
Univariate and Multivariate Statistical Analysis of Microbiome Data: An Overview

Abstract
1. Introduction
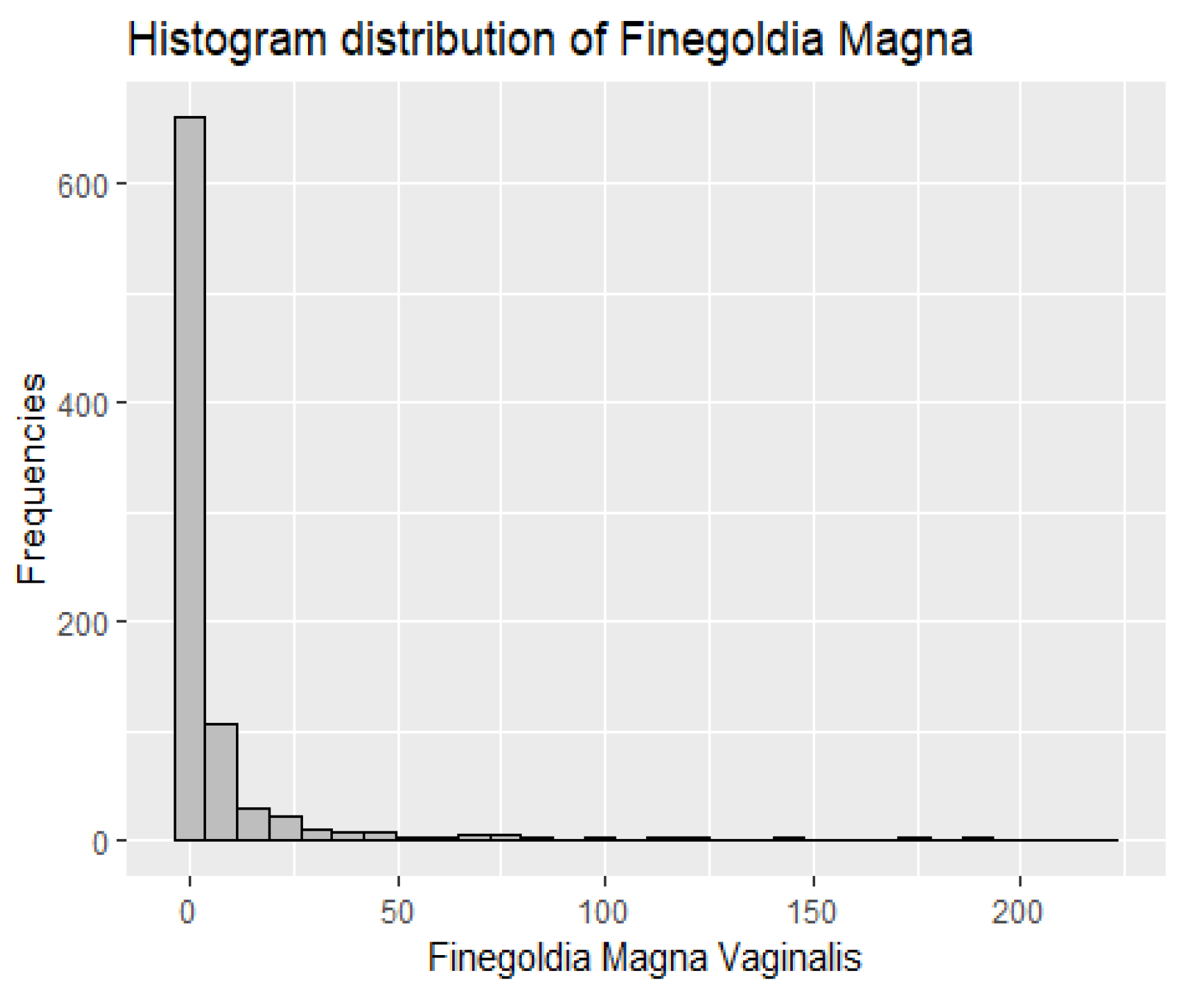
2. Microbiome Data Representation and Modeling Challenges
Differential Abundance and Normalization Methods for Microbiome Data
- Scaling Methods. The idea of the scaling method is to divide the observed abundance by a scaling (normalization) factor. More specifically, scaling is defined as follows.where is the normalized abundance for feature j within sample i, and is the scaling (normalization) factor for sample i.
- Log-ratio Methods. The most known log-ratio transformation used in microbiome data analyses is the centered log-ratio (clr) [25]. In particular, clr transforms the features by taking the log of the ratio between observed features and their geometric mean. Some common log-ratio normalization methods include centered log-ratio (CLR) transformation [26] and ALDEx [27].
