
Response of Hypolimnetic Water and Bottom Sediment Microbial Communities to Freshwater Salinization—A Microcosm Experiment


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Setup
2.2. DNA Extraction
2.3. 16S and 18S rRNA Gene Sequencing and dPCR
2.4. Bioinformatic and Statistical Analyses
3. Results
3.1. Absolute Abundance and Correlation with Salinity Concentrations
3.2. Alpha Diversity Indices and Correlation with Salinity Concentrations
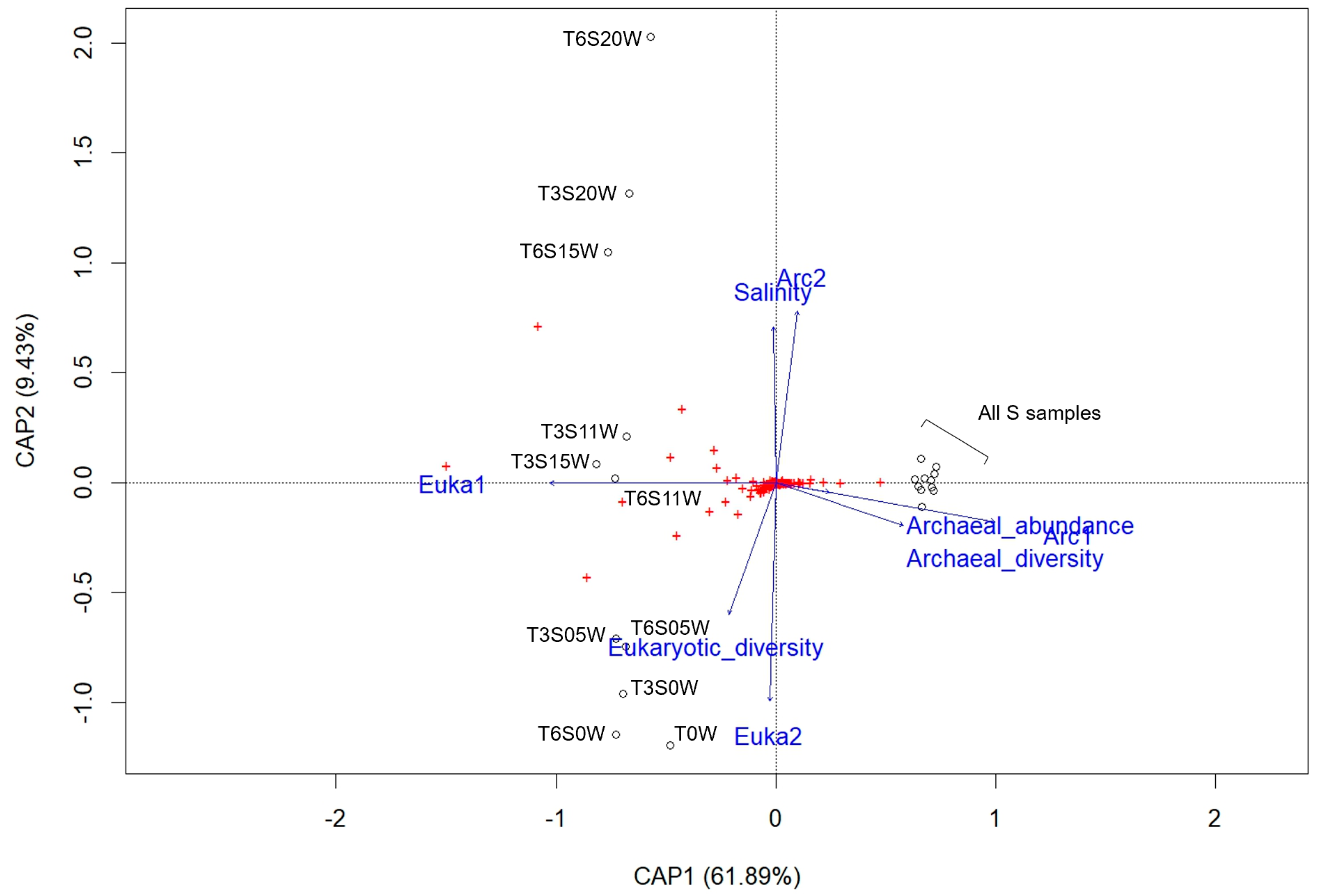
3.3. β-Diversity and Correlation with Environmental Variables
