
16S rRNA Gene Sequencing-Based Identification and Comparative Analysis of the Fecal Microbiota of Five Syntopic Lizard Species from a Low-Mountain Area in Western Bulgaria

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. Genomic DNA Extraction
2.3. PCR Amplification and Sequencing
2.4. Sequence Assembly and Taxonomic Identification
2.5. Bioinformatics and Statistical Analysis
3. Results
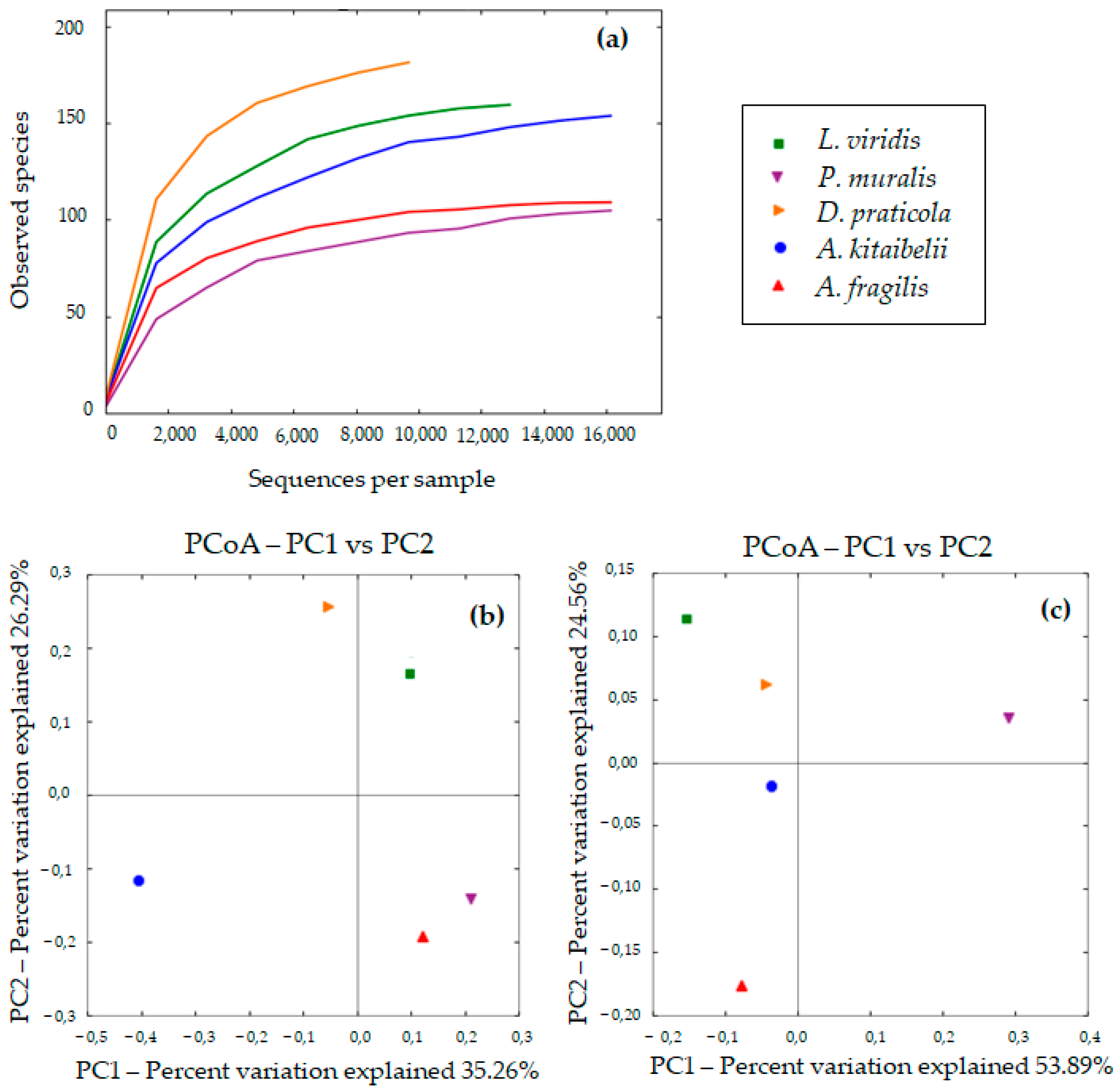
3.1. Description of the Sequencing Data
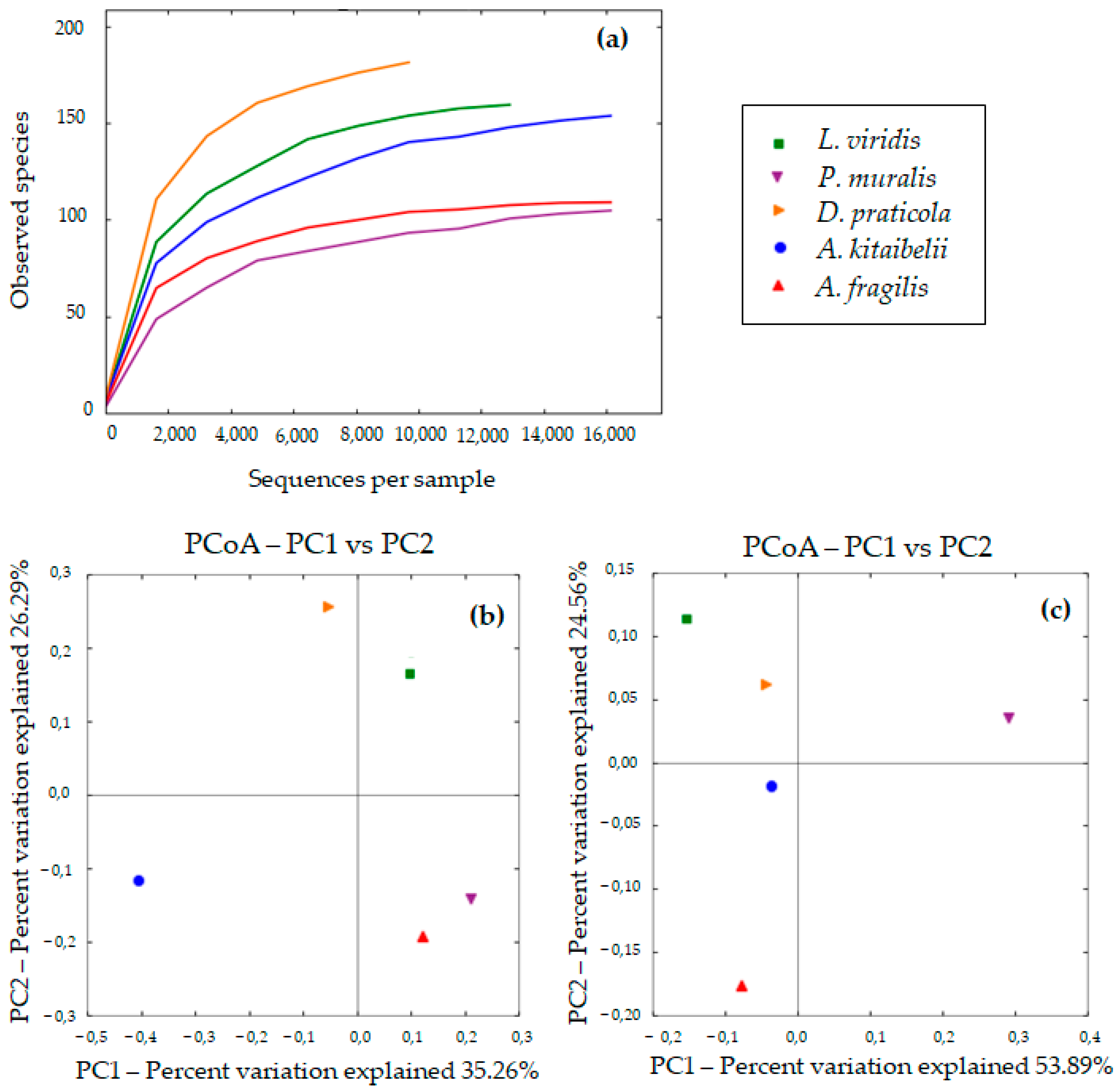
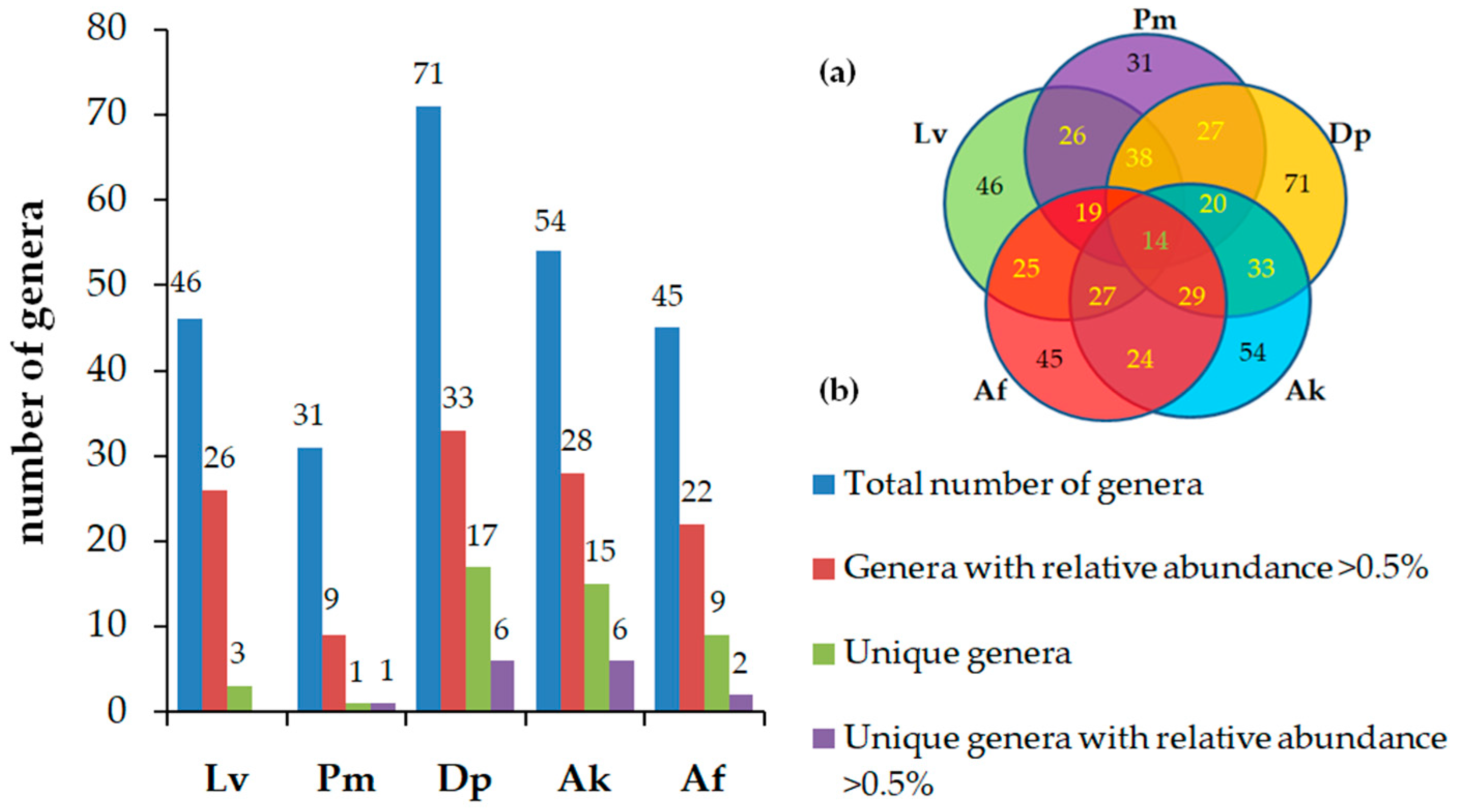
3.2. Diversity of Bacterial Communities
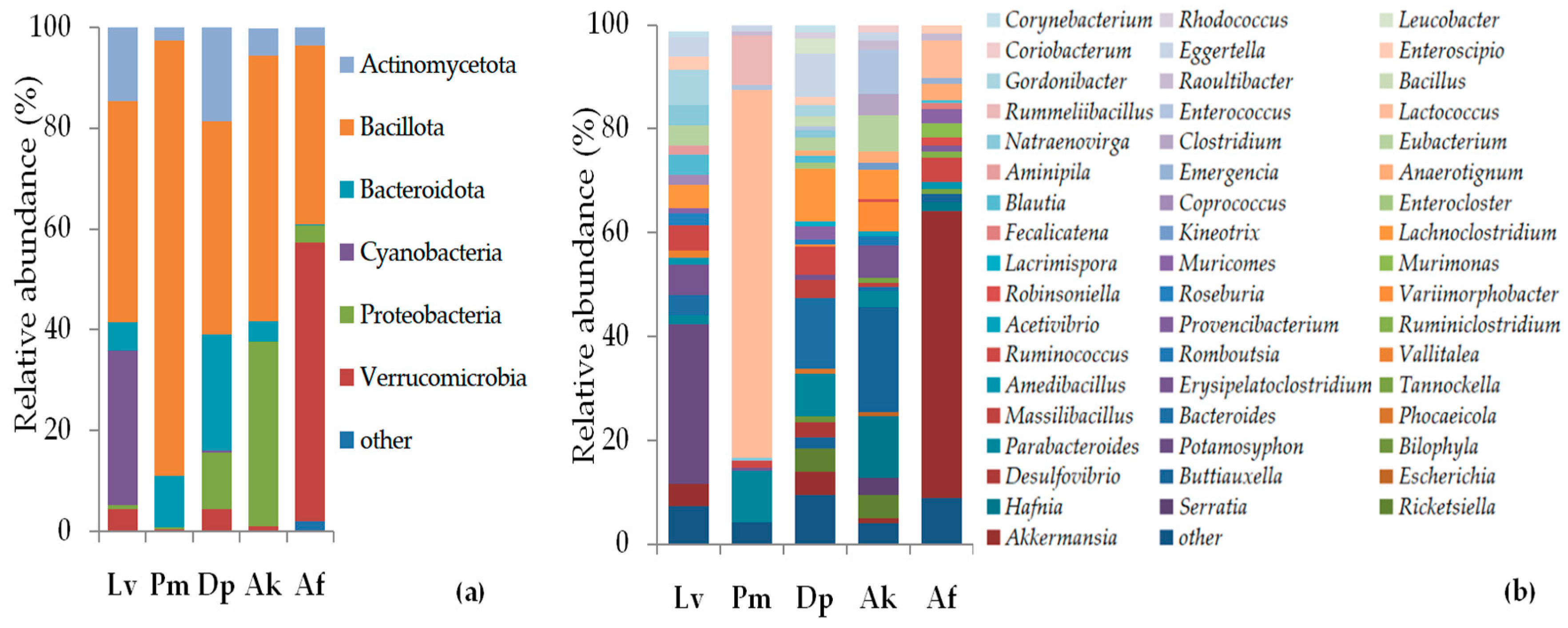
3.3. Taxonomic Composition and Abundance of Gut Microbiota
4. Discussion
4.1. Composition and Diversity of Lizard Gut Microbiota
4.2. Relationship between Gut Microbiotas of Lizard Species and Diet
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gloor, G.B.; Hummelen, R.; Macklaim, J.M.; Dickson, R.J.; Fernandes, A.D.; MacPhee, R.; Reid, G. Microbiome profiling by illumina sequencing of combinatorial sequence-tagged PCR products. PLoS ONE 2010, 5, e15406. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.; Lozupone, C.; Hamady, M.; Knight, R.; Jeffrey, I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef]
- Ficetola, G.; Manenti, R.; Taberlet, P. Environmental DNA and metabarcoding for the study of amphibians and reptiles: Species distribution, the microbiome, and much more. Amphibia-Reptilia 2019, 40, 129–148. [Google Scholar] [CrossRef]
