
Transplantation of Fecal Flora from Patients with Atherosclerosis to Mice Can Increase Serum Low-Density Lipoprotein Cholesterol and Affect Intestinal Flora and Its Metabolites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation and Transplantation of FMT Microbiota
2.2. Animals
2.3. Blood Lipid Analysis
2.4. DNA Extraction and 16S rRNA Gene Sequencing
2.5. UHPLC QE MS-Based Untargeted Metabolomics Analysis
2.6. Statistical Analysis
3. Results
3.1. The Serum Lipid Profiles Were Analyzed Between the Two Groups
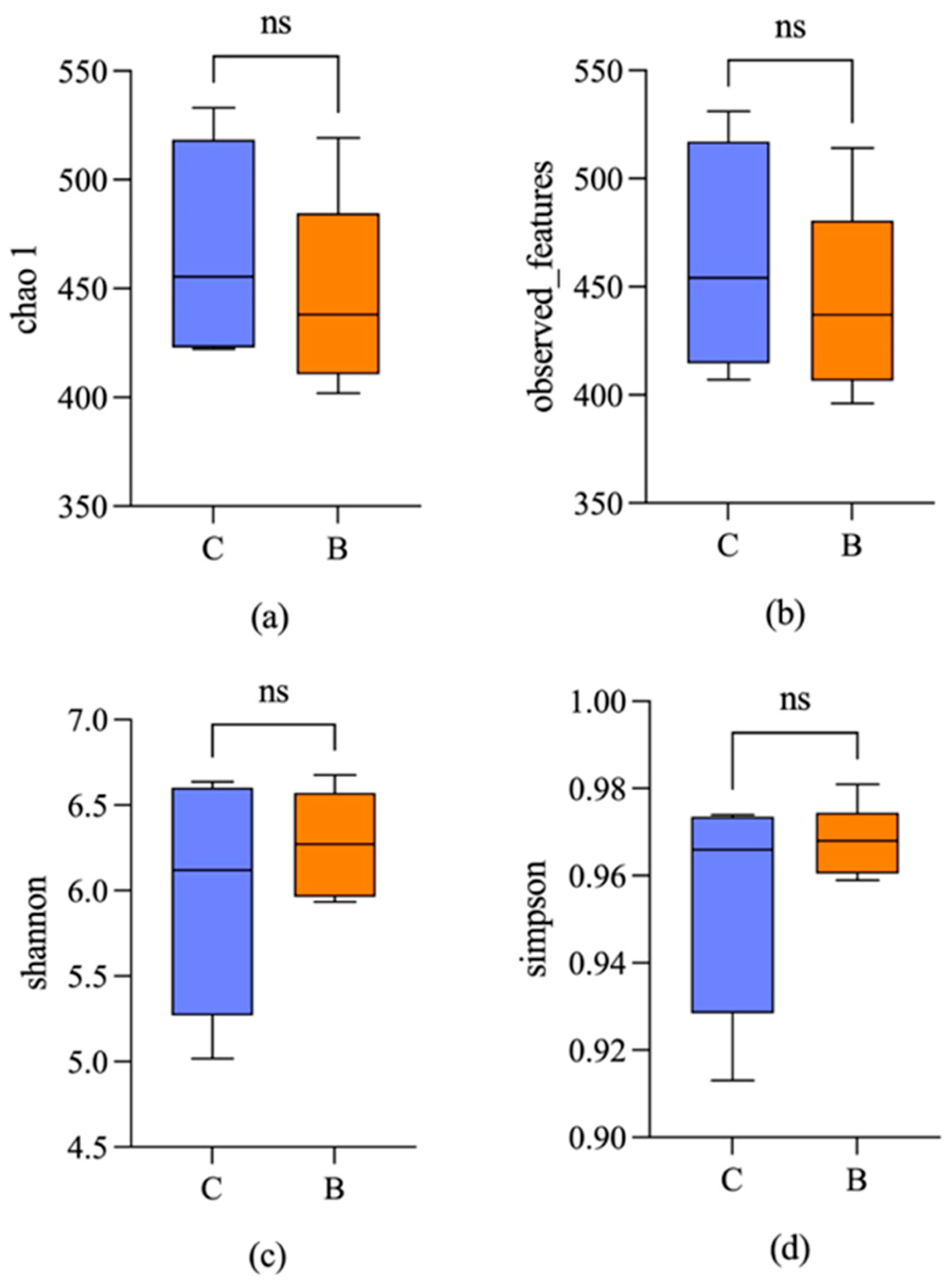
3.2. Microbial Diversity
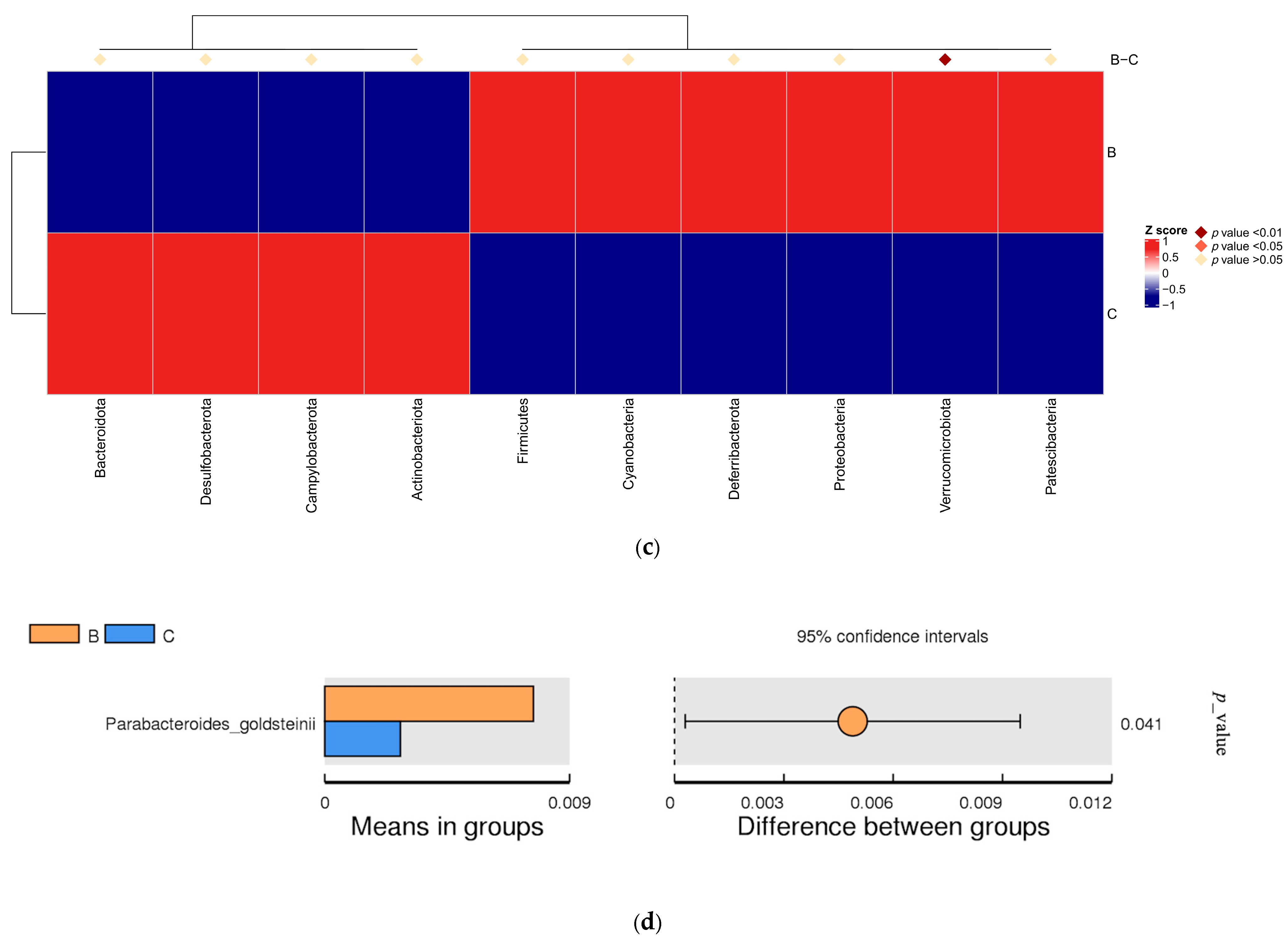
3.3. Differences in Intestinal Microbial Community Structure
3.4. Annotation and Analysis of KEGG Functional Database
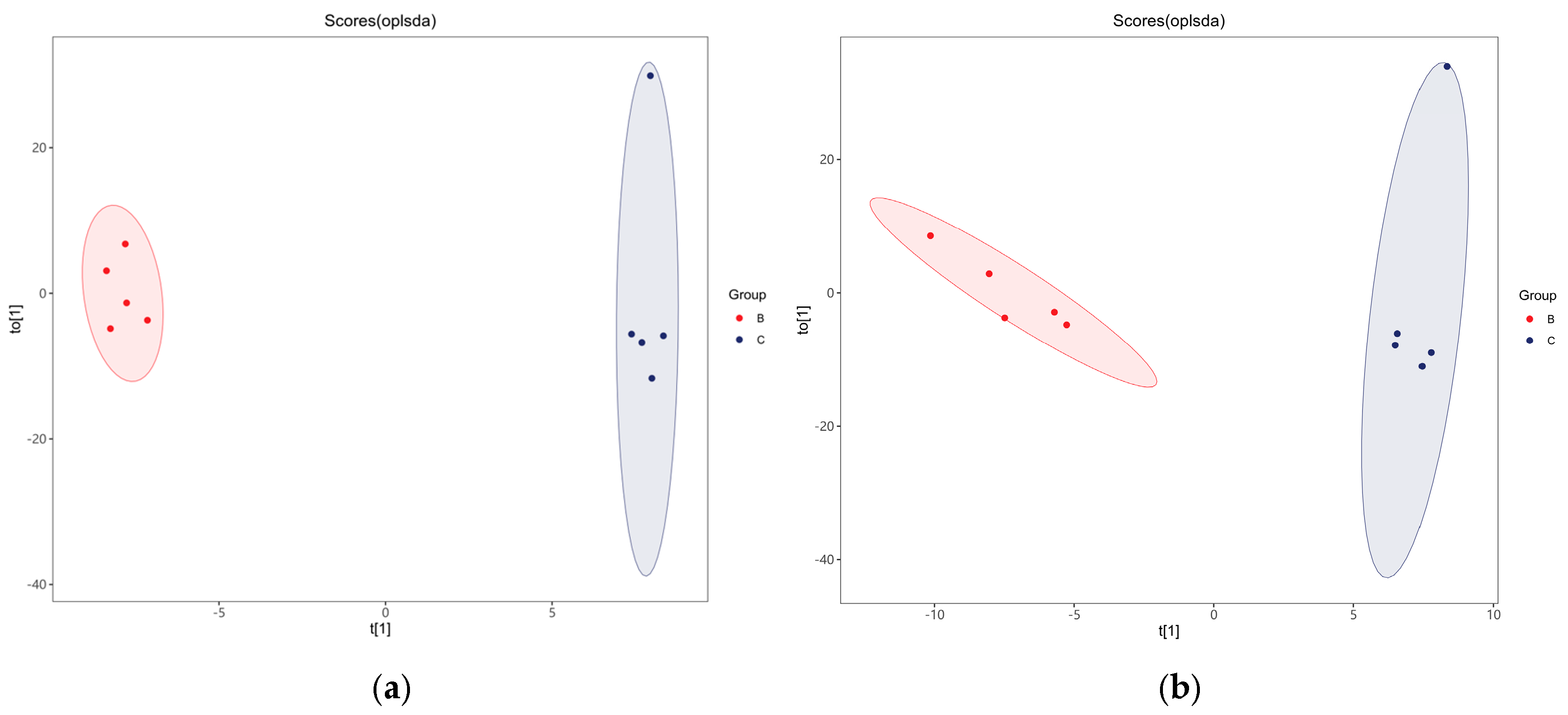
3.5. Multivariate Statistical Analysis of Fecal Metabolites
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tessler, J.; Bordoni, B. Cardiac Rehabilitation. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2023. [Google Scholar]
- Rus, M.; Ardelean, A.I.; Crisan, S.; Marian, P.; Pobirci, O.L.; Huplea, V.; Judea, P.C.; Osiceanu, G.A.; Stanis, C.E.; Andronie-Cioara, F.L. Optimizing Atrial Fibrillation Care: Comparative Assessment of Anticoagulant Therapies and Risk Factors. Clin. Pract. 2024, 14, 344–360. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Cainzos-Achirica, M.; Glassner, K.; Zawahir, H.S.; Dey, A.K.; Agrawal, T.; Quigley, E.M.; Abraham, B.P.; Acquah, I.; Yahya, T.; Mehta, N.N.; et al. Inflammatory Bowel Disease and Atherosclerotic Cardiovascular Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; DeVon, H.A.; Cantor, R.M.; Cupido, A.J.; Pan, C.; Ha, S.M.; Fernandes