3.3.1. Archaea
3.3.2. Bacteria
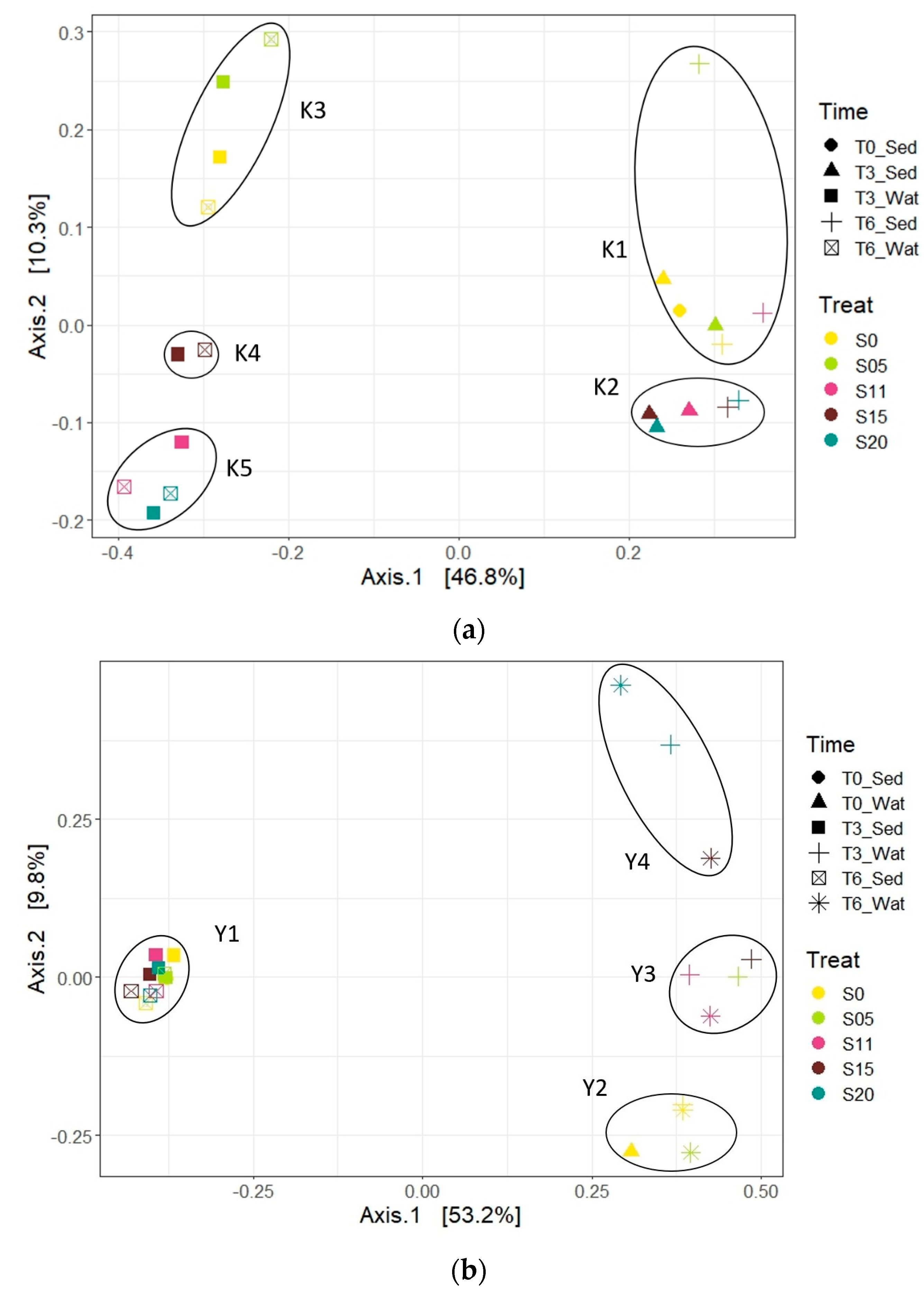
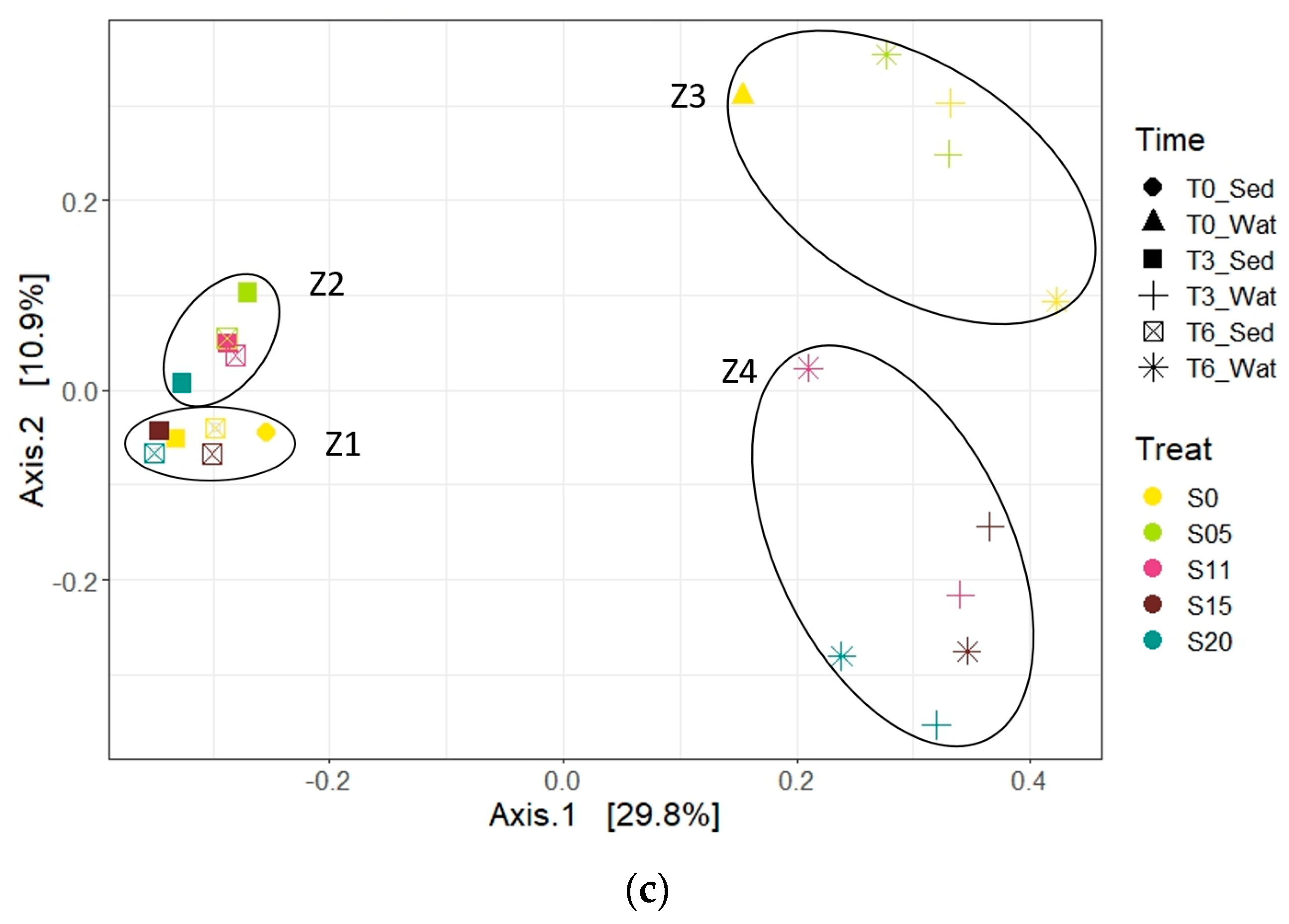
3.4. Microbial Community Structures (β-Diversity)
3.5. Correlations between the Most Abundant Taxa and NaCl Concentrations
3.6. Differential Significance of Microbial Taxa
4. Discussion
4.1. Aquatic Procaryotic Abundance and Diversity
4.2. Sedimentary Procaryotic Abundance and Diversity
4.3. Salinity Threshold for Microbial Community Transition
4.4. Contributors to the Transition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaushal, S.; Likens, G.; Pace, M.; Haq, S.; Wood, K.L.; Galella, J.G.; Morel, C.; Doody, T.; Wessel, B.; Kortelainen, P.; et al. Freshwater Salinization Syndrome: Causes, Consequences, and Management. AGU Fall Meet. Abstr. 2018, 2018, H23E-10. [Google Scholar] [CrossRef]
- USGS. WSS—Water Science School, 2018. Saline Water and Salinity. Available online: https://www.usgs.gov/special-topics/water-science-school/science/saline-water-and-salinity#overview (accessed on 7 November 2022).
