 1 at GitHub). There were 8809 (91.18%) self SCSs and 852 (8.82%) nonself SCSs in the SARS-CoV-2 proteome (
Figure 1b). Thus, the majority of SCSs in the SARS-CoV-2 proteome were considered human self SCSs. The percentages of nonself SCSs in each protein varied from 5.13% (ORF7a) to 17.65% (ORF10) (
Figure 1d;
Supplementary Table S1).
Considering that the likely minimum length of peptides presented by MHC class I molecules is 8 aa, consecutive or overlapping nonself SCSs may be able to function as epitopes more efficiently than a single SCS. Here, we define a nonself SCS cluster as two or more nonself SCSs located consecutively or overlapping without a gap. There were exactly 200 such clusters in the SARS-CoV-2 proteome (
Figure 1e;
Supplementary Table S2; Additional Data 2 at GitHub). The majority of nonself clusters were of 6-aa, 7-aa, 8-aa, and 9-aa residues, which all occurred with comparable frequency. The largest cluster in the proteome was a 27 aa segment in ORF1ab. It is to be noted that without considering clusters, the number of nonself 5-aa SCSs in the SARS-CoV-2 proteome was 852 (
Figure 1b).
3.3. Self and Nonself SCS Mapping of the Spike Protein
We next focused on the spike protein of SARS-CoV-2, which has a key role in establishing infection by binding to its receptor, angiotensin-converting enzyme 2 (ACE2) [
38,
41,
42] and has thus been a target of intensive studies for vaccine development [
43,
44,
45]. We assigned self (0) or nonself (1) identity to all SCSs in the linear sequence map of the spike protein (
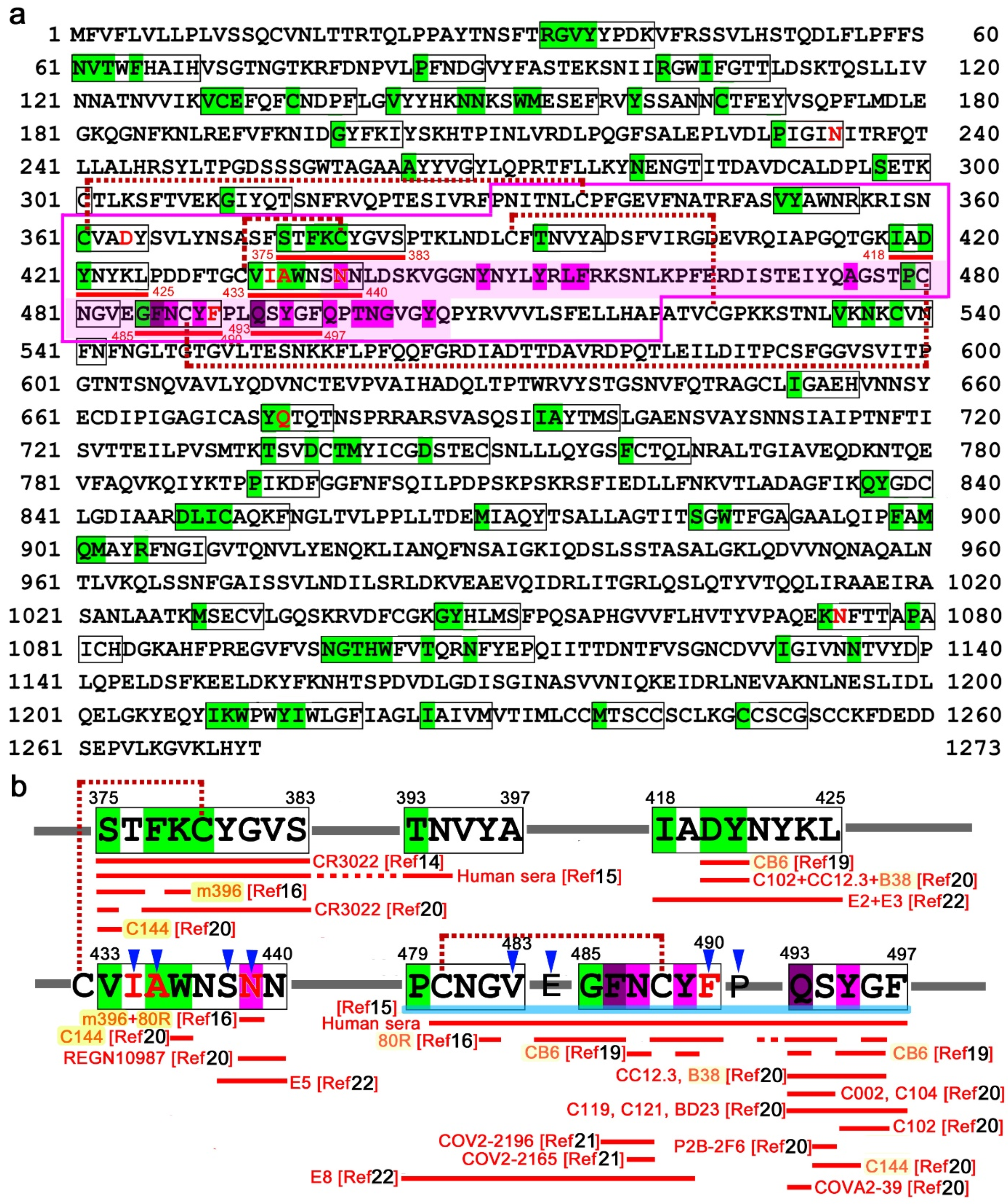
Figure 2a; Additional Data 2 at GitHub). There were 97 nonself SCSs, which was 7.64% of all SCSs in this protein. There were 22 nonself SCS clusters, together with 23 single nonself SCSs, in this protein.