Silva, L.; Hilser, J.R.; Hartiala, J.; Allayee, H.; et al. Relationships and Mendelian Randomization of Gut Microbe-Derived Metabolites with Metabolic Syndrome Traits in the METSIM Cohort. Metabolites 2024, 14, 174. [Google Scholar] [CrossRef]
- Masse, K.E.; Lu, V.B. Short-chain fatty acids, secondary bile acids and indoles: Gut microbial metabolites with effects on enteroendocrine cell function and their potential as therapies for metabolic disease. Front. Endocrinol. 2023, 14, 1169624. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Q.; Liu, Y.; Wang, L.; Ge, Z.; Li, Z.; Feng, S.; Wu, C. Gut microbiota and hypertension: Association, mechanisms, and treatment. Clin. Exp. Hypertens. 2023, 45, 2195135. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Zhang, Z.; Lv, T.; Wang, X.; Wu, M.; Zhang, R.; Yang, X.; Fu, Y.; Liu, Z. Role of the microbiota-gut-heart axis between bile acids and cardiovascular disease. Biomed. Pharmacother. 2024, 174, 116567. [Google Scholar] [CrossRef]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef]
- Luo, P.; Yin, P.; Hua, R.; Tan, Y.; Li, Z.; Qiu, G.; Yin, Z.; Xie, X.; Wang, X.; Chen, W.; et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology 2018, 67, 662–675. [Google Scholar] [CrossRef]
- Nesci, A.; Carnuccio, C.; Ruggieri, V.; D’Alessandro, A.; Di Giorgio, A.; Santoro, L.; Gasbarrini, A.; Santoliquido, A.; Ponziani, F.R. Gut Microbiota and Cardiovascular Disease: Evidence on the Metabolic and Inflammatory Background of a Complex Relationship. Int. J. Mol. Sci. 2023, 24, 9087. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, Y.; Kim, J.; Kim, J.K. Fecal Microbiota Transplantation: Indications, Methods, and Challenges. J. Microbiol. 