- RNA-seq Methods. The RNA-seq methods are parametric methods. A large part of the variability in RNA-seq data arises from the sampling of the microbial ecosystem [28]. DESeq2 and edgeR are two popular methods from RNA-seq for testing differences across study groups. Both methods model the observed abundances using the negative binomial distribution. Some recent studies have indicated the poor performance of these two methods [20,29]. MetagenomeSeq is an alternative RNA-seq method. Instead of using a negative binomial model, MetagenomeSeq is based on a zero-inflated Gaussian (ZIG). For more details about a general zero-inflated model, please read Section 3.1.3. MetagenomeSeq has been applied to different microbiome studies and shows higher powers than most of the other differential abundance methods such as DESeq2 and edgeR [30,31].
3. Modeling Single Feature
3.1. Probabilistic Models
3.1.1. Poisson Model
3.1.2. Negative Binomial Model
3.1.3. Zero-Inflated Models
3.1.4. Hurdle Models
3.2. Regression Analysis
3.2.1. Generalized Linear Models
3.2.2. Vector Generalized Linear Models
3.2.3. Bayesian Models
3.3. Longitudinal Microbiome Data
- MetaDprof [64] is a smoothing spline-based method, and a well-known method for modeling longitudinal data [65,66]. MetaDprof is used for detecting differentially abundant features from metagenomic samples by comparing different conditions across time. There is a major limitation of the MetaDprof method. It assumes consistency in longitudinal microbial samples. For example, the same number of subjects per phenotypic group, the same number of samples from each subject, and the same time points [62].
- MetaLonDA [62] is an R package that is capable of identifying significant time intervals of differentially abundant microbial features. It can be applied to any longitudinal count data such as metagenomic sequencing, 16S rRNA gene sequencing, or RNAseq. MetaLonDA relies on two modeling components. The NB distribution for modeling the features reads counts and the semi-parametric SS-ANOVA technique for modeling longitudinal profiles associated with different phenotypes. MetaLonDa is able to handle the metaDprof limitations. For example, it does not require the same number of subjects per group. The elapsed time between adjacent time points is flexible. One limitation of MetaLonDA is that when samples are sparse over time intervals, the fitted smoothing spline has a large variation.
- Zero-inflated Beta regression model with random effects (ZIBR) model [63]. Chen and Li (2016) proposed a two-part zero-inflated beta regression model with random effects (ZIBR) for testing the association between microbial abundance and clinical covariates for longitudinal microbiome data. The proposed model includes a beta regression component to model non-zero microbial abundance, and a logistic regression component to model the presence/absence of a microbe in the samples. Each component includes a random effect to account for the correlations among the repeated measurements on the same subject. Based on a real microbiome data application, the ZIBR model performed better than the commonly used models such as binomial, zero-inflated Poisson, and negative binomial regression models.
- Zero-inflated negative binomial mixed-effects (ZINBMM) model [15]. Romero et al. (2014) proposed a longitudinal vaginal microbiome study for comparing the vaginal microbiome feature (Lactobacillus) between two groups of women (pregnant and non-pregnant women). The zero-inflated negative binomial mixed-effects (ZINBLME) model was applied to model the read counts on the pregnancy status. In addition, negative binomial linear mixed effects (NBLME) and Poisson linear mixed effects (PLME) models were used in the model comparison. Based on their proposed method, the ZINBLME model provided the best fit based on AIC values. One limitation of Romero et al.’s (2014) model is that it can only be applied to count data [63].
- Long Short Term Memory Networks (LSTM) [67]. Recently, Sharma and Xu (2021) proposed a deep learning framework for the feature extraction and analysis of temporal dependency in longitudinal microbiome sequencing data along with the host’s environmental factors for disease prediction. The proposed methodology and an extensive analysis and comparison were applied to 100 simulated datasets across multiple time points and were applied to two real longitudinal human microbiome studies. The analysis showed that the proposed model significantly improves predictive accuracy.
4. Multivariate Microbiome Analysis
4.1. Microbiome–Microbiome Interaction
- Bayesian Network (BNs): BNs are directed probabilistic graphical models that represent a probabilistic relationship between multiple species via a directed acyclic graph. The nodes in BN correspond to random variables, and the directed edges correspond to conditional dependencies between them. The absence of an edge connecting two nodes indicates independence or conditional independence between them. The Bayesian network is an appropriate tool for modeling the interactions of many microbial taxa. It has been used in microbiome studies. For example, Bennett’s (2016) [70] study analysis is based on the construction of a Bayesian network using Dirichlet distributions to model the conjugate probabilities of the most common bacterial constituents in a stool sample. The results indicate that the Bayesian network adjusts the prior bacterial population distribution to more accurately reflect the transcriptionally active bacterial population.