We then focused on the receptor-binding domain (RBD) of the spike protein (
Figure 2b). Just upstream of the receptor-binding motif (RBM) within the RBD, there were two nonself SCS clusters: 375-STFKCYGVS-383 (9 aa) and 418-IADYNYKL-425 (8 aa). Between them, there was a single nonself SCS, 393-TNVYA-397 (5 aa). In the RBM, there were two nonself SCS clusters and two single nonself SCSs: 433-VIAWNSNN-440 (8 aa), 479-PCNGV-483 (5 aa), 485-GFNCYF-490 (6 aa), and 493-QSYGF-497 (5 aa). Three of them were close to one another, forming a 19-aa stretch (P479–F497), which may be considered a supercluster.
Supporting this idea, the cysteine residue in the GFNCYF cluster (C488) forms a disulfide bond with the cysteine residue in the single nonself SCS, PCNGV (C480). Similarly, the cysteine residue within the STFKCYGVS cluster (C379) forms a disulfide bond with the cysteine residue (C432) immediately before the VIAWNSNN cluster within RBM [
16], suggesting that these two clusters together may constitute another supercluster of 17 aa that may form a conformational epitope. Its C-terminal SNN is involved in direct binding to ACE2 [
17].
Other clusters were found outside the RBM and the RBD. The largest cluster, 734-TSVDCTMYICGDSTEC-749 (16 aa), was found on the more C-terminal side, and the two second largest clusters were found on the more N-terminal and C-terminal sides: 143-VYYHKNNKSWMESEF-157 (15 aa) and 1098-NGTHWFVTQRNFYEP-1112 (15 aa). These clusters were nonetheless smaller than the two superclusters in the RBD discussed above, highlighting the potential importance of the latter.
3.4. Known Epitopes of the Spike Protein Recognized by Neutralizing Antibodies
Several functional epitopes of the spike protein that are recognized by neutralizing antibodies against SARS-CoV and SARS-CoV-2 have already been identified [
14,
15,
16,
17,
18,
19,
20,
21,
22]. These epitopes were examined to determine whether they correspond to the nonself SCS clusters identified above (
Figure 2b).
The nonself SCS cluster in the RBD, STFKCYGVS (a portion of the 17-aa supercluster), corresponds to a core portion of the epitope of the neutralizing antibody CR3022 against SARS-CoV [
14,
15], although it does not neutralize SARS-CoV-2. This epitope is exposed to CR3022 only when the spike is in the open conformation and is highly conserved between SARS-CoV and SARS-CoV-2.
Using 42 chemically synthesized peptides and antisera from COVID-19 patients, 4 important epitope sequences in the RBD of the spike protein have been identified [
15]. The first epitope is from S375 to N394, which includes the STFKCYGVS nonself cluster (a portion of the 17-aa supercluster) toward the TNVYA nonself SCS. The fourth epitope is from C480 to P499, which covers the 19-aa supercluster. It appears that human antisera from COVID-19 patients preferentially, although not exclusively, recognize these two superclusters of nonself sequences. Although not neutralizing against SARS-CoV-2, m396 recognizes a few residues in the STFKCYGVS nonself cluster (a portion of the 17-aa supercluster) [
16], and 80R recognizes many residues in the 19-aa supercluster from G482 to G496 [
16].
In contrast, neutralizing antibodies that have binding sites in the core of the ACE-2-binding residues S14P5 and S21P2 [
17] and S309 [
18] all recognize self SCSs but not nonself SCSs, demonstrating that self-targeted antibodies are produced preferentially on some occasions. Another neutralizing antibody, CB6, has been thoroughly studied in terms of residues interacting with the SARS-CoV-2 RBD or with ACE2 [
19]. Most of these residues correspond to self SCSs, but two residues (D420 and Y421) in the RDB, which constitute epitopes of neutralizing antibody CB6 [
19], are located in the nonself SCS cluster, IADYNYKL. CB6 also recognizes N487 and Y489 in the GFNCYF cluster (a middle region of the 19-aa supercluster) [
19].