- Colston, T.; Jackson, C. Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Mol. Ecol. 2016, 25, 3776–3800. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.; Hamady, M.; Lozupone, C.; Turnbaugh, P.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Moeller, A.H.; Sanders, J.G. Roles of the gut microbiota in the adaptive evolution of mammalian species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190597. [Google Scholar] [CrossRef]
- Ingala, M.R.; Simmons, N.B.; Wultsch, C.; Krampis, K.; Speer, K.A.; Perkins, S.L. Comparing microbiome sampling methods in a wild mammal: Fecal and intestinal samples record different signals of host ecology, evolution. Front. Microbiol. 2018, 9, 803. [Google Scholar] [CrossRef]
- Forgacs, D.; Wallen, R.L.; Boedeker, A.L.; Derr, J.N. Evaluation of fecal samples as a valid source of DNA by comparing paired blood and fecal samples from American bison (Bison bison). BMC Genet. 2019, 20, 22. [Google Scholar] [CrossRef]
- Ilina, L.A.; Filippova, V.A.; Brazhnik, E.A.; Dubrovin, A.V.; Yildirim, E.A.; Dunyashev, T.P.; Laptev, G.Y.; Novikova, N.I.; Sobolev, D.V.; Yuzhakov, A.A.; et al. The comparative analysis of the ruminal bacterial population in reindeer (Rangifer tarandus L.) from the Russian arctic zone: Regional and seasonal effects. Animals 2021, 11, 911. [Google Scholar] [CrossRef]
- Turnbaugh, P.; Ley, R.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Walter, J.; Ley, R. The human gut microbiome: Ecology and recent evolutionary changes. Ann. Rev. Microbiol. 2011, 65, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.; Ursell, L.; Parfrey, L.