- Graphical Gaussian models (GGMs) are undirected probabilistic graphical models that identify the conditional independence relations among the nodes, where the nodes correspond to multivariate normal distributed variables, and edges between these variables represent conditional dependencies. Zhao and Duan (2019) used GGM to learn the gene interactions in 15 specific types of human cancer [71]. The networks reveal conditional dependencies among the genes, and the weights of edges indicate the strength of the dependencies. The GGM networks reveal stable conditional dependences among the genes and confirm the essential roles played by the genes that encode proteins involved in the two key signaling pathways—PI3K/AKT/mTOR and Ras/Raf/MEK/ERK—in human carcinogenesis.
- SparCC: Sparse Correlations for Compositional data (SparCC) was developed by Friedman and Alm (2012) [72]. The method is capable of estimating correlation values from compositional data. SparCC estimates the linear Pearson correlations between the log-transformed components. Since these correlations cannot be computed exactly, SparCC utilizes an approximation that is based on the assumption that the number of OTUs is large and most OTUs are not strongly correlated with each other. In Friedman and Alm’s (2012) [72] study, they infer a rich ecological network connecting hundreds of interacting species across 18 sites on the human body. SparCC shows that it can infer correlations with high accuracy even in the most challenging datasets.
- FastSpar was proposed recently by Watts et al. (2019) as a fast and parallelizable implementation of the SparCC algorithm with an unbiased P-value estimator [73]. One drawback of SparCC is the overestimated and biased p-value in some cases [74]. FastSpar produces equivalent OTU correlations as SparCC while greatly reducing run time, handling large datasets, and more accurate p-values. FastSpar has been used recently for modeling microbiome data. For example, Qiu et al. (2022) applied the FastSpar algorithm to analyze the soil and plant rhizosphere microbiome of cotton plants in the presence of some cotton-specific fungal pathogen [75]. Their statistical analysis found that Fusarium oxysporum f.sp. vasinfectum (FOV) directly and consistently changed the rhizosphere microbiome. However, the biocontrol agents enabled microbial assemblages to resist pathogenic stress. Their study is essential for understanding core microbiome responses and the existence of plant pathobiomes, which provides an excellent framework for better plant disease management.
- SPIEC-EASI: SParse InversE Covariance Estimation for Ecological Association Inference (SPIEC-EASI) was proposed by Kurtz et al. (2015). It relies on algorithms for sparse neighborhood and inverse covariance selection [76]. It can handle some technical challenges related to microbiome data analysis. For example, the abundances of OTUs are compositional (Because the Counts are normalized). Thus, microbial abundances are not independent, and traditional statistical metrics such as the correlation-based methods for the detection of OTU-OTU relationships can lead to misleading results. Moreover, microbiome data are high dimensional data in general (the number of OTUs (p) is greater than the number of samples n); thus, inference of OTU-OTU association networks is required for an accurate inference. SPIEC-EASI can address both of these issues. Kurtz et al.’s (2015) application to gut microbiome data using SPIEC-EASI produced more consistent and sparser interaction networks than SparCC and CCREPE [76].
- CCLasso: Correlation inference for Compositional data through Lasso (CCLasso) is Similar to SparCC. CCLasso explicitly considers the compositional nature of the metagenomic data in correlation analysis, and it has the advantage that the estimated correlation matrix for compositional data is positive definite [77]. The performance of CCLasso is compared with SparCC through some simulation studies and a real microbiome example from the Human Microbiome Project (HMP). The results show that CCLasso gives a more accurate estimation for the correlation matrix than SparCC as well as better edge recovery.
- Relevance Networks (RN): Relevance networks is an unsupervised learning methodology used in functional genomics and microbiome data with the principal advantages being the ability to (1) include features of more than one data type, (2) represent multiple connections between features, (3) capture both negative and positive correlations, and (4) handle missing data [78]. In the RN method, each set of p edges completely connects the n nodes, and each pair of nodes is connected by a single edge with a score. A study by Werhli et al. (2006) [79] compared three different modeling and inference paradigms, relevance networks (RNs), graphical Gaussian models (GGMs), and Bayesian networks (BNs). The result shows that on Gaussian observational data, BNs and GGMs were found to outperform RNs. There was not a significant difference between BNs and GGMs on observational data in general. However, for interventional data, BNs outperform GGMs and RNs.
- Local Similarity Analysis (LSA): There are many techniques for identifying the relationship between species and associations between species and environmental factors such as Pearson Correlation Coefficient (PCC), and canonical correlation analysis (CCA) analysis. LSA is a novel technique that can identify more complex dependence associations among species as well as associations between species and environmental factors without requiring significant data reduction [80]. Based on a marine microbial observatory dataset application, LSA identified unique, significant associations that were not detected by PCC analysis. LSA can be extended for time series data with replicates.