2024, 62, 1057–1074. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Hemker, M.; Kahn, S.A.; Steinbach, W.J.; Section on Gastroenteroloy, Hepatology, and Nutrition; Committee on Infectious Diseases. Fecal Microbiota Transplantation: Information for the Pediatrician. Pediatrics 2023, 152, e2023062922. [Google Scholar] [CrossRef]
- Karimi, M.; Shirsalimi, N.; Hashempour, Z.; Salehi Omran, H.; Sedighi, E.; Beigi, F.; Mortezazadeh, M. Safety and efficacy of fecal microbiota transplantation (FMT) as a modern adjuvant therapy in various diseases and disorders: A comprehensive literature review. Front. Immunol. 2024, 15, 1439176. [Google Scholar] [CrossRef]
- Yadegar, A.; Bar-Yoseph, H.; Monaghan, T.M.; Pakpour, S.; Severino, A.; Kuijper, E.J.; Smits, W.K.; Terveer, E.M.; Neupane, S.; Nabavi-Rad, A.; et al. Fecal microbiota transplantation: Current challenges and future landscapes. Clin. Microbiol. Rev. 2024, 37, e0006022. [Google Scholar] [CrossRef]
- Zhang, T.; Lu, G.; Zhao, Z.; Liu, Y.; Shen, Q.; Li, P.; Chen, Y.; Yin, H.; Wang, H.; Marcella, C.; et al. Washed microbiota transplantation vs. manual fecal microbiota transplantation: Clinical findings, animal studies and in vitro screening. Protein Cell 2020, 11, 251–266. [Google Scholar] [CrossRef]
- Tang, J.N.; Zeng, Z.G.; Wang, H.N.; Yang, T.; Zhang, P.J.; Li, Y.L.; Zhang, A.Y.; Fan, W.Q.; Zhang, Y.; Yang, X.; et al. An effective method for isolation of DNA from pig faeces and comparison of five different methods. J. Microbiol. Methods 2008, 75, 432–436. [Google Scholar] [CrossRef]