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.; O’Sullivan, L.A.; Meng, Y.; Williams, A.S.; Sass, A.M.; Watkins, A.J.; Parkes, R.J.; Weightman, A.J. Archaeal community diversity and abundance changes along a natural salinity gradient in estuarine sediments. FEMS Microbiol. Ecol. 2015, 91, 1–18. [Google Scholar] [CrossRef]
- Gao, J.; Hou, L.; Zheng, Y.; Liu, M.; Yin, G.; Yu, C.; Gao, D. Shifts in the Community Dynamics and Activity of Ammonia-Oxidizing Prokaryotes Along the Yangtze Estuarine Salinity Gradient. J. Geophys. Res. Biogeosci. 2018, 123, 3458–3469. [Google Scholar] [CrossRef]
- Paver, S.F.; Muratore, D.; Newton, R.J.; Coleman, M.L. Reevaluating the Salty Divide: Phylogenetic Specificity of Transitions between Marine and Freshwater Systems. Msystems 2018, 3, e00232-18. [Google Scholar] [CrossRef]
- Chapra, S.C.; Dove, A.; Warren, G.J. Long-term trends of Great Lakes major ion chemistry. J. Great Lakes Res. 2012, 38, 550–560. [Google Scholar] [CrossRef]
- Gagnon, J.-C.; Astorg, L.; Derry, A.M.; Lazar, C.S. Response of Prokaryotic Communities to Freshwater Salinization. Appl. Microbiol. 2022, 2, 330–346. [Google Scholar] [CrossRef]
- Santoro, A.E. Microbial nitrogen cycling at the saltwater–freshwater interface. Hydrogeol. J. 2010, 18, 187–202. [Google Scholar] [CrossRef]
- Pernthaler, J.; Rosenberg, E.; DeLong, E.; Lory, S. Freshwater Microbial Communities; Springer: Berlin, Germany, 2013. [Google Scholar] [CrossRef]
- Anderson, O.R. Evidence for Coupling of the Carbon and Phosphorus Biogeochemical Cycles in Freshwater Microbial Communities. Front. Mar. Sci. 2018, 5, 20. [Google Scholar] [CrossRef]
- Edmonds, J.W.; Weston, N.B.; Joye, S.B.; Mou, X.; Moran, M.A. Microbial Community Response to Seawater Amendment in Low-Salinity Tidal Sediments. Microb. Ecol. 2009, 58, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.; Morrissey, E.M.; Neubauer, S.C.; Franklin, R.B. Novel microbial community composition and carbon biogeochemistry emerge over time following saltwater intrusion in wetlands. Glob. Chang. Biol. 2019, 25, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, G.; Tang, X.; Shao, K. Can the freshwater bacterial communities shift to the “marine-like” taxa? J. Basic Microbiol. 2014, 54, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- DeVilbiss, S.E.; Steele, M.K.; Brown, B.L.; Badgley, B.D. Stream bacterial diversity peaks at intermediate freshwater salinity and varies by salt type. Sci. Total. Environ. 2022, 840, 156690. [Google Scholar] [CrossRef] [PubMed]
- Rocca, J.D.; Simonin, M.; Wright, J.P.; Washburne, A.; Bernhardt, E. Rare microbial taxa emerge when communities collide: Freshwater and marine microbiome responses to experimental seawater intrusion. bioRxiv 2019, 550756. [Google Scholar] [CrossRef]
- Kaushal, S.S.; Groffman, P.M.; Likens, G.E.; Belt, K.T.; Stack, W.P.; Kelly, V.R.; Band, L.E.; Fisher, G.T. Increased salinization of fresh water in the northeastern United States. Proc. Natl. Acad. Sci. USA 2005, 102, 13517–13520. [Google Scholar] [CrossRef]
- Kaushal, S.S. Increased Salinization Decreases Safe Drinking Water. Environ. Sci. Technol. 2016, 50, 2765–2766. [Google Scholar] [CrossRef]
- Environment Canada. Priority Substances List Assessment Report: Road Salts. In Canadian Environmental Protection Act; Environment Canada: Toronto, AB, Canada, 1999; ISBN 0-662-31018-7. [Google Scholar]
- Astorg, L.; Gagnon, J.; Lazar, C.S.; Derry, A.M. Effects of freshwater salinization on a salt-naïve planktonic eukaryote community. Limnol. Oceanogr. Lett. 2022, 8, 38–47. [Google Scholar] [CrossRef]
- Qiagen. DNeasy® PowerMax® Soil Kit Handbook; HB-2267-001 © 2023 QIAGEN, All Rights Reserved; Qiagen: Venlo, The Netherlands, 2018. [Google Scholar]
- Qiagen. DNeasy® PowerWater® Kit Handbook; HB-2267-001 © 2023 QIAGEN, All Rights Reserved; Qiagen: Venlo, The Netherlands, 2017. [Google Scholar]
- Lazar, C.S.; Stoll, W.; Lehmann, R.; Herrmann, M.; Schwab, V.F.; Akob, D.M.; Nawaz, A.; Wubet, T.; Buscot, F.; Totsche, K.-U.; et al. Archaeal Diversity and CO2 Fixers in Carbonate-/Siliciclastic-Rock Groundwater Ecosystems. Archaea 2017, 2017, 213628. [Google Scholar] [CrossRef]
- Sun, Y.; Zhong, S.; Deng, B.; Jin, Q.; Wu, J.; Huo, J.; Zhu, J.; Zhang, C.; Li, Y. Impact of Phellinus gilvus mycelia on growth, immunity and fecal microbiota in weaned piglets. PeerJ 2020, 8, e9067. [Google Scholar] [CrossRef]
- Capo, E.; Debroas, D.; Arnaud, F.; Guillemot, T.; Bichet, V.; Millet, L.; Gauthier, E.; Massa, C.; Develle, A.-L.; Pignol, C.; et al. Long-term dynamics in microbial eukaryotes communities: A palaeolimnological view based on sedimentary DNA. Mol. Ecol. 2016, 25, 5925–5943. [Google Scholar] [CrossRef]
- ThermoFisher Scientific. QuantStudio™ 3D Digital PCR System User Manual, PN MAN0007720; ThermoFisher Scientific: Waltham, MA, USA, 2015. [Google Scholar]
- ThermoFisher Scientific. Target Quantification Using SYBR® Green I Dye on the QuantStudio™ 3D Digital PCR System; Demonstrated Protocol; Applied Biosystem; CO35063 0414; ThermoFisher Scientific: Waltham, MA, USA, 2014. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Castelle, C.J.; Probst, A.J.; Zhou, Z.; Pan, J.; Liu, Y.; Banfield, J.F.; Gu, J.-D. Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 2018, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Pan, J.; Wang, F.; Gu, J.-D.; Li, M. Bathyarchaeota: Globally distributed metabolic generalists in anoxic environments. FEMS Microbiol. Rev. 2018, 42, 639–655. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2020; Available online: http://www.rstudio.com (accessed on 18 August 2023).