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, 38–44. [Google Scholar] [CrossRef]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.; Angert, E.; Montgomery, W.; Choat, J. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Gajardo, K.; Rodiles, A.; Kortner, T.; Krogdahl, A.; Bakke, A.; Merrifield, D.; Sorum, H. A high-resolution map of the gut microbiota in Atlantic salmon (Salmo salar): A basis for comparative gut microbial research. Sci. Rep. 2016, 6, 30893. [Google Scholar] [CrossRef]
- Colombo, B.; Scalvenzi, T.; Benlamara, S.; Pollet, N. Microbiota and mucosal immunity in amphibians. Front. Immunol. 2015, 6, 111. [Google Scholar] [CrossRef]
- Bletz, M.; Goedbloed, D.; Sanchez, E.; Reinhardt, T.; Tebbe, C.; Bhuju, S.; Geffers, R.; Jarek, M.; Vences, M.; Steinfartz, S. Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nat. Commun. 2016, 7, 13699. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.Q.; Liu, Y.; Zhang, J.; Chen, X.H.; Qu, Y.F. Comparative study on gut microbiota in three Anura frogs from a mountain stream. Ecol. Evol. 2022, 12, e8854. [Google Scholar] [CrossRef]
- Colston, T. The Reptile Gut Microbiome: Its Role in Host Evolution and Community Assembly. Ph.D. Thesis, University of Mississippi, Oxford, MI, USA, 2017. [Google Scholar]
- Siddique, R.; Maciver, S.; Khan, N. Gut microbiome–immune system interaction in reptiles. J. Appl. Microbiol. 2022, 132, 2558–2571. [Google Scholar] [CrossRef] [PubMed]
- Hong, P.; Wheeler, E.; Cann, I.; Mackie, R. Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galápagos Islands using 16S rRNA-based pyrosequencing. ISME J. 2011, 5, 1461–1470. [Google Scholar] [CrossRef]