- Beverly, J.K.; Budoff, M.J. Atherosclerosis: Pathophysiology of insulin resistance, hyperglycemia, hyperlipidemia, and inflammation. J. Diabetes 2020, 12, 102–104. [Google Scholar] [CrossRef]
- Gobalakrishnan, S.; Asirvatham, S.S.; Janarthanam, V. Effect of Silybin on Lipid Profile in Hypercholesterolaemic Rats. J. Clin. Diagn. Res. 2016, 10, FF01–FF05. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxidative Med. Cell. Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef] [PubMed]
- González-Navarro, B.; Segura-Egea, J.J.; Estrugo-Devesa, A.; Pintó-Sala, X.; Jane-Salas, E.; Jiménez-Sánchez, M.C.; Cabanillas-Balsera, D.; López-López, J. Relationship between Apical Periodontitis and Metabolic Syndrome and Cardiovascular Events: A Cross-Sectional Study. J. Clin. Med. 2020, 9, 3205. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Lin, C.; Cecelja, M.; Mangino, M.; Matey-Hernandez, M.L.; Keehn, L.; Mohney, R.P.; Steves, C.J.; Spector, T.D.; Kuo, C.F.; et al. Gut microbial diversity is associated with lower arterial stiffness in women. Eur. Heart J. 2018, 39, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Giakomidi, D.; Ishola, A.; Nus, M. Targeting gut microbiota to regulate the adaptive immune response in atherosclerosis. Front. Cardiovasc. Med. 2025, 12, 1502124. [Google Scholar] [CrossRef] [PubMed]
- Salvado, R.; Santos-Minguez, S.; Lugones-Sánchez, C.; Gonzalez-Sánchez, S.; Tamayo-Morales, O.; Quesada-Rico, J.A.; Benito, R.; Rodríguez-Sánchez, E.; Gómez-Marcos, M.A.; Casado-Vicente, V.; et al. Gut microbiota and its relationship with early vascular ageing in a Spanish population (MIVAS study). Eur. J. Clin. Investig. 2024, 54, e14228. [Google Scholar] [CrossRef]