- Hammer, Ø. PAST PAleontological STatistics ver. 4.03—Reference Manual. Natural History Museum. University of Oslo. 2020. Available online: https://www.nhm.uio.no/english/research/infraestructure/past (accessed on 18 August 2023).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Wagner, H. Community Ecology Package; R Package Version, 2-0; Scientific Research Publishing: Wuhan, China, 2013. [Google Scholar]
- Clarke, K.R.; Gorley, R.N. PRIMER v6. User Manual/Tutorial. Plymouth Routine in Mulitvariate Ecological Research; Plymouth Marine Laboratory: Plymouth, UK, 2006. [Google Scholar]
- Asteinberger9. asteinberger9/seq_scripts v1.1 (Version v1.1). Zenodo. 12 November 2020. Available online: https://zenodo.org/record/4270481 (accessed on 18 August 2023). [CrossRef]
- Smith, S. phylosmith: An R-package for reproducible and efficient microbiome analysis with phyloseq-objects. J. Open Source Softw. 2019, 4, 1442. [Google Scholar] [CrossRef]
- Cooper, J.I. Viruses and the Environment. Science & Business Media; Springer: Berlin/Heidelberg, Germany, 2012; ISBN 978-94-015-7701-4. [Google Scholar]
- Tang, B.-L.; Yang, J.; Chen, X.-L.; Wang, P.; Zhao, H.-L.; Su, H.-N.; Li, C.-Y.; Yu, Y.; Zhong, S.; Wang, L.; et al. A predator-prey interaction between a marine Pseudoalteromonas sp. and Gram-positive bacteria. Nat. Commun. 2020, 11, 285. [Google Scholar] [CrossRef] [PubMed]
- Weynberg, K.D. Viruses in Marine Ecosystems: From Open Waters to Coral Reefs. Adv. Virus Res. 2018, 101, 1–38. [Google Scholar] [CrossRef]
- Wang, B.; Liu, N.; Yang, M.; Wang, L.; Liang, X.; Liu, C.-Q. Co-occurrence of planktonic bacteria and archaea affects their biogeographic patterns in China’s coastal wetlands. Environ. Microbiome 2021, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-C.; Li, X.-K.; Wang, X.-W.; Liu, G.-G.; Zuo, J.-L.; Wang, S.-T.; Wang, K. Impact of salinity on anaerobic microbial community structure in high organic loading purified terephthalic acid wastewater treatment system. J. Hazard. Mater. 2020, 383, 121132. [Google Scholar] [CrossRef] [PubMed]
- Thajudeen, J.; Yousuf, J.; Veetil, V.P.; Varghese, S.; Singh, A.; Abdulla, M.H. Nitrogen fixing bacterial diversity in a tropical estuarine sediments. World J. Microbiol. Biotechnol. 2017, 33, 41. [Google Scholar] [CrossRef] [PubMed]
- Pester, M.; Bittner, N.; Deevong, P.; Wagner, M.; Loy, A. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 2010, 4, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Durà, A.; Burke, I.; Mortimer, R.; Stewart, D. Diversity patterns of benthic bacterial communities along the salinity continuum of the Humber estuary (UK). Aquat. Microb. Ecol. 2018, 81, 277–291. [Google Scholar] [CrossRef]
- Campbell, B.J.; Kirchman, D.L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 2013, 7, 210–220. [Google Scholar] [CrossRef]
- Liu, J.; Yang, H.; Zhao, M.; Zhang, X.-H. Spatial distribution patterns of benthic microbial communities along the Pearl Estuary, China. Syst. Appl. Microbiol. 2014, 37, 578–589. [Google Scholar] [CrossRef]
- Tee, H.S.; Waite, D.; Lear, G.; Handley, K.M. Microbial river-to-sea continuum: Gradients in benthic and planktonic diversity, osmoregulation and nutrient cycling. Microbiome 2021, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.L.; Brock, M.A.; Rees, G.N.; Baldwin, D.S. Effects of increasing salinity on freshwater ecosystems in Australia. Aust. J. Bot. 2003, 51, 655–665. [Google Scholar] [CrossRef]
- Pan, J.; Zhou, Z.; Béjà, O.; Cai, M.; Yang, Y.; Liu, Y.; Gu, J.-D.; Li, M. Genomic and transcriptomic evidence of light-sensing, porphyrin biosynthesis, Calvin-Benson-Bassham cycle, and urea production in Bathyarchaeota. Microbiome 2020, 8, 43. [Google Scholar] [CrossRef]