- Keenan, S.; Engel, A.; Elsey, R. The alligator gut microbiome and implications for archosaur symbioses. Sci. Rep. 2013, 3, 2877. [Google Scholar] [CrossRef] [PubMed]
- Arizza, V.; Vecchioni, L.; Caracappa, S.; Sciurba, G.; Berlinghieri, F. New insights into the gut microbiome in loggerhead sea turtles Caretta caretta stranded on the Mediterranean coast. PLoS ONE 2019, 14, e0220329. [Google Scholar] [CrossRef]
- Waite, D.; Taylor, M. Exploring the avian gut microbiota: Current trends and future directions. Front. Microbiol. 2015, 6, 673. [Google Scholar] [CrossRef]
- Hird, S.; Sanchez, C.; Carstens, B.; Brumfield, R. Comparative gut microbiota of 59 neotropical bird species. Front. Microbiol. 2015, 6, 1403. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Mourkas, E.; González-Acuna, D.; Olsen, B.; Ellström, P. Evaluation and optimization of microbial DNA extraction from fecal samples of wild Antarctic bird species. Infect. Ecol. Epidemiol. 2017, 7, 1386536. [Google Scholar] [CrossRef]
- Gregor, R.; Probst, M.; Eyal, S.; Aksenov, A.; Sasson, G.; Horovitz, I.; Dorrestein, P.C.; Meijler, M.M.; Mizrahi, I. Mammalian gut metabolomes mirror microbiome composition and host phylogeny. ISME J. 2022, 16, 1262–1274. [Google Scholar] [CrossRef]
- de Jonge, N.; Carlsen, B.; Christensen, M.H.; Pertoldi, C.; Nielsen, J.L. The gut microbiome of 54 mammalian species. Front. Microbiol. 2022, 13, 886252. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, Y.; Dai, Y.; Jiang, Y.; Lin, L.; Li, H.; Li, P.; Qu, Y.; Ji, X. Captivity affects diversity, abundance, and functional pathways of gut microbiota in the northern grass lizard Takydromus septentrionalis. Microbiol. Open 2020, 9, e1095. [Google Scholar] [CrossRef]