4.2. Host/Drug–Microbiome Interaction
4.3. Multivariate Longitudinal Data
- Dynamic Bayesian Network: A Dynamic Bayesian Network (DBN) is “a Bayesian network extended with additional mechanisms that are capable of modeling influences over time” [83]. DBN has been used recently for modeling multiple features jointly for longitudinal data. For example, Lugo-Martinez (2019) [84] proposed a study based on DBN for analyzing longitudinal microbiome data. They applied their approach to three different microbiome datasets including infant gut, vaginal, and oral cavity microbiomes. The results provide evidence that microbiome alignments coupled with DBN improve predictive performance over previous methods and enhance our ability to infer biological relationships within the microbiome and between taxa and clinical factors. In McGeachie et al.’s (2016) [85] study, DBN was applied to longitudinal infant gut microbiomes and the predictive performance was analyzed. The DBN model explicitly captured specific relationships and general trends in the data by increasing amounts of Clostridia, residual amounts of Bacilli, and increasing amounts of Gammaproteobacteria. The prediction performance of DBNs with fewer edges was accurate. DBN provided quantitative likelihood estimates for rare abruptions events. DBN was able to identify important relationships between microbiome taxa and predict future changes in microbiome composition.
- Multivariate Granger causality. The Granger causality network model was proposed by Granger (1969) [86], which was originally developed for economics but has now been used extensively in neuroscience and microbiome data analysis [87]. Variable X is the “Granger cause” of variable Y if the histories X and Y together predict the current value of Y better than the history of Y alone [88]. Several multivariate extensions of Granger causality have been developed recently [89,90,91,92,93]. For example, Mainali et al. (2019) [92] show the superiority of multivariate Granger causality over the traditional correlation methods, showing a weak negative relationship between correlation and causality, and a strong positive relationship, whereas almost all strong negative interactions. One limitation of this method is that it does not take into consideration the clinical or demographic variables when building the interaction network [94].
4.4. Multivariate Regression Analysis
- Zero-inflated generalized Dirichlet multinomial (ZIGDM) model [95]. The ZIGDM is proposed for modeling multivariate taxon counts. The ZIGDM regression model was proposed to link microbial abundances to covariates and develop a fast expectation–maximization (EM) algorithm to efficiently estimate the parameters. Based on some simulation studies and an application related to the gut microbiome dataset, the ZIGDM test is more powerful at detecting differential mean/dispersion and is more robust to the underlying distribution if the counts are zero-inflated. If the taxon counts are not zero-inflated, the generalized Dirichlet multinomial (GDM) tests are more desirable. In addition, the GDM provides a superior fit to taxon counts compared to the Dirichlet multinomial (DM), and the ZIGDM can further improve the goodness-of-fit for taxa with many zero counts.
- Bayesian nonparametric multivariate negative binomial regression with zero-inflation (BNP-ZIMNR) model [96]. BNP-ZIMNR is used to analyze multivariate count responses of microbiome data. Zero-inflated negative binomial (ZINB) distribution is used for modeling OTU counts under the assumption that OTU counts are either equal to zero or follow a negative binomial distribution. Nonparametric regression prior models were built on the probability of an OTU count being zero and the mean count of an OTU to study the effects of covariates on microbial communities. Based on some simulation studies and a real chronic wound microbiome dataset, the proposed BNP-ZIMNR model yields superior parameter estimates and model fit in various settings.
- Bayesian Dirichlet-multinomial (BDM) regression model [59]. The proposed model allows for the selection of significant associations between a set of covariates and microbiome features. The statistical inference is conducted through a Markov Chain Monte Carlo (MCMC) algorithm, and the selection of the significant covariates is based on posterior probabilities of inclusions and the thresholding of the Bayesian false discovery rate. The proposed model has been applied to simulated data and real microbiome applications. Compared to some other methods, the BDM model is more accurate and has the lowest false positive as well as false negative rates.
- Logistic Normal Multinomial (LNM) Regression Model [97]. In order to select the covariates and estimate the corresponding regression coefficients, a penalized likelihood estimation method was developed for variable selection and estimation. The Monte Carlo Expectation-Maximization algorithm was applied to implement the penalized likelihood estimation. Compared to the commonly used Dirichlet-multinomial regression model for count data, the LNM model provides a more flexible way of modeling the dependency of the bacterial composition.
- Dirichlet-multinomial (DM) regression model [98]. Because microbiome data are high dimensional data, a penalized likelihood approach was developed to estimate the regression parameters and to select the variables by imposing a sparse group penalty to encourage both group-level and within-group sparsity. A variable selection procedure and an efficient block-coordinate algorithm were developed to solve the optimization problem. Based on some extensive simulations and a real application related to the human gut microbiome, the sparse DM regression can result in better identification of the microbiome-associated covariates than models that ignore overdispersion.