In a different study [
20], epitopes were mapped for 14 neutralizing antibodies against SARS-CoV-2. The study confirmed that only CR3022 recognizes the STFKCYGVS nonself cluster (a portion of the 17-aa supercluster), except C144, which recognizes a single residue in that cluster [
20]. The IADYNYKL nonself cluster is covered by 3 antibodies, C102, CC12.3, and B38 [
20]. A single nonself SCS, QSYGF (a C-terminal region of the 19-aa supercluster), has ACE2-binding residues, which are covered by 11 antibodies (C102, CC12.3, B38, C002, C104, C119, C121, P2B-2F6, BD23, C144, and COVA2-39) [
20] in addition to CB6 [
19], suggesting that this nonself SCS may function as an efficient epitope. Neutralizing monoclonal antibodies COV2-2196 and COV2-2165 against SARS-CoV-2 recognize F486 and N487 in the GFNCYF nonself cluster (a middle region of the 19-aa supercluster) [
21]. Another extensive large-scale epitope profiling study using a triple-alanine scanning mutagenesis library and a hidden Markov model identified 12 epitopes in RBD [
22]. The IADYNYKL cluster discussed above is entirely covered by the epitopes of E2 and E3 [
22]. The C-terminal SNN of the VIAWNSNN cluster discussed above is covered by E5 [
22], and the PCNGY nonself SCS and the GFNCYF cluster (portions of the 19-aa supercluster) are recognized by E8 [
22].
3.5. 3D Locations of the Nonself SCSs in the RBD of the Spike Protein
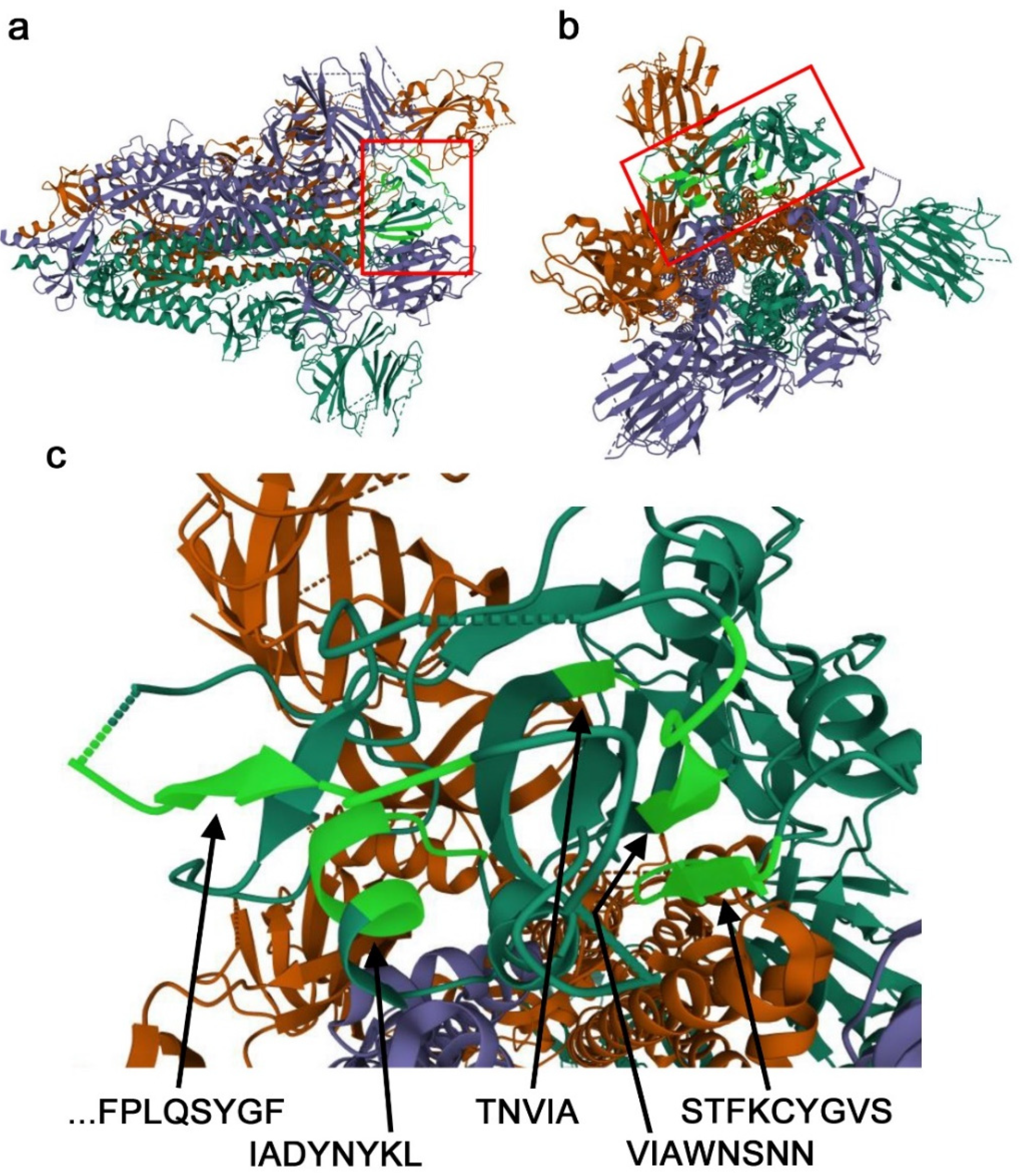
The potentially important nonself SCSs in the RBD discussed above (
Figure 2b) were mapped onto a 3D structure model of the spike protein (
Figure 3). The STFKCYGVS and VIAWNSNN clusters together form an antiparallel β-sheet, confirming that together, they form the 17-aa supercluster. They seem to be accessible in the open conformation. In contrast, a single nonself SCS, TNVYA, is a part of the β-strand embedded in surrounding residues, which does not seem to be accessible. The IADYNYKL cluster forms an α-helix, and only D420 and Y421 are located on the protein surface and may be accessible. Interestingly, the IADYNYKL cluster is closely located to the 19-aa supercluster. An N-terminal portion of the 19-aa supercluster (P479–Y489) was not structurally determined, likely because this is an intrinsically disordered region. A C-terminal portion of the 19-aa supercluster (F490–F497) forms a β-strand.