- Dong, Y.; Cheng, H.; Liu, Y.; Xue, M.; Liang, H. Red yeast rice ameliorates high-fat diet-induced atherosclerosis in Apoe-/- mice in association with improved inflammation and altered gut microbiota composition. Food Funct. 2019, 10, 3880–3889. [Google Scholar] [CrossRef]
- Xie, B.; Zu, X.; Wang, Z.; Xu, X.; Liu, G.; Liu, R. Ginsenoside Rc ameliorated atherosclerosis via regulating gut microbiota and fecal metabolites. Front. Pharmacol. 2022, 13, 990476. [Google Scholar] [CrossRef]
- Qin, M.; Luo, Y.; Lu, S.; Sun, J.; Yang, K.; Sun, G.; Sun, X. Ginsenoside F1 Ameliorates Endothelial Cell Inflammatory Injury and Prevents Atherosclerosis in Mice through A20-Mediated Suppression of NF-kB Signaling. Front. Pharmacol. 2017, 8, 953. [Google Scholar] [CrossRef]
- Tryfonos, A.; Mills, J.; Green, D.J.; Wagenmakers, A.J.; Dawson, E.A.; Cocks, M. Association between atherogenic risk-modulating proteins and endothelium-dependent flow-mediated dilation in coronary artery disease patients. Eur. J. Appl. Physiol. 2023, 123, 367–380. [Google Scholar] [CrossRef]
- Toomey, S.; Harhen, B.; Roche, H.M.; Fitzgerald, D.; Belton, O. Profound resolution of early atherosclerosis with conjugated linoleic acid. Atherosclerosis. 2006, 187, 40–49. [Google Scholar] [CrossRef]
- Kostogrys, R.B.; Franczyk, M.; Maslak, E.; Gajda, M.; Mateuszuk, L.; Chłopicki, S. Effects of margarine supplemented with t10c12 and C9T11 CLA on atherosclerosis and steatosis in apoE/LDLR -/- mice. J. Nutr. Health Aging. 