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Constructing and analyzing microbiome networks in R. In Microbiome Analysis; Springer: New York, NY, USA, 2018; pp. 243–266. [Google Scholar]
- Aldirawi, H.; Yang, J. Modeling Sparse Data Using MLE with Applications to Microbiome Data. J. Stat. Theory Pract. 2022, 16, 13. [Google Scholar] [CrossRef]
- Dousti Mousavi, N.; Yang, J.; Aldirawi, H. Variable Selection for Sparse Data with Applications to Vaginal Microbiome and Gene Expression Data. Genes 2023, 14, 403. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.M.; Dourado, M.N.; Araújo, W.L. Microbial interactions: Ecology in a molecular perspective. Braz. J. Microbiol. 2016, 47, 86–98. [Google Scholar] [CrossRef]
- Patangia, D.V.; Anthony Ryan, C.; Dempsey, E.; Paul Ross, R.; Stanton, C. Impact of antibiotics on the human microbiome and consequences for host health. MicrobiologyOpen 2022, 11, e1260. [Google Scholar] [CrossRef]
- Chowdhury, S.; Fong, S.S. Computational modeling of the human microbiome. Microorganisms 2020, 8, 197. [Google Scholar] [CrossRef]
- Palsson, B.; Zengler, K. The challenges of integrating multi-omic datasets. Nat. Chem. Biol. 2010, 6, 787–789. [Google Scholar] [CrossRef]
- Beale, D.J.; Karpe, A.V.; Ahmed, W. Beyond metabolomics: A review of multi-omics-based approaches. In Microbial Metabolomics; Springer: Berlin/Heidelberg, Germany, 2016; pp. 289–312. [Google Scholar]
- Mohan, A.M.; Bibby, K.J.; Lipus, D.; Hammack, R.W.; Gregory, K.B. The functional potential of microbial communities in hydraulic fracturing source water and produced water from natural gas extraction characterized by metagenomic sequencing. PLoS ONE 2014, 9, e107682. [Google Scholar] [CrossRef]
- Trentacoste, E.M.; Shrestha, R.P.; Smith, S.R.; Glé, C.; Hartmann, A.C.; Hildebrand, M.; Gerwick, W.H. Metabolic engineering of lipid catabolism increases microalgal lipid accumulation without compromising growth. Proc. Natl. Acad. Sci. USA 2013, 110, 19748–19753. [Google Scholar] [CrossRef]
- Jiang, R.; Sun, T.; Song, D.; Li, J.J. Statistics or biology: The zero-inflation controversy about scRNA-seq data. Genome Biol. 2022, 23, 1–24. [Google Scholar] [CrossRef]
- Silverman, J.D.; Roche, K.; Mukherjee, S.; David, L.A. Naught all zeros in sequence count data are the same. Comput. Struct. Biotechnol. J. 2020, 18, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Metwally, A.A.; Aldirawi, H.; Yang, J. A review on probabilistic models used in microbiome studies. Commun. Inf. Syst. 2018, 18, 173–191. [Google Scholar] [CrossRef]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Metwally, A.A.; Dai, Y.; Finn, P.W.; Perkins, D.L. WEVOTE: Weighted voting taxonomic identification method of microbial sequences. PLoS ONE 2016, 11, e0163527. [Google Scholar] [CrossRef] [PubMed]
- Aldirawi, H.; Yang, J.; Metwally, A.A. Identifying Appropriate Probabilistic Models for Sparse Discrete Omics Data. In Proceedings of the 2019 IEEE EMBS International Conference on Biomedical & Health Informatics (BHI), Chicago, IL, USA, 19–22 May 2019; pp. 1–4. [Google Scholar]
- He, Y.; Caporaso, J.G.; Jiang, X.T.; Sheng, H.F.; Huse, S.M.; Rideout, J.R.; Edgar, R.C.; Kopylova, E.; Walters, W.A.; Knight, R.; et al. Stability of operational taxonomic units: An important but neglected property for analyzing microbial diversity. Microbiome 2015, 3, 20. [Google Scholar] [CrossRef]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies Ecological and evolutionary microbiology. BMC Microbiol. 2015, 15, 66. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S.D. Analysis of microbial compositions: A review of normalization and differential abundance analysis. NPJ Biofilms Microbiomes 2020, 6, 60. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J. The statistical analysis of compositional data. J. R. Stat. Soc. Ser. B 1982, 44, 139–160. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, V.; Österlund, T.; Nerman, O.; Kristiansson, E. Variability in metagenomic count data and its influence on the identification of differentially abundant genes. J. Comput. Biol. 2017, 24, 311–326. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, J.; Brejnrod, A.; Mortensen, M.; Rasmussen, M.A.; Stokholm, J.; Al-Soud, W.A.; Sørensen, S.; Bisgaard, H.; Waage, J. Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome 2016, 4, 62. [Google Scholar] [CrossRef]
- Jonsson, V.; Österlund, T.; Nerman, O.; Kristiansson, E. Statistical evaluation of methods for identification of differentially abundant genes in comparative metagenomics. BMC Genom. 2016, 17, 78. [Google Scholar] [CrossRef]