- Kohl, K.; Brun, A.; Magallanes, M.; Brinkerhoff, J.; Laspiur, A.; Acosta, J.; Caviedes-Vidal, E.; Bordenstein, S. Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 2017, 26, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ma, J.; Li, J.; Zhang, X.; Li, L.; He, N.; Liu, H.; Luo, S.; Wu, Z.; Han, R.; et al. Diets alter the gut microbiome of crocodile lizards. Front. Microbiol. 2017, 8, 2073. [Google Scholar] [CrossRef]
- Tang, S.; Li, Y.; Huang, C.; Yan, S.; Li, Y.; Chen, Z.; Wu, Z. Comparison of gut microbiota diversity between captive and wild Tokay gecko (Gekko gecko). Front. Microbiol. 2022, 13, 897923. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.A.; Ramírez-Barahona, S.; Holekamp, K.E.; Theis, K.R. Host phylogeny and host ecology structure the mammalian gut microbiota at different taxonomic scales. Anim. Microbiome 2021, 3, 33. [Google Scholar] [CrossRef]
- Amato, K. Co-evolution in context: The importance of studying gut microbiomes in wild animals. Microbiome Sci. Med. 2013, 1, 10–29. [Google Scholar] [CrossRef]
- Stiers, E. The Microbiome of Southwestern Rattlesnakes. Ph.D. Thesis, Clemson University, Clemson, SC, USA, 2020. [Google Scholar]
- Uetz, P.; Freed, P.; Aguilar, R.; Reyes, F.; Kudera, J.; Hošek, J. (Eds.) The Reptile Database. 2023. Available online: http://www.reptile-database.org (accessed on 30 October 2023).