3.6. Self/Nonself Status Changes in the SARS-CoV-2 Proteome
Mutations can be categorized into two distinct groups: one causes a self-to-nonself (0-to-1) status change, and the other causes a nonself-to-self (1-to-0) status change. The former may increase the chance of being recognized and eliminated by the immune system, and the latter may decrease this chance. In the 68 SARS-CoV-2 variant proteome sequences examined, we found 19 SCSs that changed self/nonself status due to their own or surrounding mutations (
Supplementary Table S3; Additional Data 3 at GitHub). Among them, 11 were self-to-nonself (0-to-1) changes, and 8 were nonself-to-self (1-to-0) changes. Since there were 8809 self SCSs and 852 nonself SCSs in the SARS-CoV-2 proteome, 0.125% of self SCSs changed to nonself SCSs (status change rate), whereas 0.939% of nonself SCSs changed to self SCSs. The percentage ratio of nonself-to-self changes to self-to-nonself changes was 7.51, and these two frequencies were significantly different (χ
2 test:
df = 1,
t = 26.0,
p < 0.0001;
df = 1,
t = 22.0,
p < 0.0001 after Yates’s adjustment). This means that nonself-to-self changes and their associated mutations (escaping mutations or mimicry mutations) were more frequent than self-to-nonself changes and their associated mutations (exposing mutations).
Because self SCSs and nonself SCSs are not equally common in the 3.2 million SCS repertoire, probabilistically, a mutated residue may produce nonself SCSs less frequently than self SCSs. Conversion factors for this weight consideration in the human proteome were defined as ×(self/nonself) = ×(2,401,598/798,402) = ×3.0 for self-to-nonself changes and ×1.0 for nonself-to-self changes. Using these conversion factors based on the human proteome, the number of self-to-nonself changes was calculated as 33, and the number of nonself-to-self changes was calculated as 8. The self-to-nonself status change rate was 0.375%, and the nonself-to-self status change rate was 0.939%. The percentage ratio of nonself-to-self changes to self-to-nonself changes was thus 2.50. When the converted mutation frequencies between the two were subjected to the χ2 test, a significant difference was still obtained at the level of p < 0.05 (df = 1, t = 5.6, p = 0.016; df = 1, t = 4.5, p = 0.033 after Yates’s adjustment).
From an immunological point of view, nonself-to-self changes would confer a survival advantage to the coronavirus, and this result is in accordance with this interpretation, if not just a coincidence. Among the 19 SCSs that changed self/nonself status as discussed above, only a single SCS outside the RBD was found in the spike protein: a self SCS (796-DFGGF-800) that changed to a nonself SCS (796-DCGGF-800).
3.7. Self/Nonself Status Changes in the Spike Protein
A similar analysis was performed to focus on the spike protein using a set of functional mutations (48 point mutations, three of which were at redundant positions) [
46,
47], a recent England variant (B.1.1.7; WHO label Alpha) (six additional point mutations) [
48] and a South African variant (501Y.V2; WHO label Beta) (seven additional point mutations) [
49]. Due to these point mutations, we discovered 26 self-to-nonself status changes out of 1172 self SCSs (status change rate: 2.22%) and 10 nonself-to-self status changes out of 97 nonself SCSs (status change rate: 10.31%) (
Supplementary Table S4; Additional Data 4 at GitHub). The percentage ratio of nonself-to-self changes to self-to-nonself changes was 4.65. When the status change frequencies were subjected to the χ
2 test, a significant difference was obtained (
df = 1,
t = 18.9,
p < 0.0001;
df = 1,
t = 16.3,
p < 0.0001 after Yates’s adjustment).