- Cheng, M.; Cao, L.; Ning, K. Microbiome big-data mining and applications using single-cell technologies and metagenomics approaches toward precision medicine. Front. Genet. 2019, 10, 972. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG Int. J. Obstet. Gynaecol. 2017, 124, 606–611. [Google Scholar] [CrossRef]
- Hawes, S.E.; Hillier, S.L.; Benedetti, J.; Stevens, C.E.; Koutsky, L.A.; Wølner-Hanssen, P.; Holmes, K.K. Hydrogen peroxide—producing lactobacilli and acquisition of vaginal infections. J. Infect. Dis. 1996, 174, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.J.; Prabhu, K.S.; Vijay-Kumar, M. The microbiome and obesity—An established risk for certain types of cancer. Cancer J. 2014, 20, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Stratigou, T.; Tsagarakis, S. Microbiome and diabetes: Where are we now? Diabetes Res. Clin. Pract. 2018, 146, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Lo, M.T.; Liu, A.; Bettencourt, R.; Ajmera, V.H.; Bassirian, S.; Hooker, J.; Sy, E.; Richards, L.; et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018, 68, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Vuong, H.E.; Hsiao, E.Y. Emerging roles for the gut microbiome in autism spectrum disorder. Biol. Psychiatry 2017, 81, 411–423. [Google Scholar] [CrossRef]
- Di Costanzo, M.; Carucci, L.; Berni Canani, R.; Biasucci, G. Gut microbiome modulation for preventing and treating pediatric food allergies. Int. J. Mol. Sci. 2020, 21, 5275. [Google Scholar] [CrossRef]
- Peng, J.; Xiao, X.; Hu, M.; Zhang, X. Interaction between gut microbiome and cardiovascular disease. Life Sci. 2018, 214, 153–157. [Google Scholar] [CrossRef]
- Peirce, J.M.; Alviña, K. The role of inflammation and the gut microbiome in depression and anxiety. J. Neurosci. Res. 2019, 97, 1223–1241. [Google Scholar] [CrossRef]
- Ohtani, N. Microbiome and cancer. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 37, pp. 65–72. [Google Scholar]
- Sekirov, I.; Finlay, B.B. The role of the intestinal microbiota in enteric infection. J. Physiol. 2009, 587, 4159–4167. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Sun, J.; Chen, D.G. Statistical Analysis of Microbiome Data with R; Springer: Singapore, 2018. [Google Scholar]
- Cameron, A.C. Regression Analysis of Count Data; Cambridge University Press: New York, NY, USA, 2013. [Google Scholar]
- Tipton, L.; Müller, C.L.; Kurtz, Z.D.; Huang, L.; Kleerup, E.; Morris, A.; Bonneau, R.; Ghedin, E. Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Dousti Mousavi, N.; Aldirawi, H.; Yang, J. AZIAD: Analyzing Zero-Inflated and Zero-Altered Data. R Package Version 0.0.2. 2022. Available online: https://arxiv.org/pdf/2205.01294.pdf (accessed on 25 October 2022).
- McCullagh, P.; Nelder, J.A. Generalized Linear Models; Routledge: Boca Raton, FL, USA, 2019. [Google Scholar]
- Yee, T.W.; Stephenson, A.G. Vector generalized linear and additive extreme value models. Extremes 2007, 10, 1–19. [Google Scholar] [CrossRef]
- Welsh, A.H.; Cunningham, R.B.; Donnelly, C.; Lindenmayer, D.B. Modelling the abundance of rare species: Statistical models for counts with extra zeros. Ecol. Model. 1996, 88, 297–308. [Google Scholar] [CrossRef]
- Yee, T. Vector Generalized Linear and Additive Models: With an Implementation in R; Springer: New York, NY, USA, 2015. [Google Scholar]
- Aldirawi, H. Model Selection and Regression Analysis for Sparse Discrete Data. Ph.D. Thesis, University of Illinois at Chicago, Chicago, IL, USA, 2020. [Google Scholar]
- Hu, T.; Gallins, P.; Zhou, Y.H. A zero-inflated beta-binomial model for microbiome data analysis. Stat 2018, 7, e185. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Paterson, A.D.; Turpin, W.; Xu, W. Assessment and selection of competing models for zero-inflated microbiome data. PLoS ONE 2015, 10, e0129606. [Google Scholar] [CrossRef] [PubMed]
- Van den Elskamp, I.; Knol, D.; Uitdehaag, B.; Barkhof, F. The distribution of new enhancing lesion counts in multiple sclerosis: Further explorations. Mult. Scler. J. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, W.D.; Argiento, R.; Guindani, M.; Galloway-Pena, J.; Shelburne, S.A.; Vannucci, M. An integrative Bayesian Dirichlet-multinomial regression model for the analysis of taxonomic abundances in microbiome data. BMC Bioinform. 2017, 18, 94. [Google Scholar]
- Koslovsky, M.D.; Hoffman, K.L.; Daniel, C.R.; Vannucci, M. A Bayesian Model of Microbiome Data for Simultaneous Identification of Covariate Associations and Prediction of Phenotypic Outcomes. Submitted to “Annals of Applied Statistics”. 2020. Available online: https://arxiv.org/pdf/2004.14817.pdf (accessed on 25 October 2022).