- Kohl, K.; Brun, A.; Magallanes, M.; Brinkerhoff, J.; Laspiur, A.; Acosta, J.; Bordenstein, S.; Caviedes-Vidal, E. Physiological and microbial adjustments to diet quality permit facultative herbivory in an omnivorous lizard. J. Exp. Biol. 2016, 219, 1903–1912. [Google Scholar] [CrossRef]
- Ren, T.; Kahrl, A.F.; Wu, M.; Cox, R.M. Does adaptive radiation of a host lineage promote ecological diversity of its bacterial communities? A test using gut microbiota of Anolis lizards. Mol. Ecol. 2016, 25, 4793–4804. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, F.; Li, N.; Dayananda, B. Environment-dependent variation in gut microbiota of an oviparous lizard (Calotes versicolor). Animals 2021, 11, 2461. [Google Scholar] [CrossRef]
- Montoya-Ciriaco, N.; Gómez-Acata, S.; Muñoz-Arenas, L.; Dendooven, L.; Estrada-Torres, A.; de la Vega-Perez, A.; Navarro-Noya, Y. Dietary effects on gut microbiota of the mesquite lizard Sceloporus grammicus (Wiegmann, 1828) across different altitudes. Microbiome 2020, 8, 6. [Google Scholar] [CrossRef]
- Chen, J.Q.; Zhang, L.W.; Zhao, R.M.; Wu, H.X.; Lin, L.H.; Li, P.; Li, H.; Qu, Y.F.; Ji, X. Gut microbiota differs between two cold-climate lizards distributed in thermally different regions. BMC Ecol. Evol. 2022, 22, 120. [Google Scholar] [CrossRef]
- Vacheva, E. Relationships between Representatives of Three Lizard Families—Anguidae, Lacertidae and Scincidae (Reptilia: Squamata: Sauria), in Regard to Their Spatial and Dietary Niches in Western Bulgaria. Ph.D. Thesis, National Museum of Natural History—Bulgarian Academy of Sciences, Sofia, Bulgaria, 2021. (In Bulgarian). [Google Scholar]
- Bestion, E.; Jacob, S.; Zinger, L.; Di Gesu, L.; Richard, M.; White, J.; Cote, J. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat. Ecol. Evol. 2017, 1, 161. [Google Scholar] [CrossRef]
- Dudek, K.; Koczura, R.; Dudek, M.; Sajkowska, Z.; Ekner-Grzyb, A. Detection of Salmonella enterica in a sand lizard (Lacerta agilis, Linnaeus, 1758) city population. Herpetol. J. 2016, 26, 57–60. [Google Scholar]
- Bassitta, M.; Alemany, I.; Pérez-Mellado, V.; Pérez-Cembranos, A.; Navarro, P.; Lluch, J.; Jurado-Rivera, J.; Castro, J.; Picornell, A.; Ramon, C. Phylogenetic, microbiome, and diet characterisation of wall lizards in the Columbretes Archipelago (Spain): Clues for their conservation. Diversity 2022, 14, 408. [Google Scholar] [CrossRef]
- Alemany, I.; Pérez-Cembranos, A.; Pérez-Mellado, V.; Castro, J.; Picornell, A.; Ramon, C.; Jurado-Rivera, J. Faecal microbiota divergence in allopatric populations of Podarcis lilfordi and P. pityusensis, two lizard species endemic to the Balearic Islands. Microb. Ecol. 2023, 85, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, D.S.; Harris, D.J.; Damas-Moreira, I.; Pereira, A.; Xavier, R. Factors shaping the gut microbiome of five species of lizards from different habitats. PeerJ 2023, 11, e15146. [Google Scholar] [CrossRef]
- Baldo, L.; Tavecchia, G.; Rotger, A.; Igual, J.; Riera, J. Insular holobionts: Persistence and seasonal plasticity of the Balearic wall lizard (Podarcis lilfordi) gut microbiota. PeerJ 2023, 11, e14511. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.; Ryan, P. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaentol. Electron. 2001, 4, 1. [Google Scholar]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Coletto, E.; Latousakis, D.; Pontifex, M.G.; Crost, E.H.; Vaux, L.; Perez Santamarina, E.; Goldson, A.; Brion, A.; Hajihosseini, M.K.; Vauzour, D.; et al. The role of the mucin-glycan foraging Ruminococcus gnavus in the communication between the gut and the brain. Gut Microbes 2022, 14, 2073784. [Google Scholar] [CrossRef]
- Mukherjee, A.; Lordan, C.; Ross, R.P.; Cotter, P.D. Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microbes 2020, 12, 1802866. [Google Scholar] [CrossRef] [PubMed]