2012, 16, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Ramos, K.S.; Chapkin, R.S. Dietary gamma-linolenic acid suppresses aortic smooth muscle cell proliferation and modifies atherosclerotic lesions in apolipoprotein E knockout mice. J. Nutr. 2001, 131, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Zheng, X.; Zhou, L.; Li, H. Prostaglandin E1 dose-dependently promotes stability of atherosclerotic plaque in a rabbit model. Can. J. Physiol. Pharmacol. 2012, 90, 131–139. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, L.; Feng, J.; He, L.; Chen, F.; Feng, X.; Wang, S. Transplantation of Fecal Flora from Patients with Atherosclerosis to Mice Can Increase Serum Low-Density Lipoprotein Cholesterol and Affect Intestinal Flora and Its Metabolites. Appl. Microbiol. 2025, 5, 29. https://doi.org/10.3390/applmicrobiol5010029
Feng L, Feng J, He L, Chen F, Feng X, Wang S. Transplantation of Fecal Flora from Patients with Atherosclerosis to Mice Can Increase Serum Low-Density Lipoprotein Cholesterol and Affect Intestinal Flora and Its Metabolites. Applied Microbiology. 2025; 5(1):29. https://doi.org/10.3390/applmicrobiol5010029
Chicago/Turabian StyleFeng, Liang, Jianting Feng, Li He, Fu Chen, Xin Feng, and Suwen Wang. 2025. "Transplantation of Fecal Flora from Patients with Atherosclerosis to Mice Can Increase Serum Low-Density Lipoprotein Cholesterol and Affect Intestinal Flora and Its Metabolites" Applied Microbiology 5, no. 1: 29. https://doi.org/10.3390/applmicrobiol5010029
APA StyleFeng, L., Feng, J., He, L., Chen, F., Feng, X., & Wang, S. (2025). Transplantation of Fecal Flora from Patients with Atherosclerosis to Mice Can Increase Serum Low-Density Lipoprotein Cholesterol and Affect Intestinal Flora and Its Metabolites. Applied Microbiology, 5(1), 29. https://doi.org/10.3390/applmicrobiol5010029