Using conversion factors for weight adjustment based on the human proteome (×3.0 for self-to-nonself changes and ×1.0 for nonself-to-self changes), the number of self-to-nonself changes was now calculated as 78 (status change rate: 6.66%), and the number of nonself-self changes was calculated as 10 (status change rate: 10.31%). The percentage ratio of nonself-to-self changes to self-to-nonself changes was 1.55. When the converted number of changes was subjected to the χ2 test, no significant difference was obtained (df = 1, t = 1.57, p = 0.21; df = 1, t = 1.10, p = 0.29 after Yates’s adjustment). This result suggests that the ongoing spike protein evolution in humans may not be driven by mimicry to escape immunological recognition and elimination.
Among the mutations that caused the nonself-to-self status changes detected above, 4 mutation sites were directly located within the nonself SCS clusters in the RBD discussed above (
Figure 2b;
Supplementary Table S4). Three of them (I434K, A435S, and N439K) were located in the VIAWNSNN cluster of the 17-aa supercluster. An additional mutation site that does not change the self/nonself status was located in the same cluster, suggesting that this is an unstable nonself cluster. In contrast, only one status-changing site (F490L) was located in the 19-aa supercluster. This supercluster contained three additional mutation sites, but they did not change the self/nonself status and were at the self/nonself boundaries.
4. Discussion
4.1. Self/Nonself SCSs in the Proteome of SARS-CoV-2
Here, we identified self and nonself SCSs throughout the proteome of SARS-CoV-2. This study is based on the theoretical concept that nonself SCSs may be better suited than self SCSs as epitopes for the immune system to boost both T-cell and B-cell responses and not to cause autoimmune diseases in the long term. Self/nonself discrimination in vivo is achieved by the complex functions of DCs, T
reg cells, and other cell types [
5,
6,
7,
23,
24] but can be attained relatively simply in silico by SCS-based computation when both host and parasite proteomes are available. From an evolutionary perspective, this concept leads to the sequence mimicry hypothesis.
We examined the SCS distribution in the human proteome (
Figure 1a–c), which suggested a scale-free distribution in the rank–frequency plot, following Zipf’s law (
Figure 1c). Since Zipf’s law is applicable to natural languages, this result justifies the application of SCS-based frequency analysis to human protein “language”, similar to linguistic frequency analyses [
35,
36]. The breakdown of linearity in the plot at the largest ranks probably reflects the fact that there are many zero-count SCSs. The zero-count SCSs in the human proteome are nonself SCSs themselves, and they are outside the human proteome vocabulary. In other words, the human proteome is composed of a mathematically coordinated collection of words (i.e., SCS vocabulary), which may make the identification of nonself SCSs (and hence foreign proteins) practically attainable for the immune system.
To our knowledge, most SARS-CoV-2 vaccines available at present are based on the antigenicity of the spike protein [
43,
44,
45]. The current mRNA vaccines are highly effective, demonstrating that the use of spike protein for vaccines has probably been the correct choice. Further efforts to search for epitopes continue; studies using neutralizing antibodies and synthetic peptides have identified several epitope sequences in spike proteins [
14,
15,
16,
17,
18,
19,
20,
21,
22]. Numerous search efforts for epitopes for peptide vaccines based on bioinformatics have been performed [
50,
51,
52,
53]. Potential CTL (cytotoxic T lymphocyte) epitopes have been identified in silico and in mice [
54,
55,
56,
57]. However, the concept of self/nonself discrimination has not been incorporated. The present study is a novel attempt to incorporate this concept.
4.2. Limitations of This Study
On the other hand, we admit that the current study has some methodological limitations. First, only the human reference proteome was used, but the human proteomes are variable. Use of the UniprotKB human proteome datasets (UP000005640) may also improve our results because the datasets are curated well. If the variability is fully considered, it may be possible to obtain candidate epitopes specific for various human populations. Personally tailored vaccines may also be prepared based on an individual proteome (genome) sequence from each person.
Another potential limitation of this study may be that self/nonself distributions of SARS-CoV-2 SCSs were not examined using nonhuman proteomes. Cross-species comparisons using other mammalian proteomes may validate the current methodology and suggest infectivity differences among species. Whether the high frequencies of self SCSs in the SARS-CoV-2 proteome are beyond probabilistically expected frequencies may be an additional concern. The self/nonself SCS assignments may be executed using randomized human proteome sequences from its constituent amino acids. This is a concept similar to “availability scores” [
27,
28].
4.3. Self SCSs and Autoimmunity
We discovered that most parts of the SARS-CoV-2 proteome are occupied by self SCSs and that nonself SCSs occupied only 8.82% of the proteome and 7.64% of the spike protein. These results may not be surprising, considering that a single SCS in this study contains just 5 aa and that all proteins on Earth may have a common set of SCS distributions [
27,
28]. However, this high “similarity” may be surprising, considering that the SARS-CoV-2 proteome and its proteins are totally foreign for humans. Theoretically, these results suggest that the human immune system must search for nonself SCSs that are embedded within a sea of self SCSs to avoid the development of autoimmune diseases over the long term. This view is consistent with the recent finding that various autoantibodies are produced in COVID-19 patients [
8,
9,
10].