- Rettenmaier, R.; Thieme, N.; Streubel, J.; Di Bello, L.; Kowollik, M.L.; Huang, L.; Maus, I.; Klingl, A.; Liebl, W.; Zverlov, V.V. Variimorphobacter saccharofermentans gen. nov., sp. nov., a new member of the family Lachnospiraceae, isolated from a maize-fed biogas fermenter. Int. J. Syst. Evol. Microbiol. 2021, 71, 005044. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, B.; Wityk, P.; Gałęcka, M.; Michalik, M. The many faces of Enterococcus spp.-commensal, probiotic and opportunistic pathogen. Microorganisms 2021, 9, 1900. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Hehemann, J.H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut Bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef] [PubMed]
- Bensch, H.M.; Tolf, C.; Waldenström, J.; Lundin, D.; Zotl, M. Bacteroidetes to Firmicutes: Captivity changes the gut microbiota composition and diversity in a social subterranean rodent. Anim. Microbiome 2023, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Cholewińska, P.; Wołoszyńska, M.; Michalak, M.; Czyż, K.; Rant, W.; Janczak, M. Evaluation of changes in the levels of Firmicutes and Bacteroidetes phyla of sheep feces depending on the breed. Animals 2020, 10, 1901. [Google Scholar] [CrossRef]
- Costello, E.K.; Gordon, J.I.; Secor, S.M.; Knight, R. Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 2010, 4, 1375–1385. [Google Scholar] [CrossRef]
- Zhang, W.; Li, N.; Tang, X.; Liu, N.; Zhao, W. Changes in intestinal microbiota across an altitudinal gradient in the lizard Phrynocephalus vlangalii. Ecol. Evol. 2018, 8, 4695–4703. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Qin, T.; Yu, T.; Zhang, G. Improvement of the gut microbiota in vivo by a short-chain fatty acids-producing strain Lactococcus garvieae CF11. Processes 2022, 10, 604. [Google Scholar] [CrossRef]
- Suresh, L.; Nagaraju, A.; Chouhan, R.; Podeti, S. Isolation and molecular characterization of Rummelii Bacillus Stabekisii: An efficient protease producing bacterial strain identified from environmental waste samples of warangal District in Telangana. J. Pure Appl. Microbiol. 2020, 14, 461–472. [Google Scholar] [CrossRef]
- Moon, C.D.; Young, W.; Maclean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic insights into the roles of Proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. Microbiologyopen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed]
- Givens, C.; Ransom, B.; Bano, N.; Hollibaugh, J. Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser. 2015, 518, 209–223. [Google Scholar] [CrossRef]
- Xie, Y.; Xia, P.; Wang, H.; Yu, H.; Giesy, J.; Zhang, Y.; Mora, M.; Zhang, X. Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 2016, 6, 33350. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. The genus Hafnia: From soup to nuts. Clin. Microbiol. Rev. 2006, 19, 12–18. [Google Scholar] [CrossRef]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef]
- Saini, N.; Gupta, R.S. A robust phylogenetic framework for members of the order Legionellales and its main genera (Legionella, Aquicella, Coxiella and Rickettsiella) based on phylogenomic analyses and identification of molecular markers demarcating different clades. Antonie Van Leeuwenhoek 2021, 114, 957–982. [Google Scholar] [CrossRef]
- Garcia-Vozmediano, A.; Tomassone, L.; Fonville, M.; Bertolotti, L.; Heylen, D.; Fabri, N.D.; Medlock, J.M.; Nijhof, A.M.; Hansford, K.M.; Sprong, H.; et al. The genetic diversity of Rickettsiella symbionts in Ixodes ricinus throughout Europe. Microb. Ecol. 2022, 84, 613–626. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Davids, M.; Suarez-Diez, M.; Boeren, S.; Schaap, P.J.; Martins Dos Santos, V.A.P.; Smidt, H.; Belzer, C.; de Vos, W.M. Genome-scale model and omics analysis of metabolic capacities of Akkermansia muciniphila reveal a preferential mucin-degrading lifestyle. Appl. Environ. Microbiol. 2017, 83, e01014-17. [Google Scholar] [CrossRef] [PubMed]
- Kosciow, K.; Deppenmeier, U. Characterization of a phospholipid-regulated β-galactosidase from Akkermansia muciniphila involved in mucin degradation. Microbiol. Open 2019, 8, e00796. [Google Scholar] [CrossRef] [PubMed]
- Kosciow, K.; Deppenmeier, U. Characterization of three novel β-galactosidases from Akkermansia muciniphila involved in mucin degradation. Int. J. Biol. Macromol. 2020, 149, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Hagi, T.; Belzer, C. The interaction of Akkermansia muciniphila with host-derived substances, bacteria and diets. Appl. Microbiol. Biotechnol. 2021, 105, 4833–4841. [Google Scholar] [CrossRef] [PubMed]