- Zou, D.; Liu, H.; Li, M. Community, Distribution, and Ecological Roles of Estuarine Archaea. Front. Microbiol. 2020, 11, 2060. [Google Scholar] [CrossRef]
- Zou, D.; Pan, J.; Liu, Z.; Zhang, C.; Liu, H.; Li, M. The Distribution of Bathyarchaeota in Surface Sediments of the Pearl River Estuary Along Salinity Gradient. Front. Microbiol. 2020, 11, 285. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Thakur, N.; El-Dalatony, M.M.; Salama, E.-S.; Li, X. Protein biomethanation: Insight into the microbial nexus. Trends Microbiol. 2022, 30, 69–78. [Google Scholar] [CrossRef]
- Laskar, F.; Das Purkayastha, S.; Sen, A.; Bhattacharya, M.K.; Misra, B.B. Diversity of methanogenic archaea in freshwater sediments of lacustrine ecosystems. J. Basic Microbiol. 2018, 58, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xia, K.; Yang, X.; Tang, C. Growth strategy of microbes on mixed carbon sources. Nat. Commun. 2019, 10, 1279. [Google Scholar] [CrossRef]
- Lerman, A.; Weiler, R.R. Diffusion and accumulation of chloride and sodium in Lake Ontario sediment. Earth Planet. Sci. Lett. 1970, 10, 150–156. [Google Scholar] [CrossRef]
- Gagliano, M.; Ismail, S.; Stams, A.; Plugge, C.; Temmink, H.; Van Lier, J. Biofilm formation and granule properties in anaerobic digestion at high salinity. Water Res. 2017, 121, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Sudmalis, D.; Gagliano, M.C.; Pei, R.; Grolle, K.; Plugge, C.M.; Rijnaarts, H.H.M.; Zeeman, G.; Temmink, H. Fast anaerobic sludge granulation at elevated salinity. Water Res. 2018, 128, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, M.C.; Sudmalis, D.; Pei, R.; Temmink, H.; Plugge, C.M. Microbial Community Drivers in Anaerobic Granulation at High Salinity. Front. Microbiol. 2020, 11, 235. [Google Scholar] [CrossRef]
- Chen, S.; Wang, P.; Liu, H.; Xie, W.; Wan, X.S.; Kao, S.-J.; Phelps, T.J.; Zhang, C. Population dynamics of methanogens and methanotrophs along the salinity gradient in Pearl River Estuary: Implications for methane metabolism. Appl. Microbiol. Biotechnol. 2020, 104, 1331–1346. [Google Scholar] [CrossRef]
- Liu, Y.; Priscu, J.C.; Xiong, J.; Conrad, R.; Vick-Majors, T.; Chu, H.; Hou, J. Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiol. Ecol. 2016, 92, fiw033. [Google Scholar] [CrossRef]
- Tong, C.; Cadillo-Quiroz, H.; Zeng, Z.; She, C.; Yang, P.; Huang, J. Changes of community structure and abundance of methanogens in soils along a freshwater–brackish water gradient in subtropical estuarine marshes. Geoderma 2017, 299, 101–110. [Google Scholar] [CrossRef]
- Bertrand, J.C.; Caumette, P.; Lebaron, P.; Matheron, R.; Normand, P.; Ngando, T.S. (Eds.) Environmental Microbiology: Fundamentals and Applications; Springer: Dordrecht, The Netherlands, 2015; pp. 3–7. ISBN 9789401791182. [Google Scholar]
- Takeshita, K.; Yamada, T.; Kawahara, Y.; Narihiro, T.; Ito, M.; Kamagata, Y.; Shinzato, N. Tripartite Symbiosis of an Anaerobic Scuticociliate with Two Hydrogenosome-Associated Endosymbionts, a Holospora—Related Alphaproteobacterium and a Methanogenic Archaeon. Appl. Environ. Microbiol. 2019, 85, e00854-19. [Google Scholar] [CrossRef] [PubMed]
- CCME—Canadian Council of Ministers of the Environment. Scientific Criteria Document for the Development of the Canadian Water Quality Guidelines for the Protection of Aquatic Life: Chloride ion; Canadian Council of Ministers of the Environment: Winnepeg, MB, Canada, 2011; PN 1460; ISBN 9781-896997-77-3. [Google Scholar]
- Schuler, M.S.; Cañedo-Argüelles, M.; Hintz, W.D.; Dyack, B.; Birk, S.; Relyea, R.A. Regulations are needed to protect freshwater ecosystems from salinization. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180019. [Google Scholar] [CrossRef] [PubMed]