On the other hand, the immune system produces antibodies against self SCSs as well as against nonself SCSs. Based on a literature survey, we found that COVID-19 patients produced antisera against both self and nonself sequences [
14,
15,
16,
17,
18,
19,
20,
21,
22]. This is not surprising, because nonself SCS regions are relatively infrequent and because an antibody often recognizes a few different short sequences simultaneously in a 3D space, as demonstrated in the case of anti-spike antibodies [
14,
15,
16,
17,
18,
19,
20,
21,
22]. Furthermore, T
reg cells may change the level of the self/nonself discrimination threshold to allow the production of self-targeted antibodies under various conditions [
23,
24].
A similar discussion may be valid regarding the activation of CTLs via MHC class I molecules. Consider a self SCS cluster of 8 aa residues from SARS-CoV-2 that is composed of 4 consecutive self SCSs, which can be fully presented by MHC class I. This means that its N-terminal 5-aa SCS is identical to an SCS from a human protein and that its C-terminal 5-aa SCS is also identical to a different SCS from another human protein. Moreover, the two 5-aa SCSs in the middle are also identical to yet different SCSs from different human proteins. These 5-aa SCSs are all self SCSs, but their combination is novel to humans. In this way, a self SCS cluster can behave as a nonself cluster combinatorially. However, there is a possibility that a single self SCS may be able to function as an epitope.
In any case, various self and nonself epitopes are likely targeted simultaneously during acute infection, and we believe that linear self epitopes are mostly, although not completely, “benign” in terms of autoimmunity. A similar discussion may be valid in immunological memory. If self epitopes are not completely safe in terms of autoimmunity, once pathogenic antigens are eliminated, the immune system should not retain memories of self epitopes of acute pathogens. In contrast, immunological memory for nonself epitopes may safely be retained for life. This may be one of the reasons why it is difficult to establish immunological memory for relatively benign pathogens such as the common cold and influenza. In this sense, establishing a life-long immunological memory for SARS-CoV-2 using vaccines may not be straightforward. The potential risks of autoimmune responses, although not substantial, should not be ignored in the context of worldwide immunization. Potentially safer and more effective vaccines, from the viewpoint of self/nonself immunological recognition of epitopes, are encouraged in the COVID-19 pandemic era.
4.4. Self/Nonself SCSs in the RBD of the Spike Protein
Although we found many nonself SCSs and their clusters throughout the SARS-CoV-2 proteome (
Figure 1d,e), we focused on the RBD of the spike protein to narrow our focus to practically important epitopes (
Figure 2a). We indeed discovered nonself SCSs and their clusters in the RBD. All of them, except the single TNVYA nonself SCS, have already been demonstrated to be parts of epitopes of existing neutralizing antibodies in previous studies [
14,
15,
16,
17,
18,
19,
20,
21] (
Figure 2b). Two superclusters were identified. The 17-aa supercluster is composed of the STFKCYGVS and VIAWNSNN clusters, and together they form an antiparallel β-sheet (
Figure 3). The self sequences between these two clusters should be eliminated when designing candidate epitopes for vaccine targets, but their elimination would disrupt the conformational relationship between these two clusters. In this sense, the use of this conformational epitope without the inclusion of self SCSs might not be practical. An additional drawback of the VIAWNSNN cluster is that it contains four point mutation sites, three of which cause a nonself-to-self status change. This cluster thus may be relatively prone to mutagenesis that allows it to become “invisible”.
In contrast, the 19-aa nonself supercluster, PCNGV-GFNCYF-QSYGF, may be more suitable as a vaccine target. This 19-aa sequence contains four point-mutation sites, but they are all at boundaries between nonself and self SCSs (two of them are located in the gap between two nonself SCSs). The structure of the PCNGV nonself SCS (the first part of the 19-aa supercluster) has not been determined, suggesting that it may be within an intrinsically disordered region (
Figure 3). Probably reflecting this fact, this region of the 19-aa supercluster is recognized by just a few neutralizing antibodies, whereas its C-terminal region is recognized by many existing neutralizing antibodies (
Figure 2b). Indeed, this region is the most targeted epitope. Among them, CB6 and B38 recognize not only the C-terminal region of the 19-aa supercluster (forming a β-strand) but also the IADYNYKL cluster (forming an α-helix), indicating that this cluster may join the 19-aa supercluster to constitute a conformational epitope. However, only one side of the α-helix of the IADYNYKL cluster (i.e., D420 and Y421) is likely accessible, suggesting that the contribution of the IADYNYKL cluster to the antigenicity of this epitope is not large. Therefore, the 19-aa supercluster or its C-terminal region alone may be sufficient for vaccines. As an exception, one neutralizing antibody, C144, appears to recognize both superclusters [
20].