- Gerber, G.K. Longitudinal microbiome data analysis. In Metagenomics for Microbiology; Elsevier: San Diego, CA, USA, 2015; pp. 97–111. [Google Scholar]
- Metwally, A.A.; Yang, J.; Ascoli, C.; Dai, Y.; Finn, P.W.; Perkins, D.L. MetaLonDA: A flexible R package for identifying time intervals of differentially abundant features in metagenomic longitudinal studies. Microbiome 2018, 6, 32. [Google Scholar] [CrossRef]
- Chen, E.Z.; Li, H. A two-part mixed-effects model for analyzing longitudinal microbiome compositional data. Bioinformatics 2016, 32, 2611–2617. [Google Scholar] [CrossRef]
- Luo, D.; Ziebell, S.; An, L. An informative approach on differential abundance analysis for time-course metagenomic sequencing data. Bioinformatics 2017, 33, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Gu, C. Smoothing Spline ANOVA Models, 2nd ed.; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Wang, Y. Smoothing Splines: Methods and Applications; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Sharma, D.; Xu, W. phyLoSTM: A novel deep learning model on disease prediction from longitudinal microbiome data. Bioinformatics 2021, 37, 3707–3714. [Google Scholar] [CrossRef] [PubMed]
- Nadal Jimenez, P.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Todd, J.A. Metagenomics and personalized medicine. Cell 2011, 147, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.E. Bayesian Networks to Assess the Human Newborn Stool Metatranscriptome. Ph.D. Thesis, Washington University, Washington, DC, USA, 2016. [Google Scholar]
- Zhao, H.; Duan, Z.H. Cancer genetic network inference using gaussian graphical models. Bioinform. Biol. Insights 2019, 13, 1177932219839402. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed]
- Watts, S.C.; Ritchie, S.C.; Inouye, M.; Holt, K.E. FastSpar: Rapid and scalable correlation estimation for compositional data. Bioinformatics 2019, 35, 1064–1066. [Google Scholar] [CrossRef]
- Phipson, B.; Smyth, G.K. Permutation P-values should never be zero: Calculating exact P-values when permutations are randomly drawn. Stat. Appl. Genet. Mol. Biol. 2010, 9. [Google Scholar] [CrossRef]
- Qiu, Z.; Verma, J.P.; Liu, H.; Wang, J.; Batista, B.D.; Kaur, S.; de Araujo Pereira, A.P.; Macdonald, C.A.; Trivedi, P.; Weaver, T.; et al. Response of the plant core microbiome to Fusarium oxysporum infection and identification of the pathobiome. Environ. Microbiol. 2022, 24, 4652–4669. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Huang, C.; Zhao, H.; Deng, M. CCLasso: Correlation inference for compositional data through Lasso. Bioinformatics 2015, 31, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, G.; Garrett, E.S.; Irizarry, R.A.; Zeger, S.L. The Analysis of Gene Expression Data: An Overview of Methods and Software; Springer: New York, NY, USA, 2003; pp. 1–45. [Google Scholar]
- Werhli, A.V.; Grzegorczyk, M.; Husmeier, D. Comparative evaluation of reverse engineering gene regulatory networks with relevance networks, graphical Gaussian models and Bayesian networks. Bioinformatics 2006, 22, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Dutta, D.; Schwalbach, M.S.; Steele, J.A.; Fuhrman, J.A.; Sun, F. Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 2006, 22, 2532–2538. [Google Scholar] [CrossRef]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Murphy, K.P. Dynamic Bayesian Networks: Representation, Inference and Learning; University of California: Berkeley, CA, USA, 2002. [Google Scholar]
- Lugo-Martinez, J.; Ruiz-Perez, D.; Narasimhan, G.; Bar-Joseph, Z. Dynamic interaction network inference from longitudinal microbiome data. Microbiome 2019, 7, 54. [Google Scholar] [CrossRef]
- McGeachie, M.J.; Sordillo, J.E.; Gibson, T.; Weinstock, G.M.; Liu, Y.Y.; Gold, D.R.; Weiss, S.T.; Litonjua, A. Longitudinal prediction of the infant gut microbiome with dynamic bayesian networks. Sci. Rep. 2016, 6, 20359. [Google Scholar] [CrossRef]