- Stefanidou, N.; Genitsaris, S.; Lopez-Bautista, J.; Sommer, U.; Moustaka-Gouni, M. Unicellular Eukaryotic Community Response to Temperature and Salinity Variation in Mesocosm Experiments. Front. Microbiol. 2018, 9, 2444. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, D.S.; Rees, G.N.; Mitchell, A.M.; Watson, G.; Williams, J. The short-term effects of salinization on anaerobic nutrient cycling and microbial community structure in sediment from a freshwater wetland. Wetlands 2006, 26, 455–464. [Google Scholar] [CrossRef]
- Yang, J.; Ma, L.; Jiang, H.; Wu, G.; Dong, H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 2016, 6, 25078. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dong, H.; Zhang, G.; Yu, B.; Chapman, L.R.; Fields, M.W. Microbial Diversity in Water and Sediment of Lake Chaka, an Athalassohaline Lake in Northwestern China. Appl. Environ. Microbiol. 2006, 72, 3832–3845. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.R.; Vallaire, S.C. Effects of salinity and nutrients on microbial assemblages in Louisiana wetland sediments. Wetlands 2009, 29, 277–287. [Google Scholar] [CrossRef]
- Ikenaga, M.; Guevara, R.; Dean, A.L.; Pisani, C.; Boyer, J.N. Changes in Community Structure of Sediment Bacteria Along the Florida Coastal Everglades Marsh–Mangrove–Seagrass Salinity Gradient. Microb. Ecol. 2010, 59, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Freitag, T.E.; Chang, L.; Prosser, J.I. Changes in the community structure and activity of betaproteobacterial ammonia-oxidizing sediment bacteria along a freshwater-marine gradient. Environ. Microbiol. 2006, 8, 684–696. [Google Scholar] [CrossRef]
- Walsh, E.A.; Kirkpatrick, J.B.; Rutherford, S.D.; Smith, D.C.; Sogin, M.; D’Hondt, S. Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 2016, 10, 979–989. [Google Scholar] [CrossRef]
- Yan, N.; Marschner, P.; Cao, W.; Zuo, C.; Qin, W. Influence of salinity and water content on soil microorganisms. Int. Soil Water Conserv. Res. 2015, 3, 316–323. [Google Scholar] [CrossRef]
- Mani, K.; Taib, N.; Hugoni, M.; Bronner, G.; Bragança, J.M.; Debroas, D. Transient Dynamics of Archaea and Bacteria in Sediments and Brine Across a Salinity Gradient in a Solar Saltern of Goa, India. Front. Microbiol. 2020, 11, 1891. [Google Scholar] [CrossRef]
- Lv, J.; Niu, Y.; Yuan, R.; Wang, S. Different Responses of Bacterial and Archaeal Communities in River Sediments to Water Diversion and Seasonal Changes. Microorganisms 2021, 9, 782. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.; Whitfield, A. Challenging paradigms in estuarine ecology and management. Estuar. Coast. Shelf Sci. 2011, 94, 306–314. [Google Scholar] [CrossRef]
- Wisnoski, N.I.; Lennon, J.T. Stabilising role of seed banks and the maintenance of bacterial diversity. Ecol. Lett. 2021, 24, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Rath, J. Methods in Methane Metabolism, Part A. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2011; Volume 494, pp. 338–339. [Google Scholar] [CrossRef]
- Karakashev, D.; Batstone, D.J.; Trably, E.; Angelidaki, I. Acetate Oxidation Is the Dominant Methanogenic Pathway from Acetate in the Absence of Methanosaetaceae. Appl. Environ. Microbiol. 2006, 72, 5138–5141. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Fotidis, I.A.; Kissas, K.; Angelidaki, I. Effect of different ammonia sources on aceticlastic and hydrogenotrophic methanogens. Bioresour. Technol. 2018, 250, 390–397. [Google Scholar] [CrossRef]
- Fenchel, T.; King, G.; Blackburn, T. Aquatic Sediments. In Bacterial Biogeochemistry, 3rd ed.; Academic Press: Boston, MA, USA, 2012; pp. 121–142. ISBN 978-0-12-415836-8. [Google Scholar]
- Zheng, P.-F.; Wei, Z.; Zhou, Y.; Li, Q.; Qi, Z.; Diao, X.; Wang, Y. Genomic Evidence for the Recycling of Complex Organic Carbon by Novel Thermoplasmatota Clades in Deep-Sea Sediments. mSystems 2022, 7, e0007722. [Google Scholar] [CrossRef]