4.5. Self/Nonself Status Changes in Mutants
After infection, pathogenic genomes mutate under strong immunological pressure from the host. One consequence of accumulated mutations is CTL escape [
58,
59]. Although the mechanisms of CTL escape are elusive and may be multifaceted, CTL escape may be triggered when pathogens continuously mutate to the point that they contain an insufficient number of nonself epitopes for the human immune system to recognize in comparison to the number of self epitopes. The present study suggests that upon a host change of SARS-CoV-2, probably from bats to humans, in December 2019, the proteome of SARS-CoV-2 may be evolving to contain fewer nonself and more self sequences to escape recognition and elimination by the immune system, including CTLs, in accordance with the sequence mimicry hypothesis. The use of relatively invariant nonself SCSs, such as the 19-aa supercluster identified in the present study, as vaccine targets may alleviate this problem.
Consistent with the discussion above, throughout the proteome of SARS-CoV-2, nonself-to-self status changes were significantly greater than self-to-nonself status changes even after weighting, supporting the sequence mimicry hypothesis of host-parasite interactions; however, the sample size was small, and the conclusion here may thus be considered tentative. Similarly, the self/nonself status changes in SARS-CoV-2 spike protein also showed significant differences but only without a weight consideration. Spike protein evolution may not be driven much by sequence mimicry at present. Nonetheless, it is also true that nonself-to-self changes indeed occur in the spike protein at a probabilistically reasonable rate.
4.6. Epidemiological Dynamics Based on Mimicry Mutations
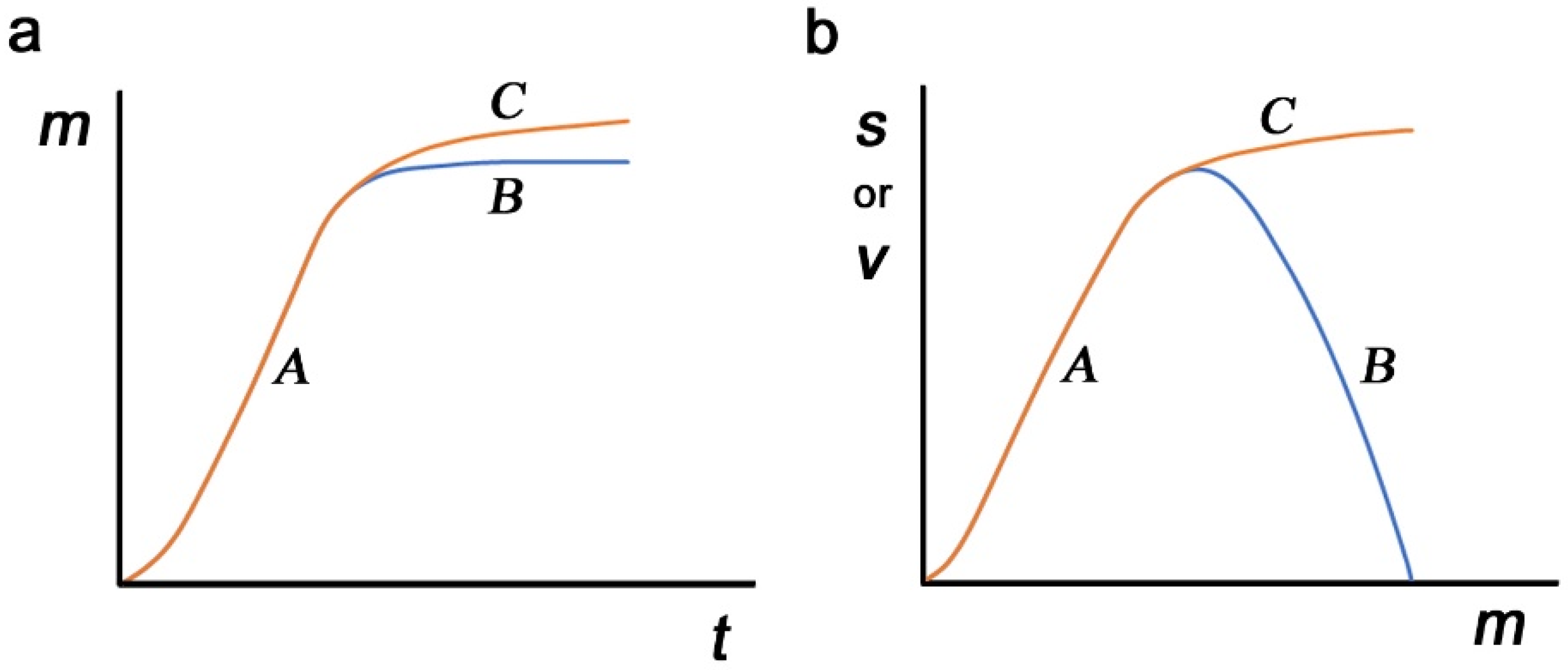
The results above describe the real-time evolution of the virus, which may provide a hint at epidemiological dynamics. The accumulation of nonself-to-self mutations (hereafter called mimicry mutations) may be an unavoidable evolutionary route for any pathogen after its host change as a consequence of immunological escape. These mutations would occur independently of those that increase the virulence of the virus. However, for the sake of discussion, we assume here that mutations occur exclusively for sequence mimicry and that viral virulence is determined by the mimicry level. First, the number of mimicry mutations is considered a function of time after a host change. Mimicry mutations are advantageous for any pathogen and will accumulate rapidly until a saturation point at which further accumulation of such mutations harms the molecular functions of viral proteins (route
A to
B in
Figure 4a). Alternatively, harmful mutations may gradually accumulate to increase the mimicry level (routes
A to
C in
Figure 4a). These harmful mutations will be eliminated by natural selection when they reduce the survival of the virus.