- Granger, C.W. Investigating causal relations by econometric models and cross-spectral methods. Econom. J. Econom. Soc. 1969, 37, 424–438. [Google Scholar] [CrossRef]
- Gourévitch, B.; Bouquin-Jeannès, R.L.; Faucon, G. Linear and nonlinear causality between signals: Methods, examples and neurophysiological applications. Biol. Cybern. 2006, 95, 349–369. [Google Scholar] [CrossRef]
- Dohlman, A.B.; Shen, X. Mapping the microbial interactome: Statistical and experimental approaches for microbiome network inference. Exp. Biol. Med. 2019, 244, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Siggiridou, E.; Kugiumtzis, D. Granger causality in multivariate time series using a time-ordered restricted vector autoregressive model. IEEE Trans. Signal Process. 2015, 64, 1759–1773. [Google Scholar] [CrossRef]
- Basu, S.; Shojaie, A.; Michailidis, G. Network granger causality with inherent grouping structure. J. Mach. Learn. Res. 2015, 16, 417–453. [Google Scholar]
- Lozano, A.C.; Abe, N.; Liu, Y.; Rosset, S. Grouped graphical Granger modeling methods for temporal causal modeling. In Proceedings of the 15th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Paris, France, 28 June–1 July 2009; pp. 577–586. [Google Scholar]
- Mainali, K.; Bewick, S.; Vecchio-Pagan, B.; Karig, D.; Fagan, W.F. Detecting interaction networks in the human microbiome with conditional Granger causality. PLoS Comput. Biol. 2019, 15, e1007037. [Google Scholar] [CrossRef]
- Shojaie, A.; Michailidis, G. Discovering graphical Granger causality using the truncating lasso penalty. Bioinformatics 2010, 26, i517–i523. [Google Scholar] [CrossRef] [PubMed]
- Kodikara, S.; Ellul, S.; Lê Cao, K.A. Statistical challenges in longitudinal microbiome data analysis. Brief. Bioinform. 2022, 23, bbac273. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Chen, G. Zero-inflated generalized Dirichlet multinomial regression model for microbiome compositional data analysis. Biostatistics 2019, 20, 698–713. [Google Scholar] [CrossRef]
- Shuler, K.; Verbanic, S.; Chen, I.A.; Lee, J. A Bayesian nonparametric analysis for zero-inflated multivariate count data with application to microbiome study. J. R. Stat. Soc. Ser. C 2021, 70, 961–979. [Google Scholar] [CrossRef]
- Xia, F.; Chen, J.; Fung, W.K.; Li, H. A logistic normal multinomial regression model for microbiome compositional data analysis. Biometrics 2013, 69, 1053–1063. [Google Scholar] [CrossRef]
- Chen, J.; Li, H. Variable selection for sparse Dirichlet-multinomial regression with an application to microbiome data analysis. Ann. Appl. Stat. 2013, 7, 418–442. [Google Scholar] [CrossRef]


{kind=link}
| Sample/Species | OTU 1 | OTU 2 | OTU 3 | ... | OTU m | Total Reads |
|---|---|---|---|---|---|---|
| Sample 1 | ... | |||||
| Sample 2 | ... | |||||
| ... | ... | ... | ... | ... | ... | ... |
| Sample n | ... |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldirawi, H.; Morales, F.G. Univariate and Multivariate Statistical Analysis of Microbiome Data: An Overview. Appl. Microbiol. 2023, 3, 322-338. https://doi.org/10.3390/applmicrobiol3020023
Aldirawi H, Morales FG. Univariate and Multivariate Statistical Analysis of Microbiome Data: An Overview. Applied Microbiology. 2023; 3(2):322-338. https://doi.org/10.3390/applmicrobiol3020023
Chicago/Turabian StyleAldirawi, Hani, and Franceskrista G. Morales. 2023. "Univariate and Multivariate Statistical Analysis of Microbiome Data: An Overview" Applied Microbiology 3, no. 2: 322-338. https://doi.org/10.3390/applmicrobiol3020023
APA StyleAldirawi, H., & Morales, F. G. (2023). Univariate and Multivariate Statistical Analysis of Microbiome Data: An Overview. Applied Microbiology, 3(2), 322-338. https://doi.org/10.3390/applmicrobiol3020023