- Bianchi, T.S.; Bauer, J.E. 5.03–Particulate Organic Carbon Cycling and Transformation. Treatise Estuar. Coast. Sci. 2011, 5, 69–117. [Google Scholar] [CrossRef]
- Pavlostathis, S.G. Environmental Biotechnology and Safety. In Comprehensive Biotechnology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2011; ISBN 9780080885049. [Google Scholar]
- Smith, K.S.; Ingram-Smith, C. Methanosaeta, the forgotten methanogen? Trends Microbiol. 2007, 15, 150–155. [Google Scholar] [CrossRef]
- Chen, S.; He, Q. Persistence of Methanosaeta populations in anaerobic digestion during process instability. J. Ind. Microbiol. Biotechnol. 2015, 42, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, G.M.F.; Domingues, M.R.; de Jesus, T.A.; Coelho, L.H.G.; Hanisch, W.S.; Pompêo, M.L.M.; Saia, F.T.; Gregoracci, G.B.; Benassi, R.F. Higher Abundance of Sediment Methanogens and Methanotrophs Do Not Predict the Atmospheric Methane and Carbon Dioxide Flows in Eutrophic Tropical Freshwater Reservoirs. Front. Microbiol. 2021, 12, 647921. [Google Scholar] [CrossRef] [PubMed]
- Fischer, P.Q.; Sánchez-Andrea, I.; Stams, A.J.M.; Villanueva, L.; Sousa, D.Z. Anaerobic microbial methanol conversion in marine sediments. Environ. Microbiol. 2021, 23, 1348–1362. [Google Scholar] [CrossRef] [PubMed]
- Pedrós-Alió, C.; Calderón-Paz, J.I.; Gasol, J.M. Comparative analysis shows that bacterivory, not viral lysis, controls the abundance of heterotrophic prokaryotic plankton. FEMS Microbiol. Ecol. 2000, 32, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, J.P. Les Sels de Voirie au Québec: Proposition D’une Démarche de Gestion Environnementale Spécifique aux Zones Vulnérables. Doctoral Dissertation, Université de Sherbrooke, Sherbrooke, QC, Canada, 2011. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Time | R2 | p Value |
|---|---|---|---|
| Water archaea absolute abundance × Salinity | T3 | 0.2342 | 0.4088 |
| T6 | 0.1831 | 0.4723 | |
| Sediment archaea absolute abundance × Salinity | T3 | 0.6703 | 0.09011 |
| T6 | 0.5589 | 0.1463 | |
| Water bacterial absolute abundance × Salinity | T3 | 0.5555 | 0.1482 |
| T6 | 0.4064 | 0.2473 | |
| Sediment bacterial absolute abundance × Salinity | T3 | 0.0336 | 0.0768 |
| T6 | 0.0613 | 0.688 |
| Variables | Time | R2 | p Value |
|---|---|---|---|
| Water archaeal α-diversity × Salinity | T3 | 0.2637 | 0.3762 |
| T6 | 0.2605 | 0.3796 | |
| Sediment archaeal α-diversity × Salinity | T3 | 0.0088 | 0.8808 |
| T6 | 0.5186 | 0.1701 | |
| Water bacterial α-diversity × Salinity | T3 | 0.0418 | 0.7414 |
| T6 | 0.9196 | 0.00992 ** | |
| Sediment bacterial α-diversity × Salinity | T3 | 0.0893 | 0.62525 |
| T6 | 0.0004 | 0.97385 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagnon, J.-C.; Blais, V.T.; Lazar, C.S. Response of Hypolimnetic Water and Bottom Sediment Microbial Communities to Freshwater Salinization—A Microcosm Experiment. Appl. Microbiol. 2023, 3, 915-934. https://doi.org/10.3390/applmicrobiol3030063
Gagnon J-C, Blais VT, Lazar CS. Response of Hypolimnetic Water and Bottom Sediment Microbial Communities to Freshwater Salinization—A Microcosm Experiment. Applied Microbiology. 2023; 3(3):915-934. https://doi.org/10.3390/applmicrobiol3030063
Chicago/Turabian StyleGagnon, Jean-Christophe, Valérie Turcotte Blais, and Cassandre Sara Lazar. 2023. "Response of Hypolimnetic Water and Bottom Sediment Microbial Communities to Freshwater Salinization—A Microcosm Experiment" Applied Microbiology 3, no. 3: 915-934. https://doi.org/10.3390/applmicrobiol3030063
APA StyleGagnon, J.-C., Blais, V. T., & Lazar, C. S. (2023). Response of Hypolimnetic Water and Bottom Sediment Microbial Communities to Freshwater Salinization—A Microcosm Experiment. Applied Microbiology, 3(3), 915-934. https://doi.org/10.3390/applmicrobiol3030063