- Chia, L.W.; Hornung, B.V.H.; Aalvink, S.; Schaap, P.J.; de Vos, W.M.; Knol, J.; Belzer, C. Deciphering the trophic interaction between Akkermansia muciniphila and the butyrogenic gut commensal Anaerostipes caccae using a metatranscriptomic approach. Antonie Van Leeuwenhoek 2018, 111, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Mohd Salleh, M.; Esa, Y.; Ngalimat, M.; Chen, P. Faecal DNA metabarcoding reveals novel bacterial community patterns of critically endangered Southern River Terrapin, Batagur affinis. PeerJ 2019, 10, e12970. [Google Scholar] [CrossRef]
- Glover, J.S.; Ticer, T.D.; Engevik, M.A. Characterizing the mucin-degrading capacity of the human gut microbiota. Sci. Rep. 2022, 12, 8456. [Google Scholar] [CrossRef]
- Hu, C.; Rzymski, P. Non-photosynthetic melainabacteria (Cyanobacteria) in human gut: Characteristics and association with health. Life 2022, 12, 476. [Google Scholar] [CrossRef]
- Utami, Y.D.; Kuwahara, H.; Murakami, T.; Morikawa, T.; Sugaya, K.; Kihara, K.; Yuki, M.; Lo, N.; Deevong, P.; Hasin, S.; et al. Phylogenetic diversity and single-cell genome analysis of “Melainabacteria”, a non-photosynthetic cyanobacterial group, in the termite gut. Microbes Environ. 2018, 33, 50–57. [Google Scholar] [CrossRef]
- Chiang, E.; Deblois, C.L.; Carey, H.V.; Suen, G. Characterization of captive and wild 13-lined ground squirrel cecal microbiotas using Illumina-based sequencing. Anim. Microbiome 2022, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yang, D.; Chang, L.; Zhang, M.; Zhu, L.; Jiang, J. Animal gut microbiome mediates the effects of antibiotic pollution on an artificial freshwater system. J. Hazard Mater. 2022, 425, 127968. [Google Scholar] [CrossRef] [PubMed]
- McGregor, G.; Sendall, B. Potamosiphon australiensis gen. nov., sp nov. (Oscillatoriales), a new filamentous cyanobacterium from subtropical north-eastern Australia. Phytotaxa 2019, 387, 77–93. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Sample | OTUs | Chao1 | Shannon | Gini–Simpson | Good’s Coverage |
|---|---|---|---|---|---|
| L. viridis | 161.0 | 165.71 | 4.54 | 0.886 | 0.999 |
| P. muralis | 108.0 | 112.0 | 1.92 | 0.493 | 0.999 |
| D. praticola | 185.0 | 202.76 | 5.33 | 0.955 | 0.998 |
| A. kitaibelii | 158.0 | 160.65 | 4.52 | 0.919 | 0.999 |
| A. fragilis | 109.0 | 111.5 | 3.14 | 0.683 | 0.999 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarkevich, I.; Engibarov, S.; Mitova, S.; Vacheva, E.; Popova, S.; Stanchev, N.; Eneva, R.; Gocheva, Y.; Boyadzhieva, I.; Gerginova, M. 16S rRNA Gene Sequencing-Based Identification and Comparative Analysis of the Fecal Microbiota of Five Syntopic Lizard Species from a Low-Mountain Area in Western Bulgaria. Appl. Microbiol. 2024, 4, 181-193. https://doi.org/10.3390/applmicrobiol4010013
Lazarkevich I, Engibarov S, Mitova S, Vacheva E, Popova S, Stanchev N, Eneva R, Gocheva Y, Boyadzhieva I, Gerginova M. 16S rRNA Gene Sequencing-Based Identification and Comparative Analysis of the Fecal Microbiota of Five Syntopic Lizard Species from a Low-Mountain Area in Western Bulgaria. Applied Microbiology. 2024; 4(1):181-193. https://doi.org/10.3390/applmicrobiol4010013
Chicago/Turabian StyleLazarkevich, Irina, Stephan Engibarov, Simona Mitova, Emiliya Vacheva, Steliyana Popova, Nikola Stanchev, Rumyana Eneva, Yana Gocheva, Ivanka Boyadzhieva, and Maria Gerginova. 2024. "16S rRNA Gene Sequencing-Based Identification and Comparative Analysis of the Fecal Microbiota of Five Syntopic Lizard Species from a Low-Mountain Area in Western Bulgaria" Applied Microbiology 4, no. 1: 181-193. https://doi.org/10.3390/applmicrobiol4010013
APA StyleLazarkevich, I., Engibarov, S., Mitova, S., Vacheva, E., Popova, S., Stanchev, N., Eneva, R., Gocheva, Y., Boyadzhieva, I., & Gerginova, M. (2024). 16S rRNA Gene Sequencing-Based Identification and Comparative Analysis of the Fecal Microbiota of Five Syntopic Lizard Species from a Low-Mountain Area in Western Bulgaria. Applied Microbiology, 4(1), 181-193. https://doi.org/10.3390/applmicrobiol4010013