Second, possible contributions of mimicry mutations to survival and virulence are considered. At a relatively low level, an increase in mimicry mutations simply means that the virus can escape the host immune system better, directly contributing to viral virulence and simultaneously contributing to viral survival (route
A for both survival and virulence in
Figure 4b). Here, survival and virulence may show similar dynamics, and mimicry mutations may reach the maximum level of virulence without harming molecular functions. The level of mimicry mutations may become stable at the maximum point. A further increase in mimicry mutations, if it occurs, may negatively impact the molecular functions of viral proteins, reducing virulence, and this may result in a simultaneous decrease in the survival of the virus (routes
A to
B for both survival and virulence in
Figure 4b). This excessive mimicry is not favorable for viral existence. Alternatively, such an increase in mimicry mutations may enhance the survival of the virus at the expense of virulence or molecular functionality (routes
A to
C for survival and routes
A to
B for virulence in
Figure 4b). In this way, survival and virulence may show two different dynamics in response to mimicry mutations. This scenario may be more likely to sustain viral existence due to a higher level of survival, and such mutations may open the possibility of a benign life cycle in response to a decrease in immunological selection pressure.
Over time, the virus may find an equilibrium between functional compromise (low virulence) and immunological escape (high invisibility) for coexistence. When the benign life cycle of the virus is established, SARS-CoV-2 may become just one of the agents of the common cold or an agent that may occasionally manifest small degrees of characteristic symptoms of COVID-19. The present study on self/nonself status changes in variants suggests that the SARS-CoV-2 proteome (although not spike protein) may currently be under such an evolutionary process, and we predict that the current pandemic of SARS-CoV-2 may cease over time due to the accumulation of mimicry mutations for immunological escape. This scenario also indicates that a complete eradiation of SARS-CoV-2 is difficult, if not impossible, and may not be necessary. Unfortunately, we are unable to predict when this might occur. Large-scale bioinformatics studies may make such predictions possible.
Although we have no evidence, the time for coexistence due to the accumulation of mimicry mutations may have already come in Japan. After the spread of the Delta lineage [
60], a sharp decrease in the number of new SARS-CoV-2 positive cases has been reported throughout Japan since the end of the Tokyo Olympics [
61]. There may be several factors for this decrease [
61], but if the viral mimicry evolution with a compromised virulence is a major contributor, a subsequent increase in new positive cases will not be substantial.
4.7. Future Directions
Additionally, many further studies remain unexplored. For example, the nonself SCSs in the SARS-CoV-2 proteome (other than spike proteins) identified in this study have not been examined thoroughly in terms of the possibilities of their functioning as epitopes. An analysis of human proteome variants may reveal differences in infectivity among human individuals and could predict prognosis (see
Section 4.2). An analysis of other pathogens in relation to the human proteome may predict viral infectivity or virulence in humans and may further test the sequence mimicry hypothesis. This hypothesis may be widely applicable not only to a host-pathogen relationship but also to a host-parasite relationship widely seen in biological mutualism. The nonself definition in the present study (i.e., zero-count SCSs) may vary under different genetic backgrounds and environmental conditions, which may trigger or prevent autoimmune diseases. Reflecting the worldwide pandemic, numerous mutations in SARS-CoV-2 have been detected [
62,
63], and these mutations should be analyzed more comprehensively in silico in the future. Such studies will be able to contribute to improved vaccines. Other lines of preventive and treatment measures, such as nutritional balance that could induce nitric oxide (NO) production [
64] and traditional complementary medicine [
65,
66], should also be encouraged.
The present study can be considered an application of the SCS concept to physiological problems. The SCS concept is simple, and its applications are diverse [
36]. This field of study has expanded in silico [
35,
36,
37,
67,
68,
69,
70,
71,
72,
73,
74,
75], but the SCS concept has not yet been sufficiently explored to understand physiological systems. To our knowledge, the first physiological application in this field is the use of a group of peptides as immunological adjuvants [
76,
77]. We anticipate that the present study will expand important frontiers of physiological studies from the viewpoint of the SCS concept.




{kind=link}
{kind=link}
{kind=link}
{kind=link}



