
Cambisol Mycobiome in a Long-Term Field Experiment with Korean Pine as a Sole Edificator: A Case Study



Abstract
:1. Introduction
2. Materials and Methods
2.1. The Study Site
2.2. Soil Sampling and Properties
2.3. Chemical Analyses
2.4. Extraction of Total Nucleic Acid from Soil
2.5. ITS Fragment and 16S rRNA Gene Amplification and Sequencing
2.6. Bioinformatic and Statistical Analyses
3. Results
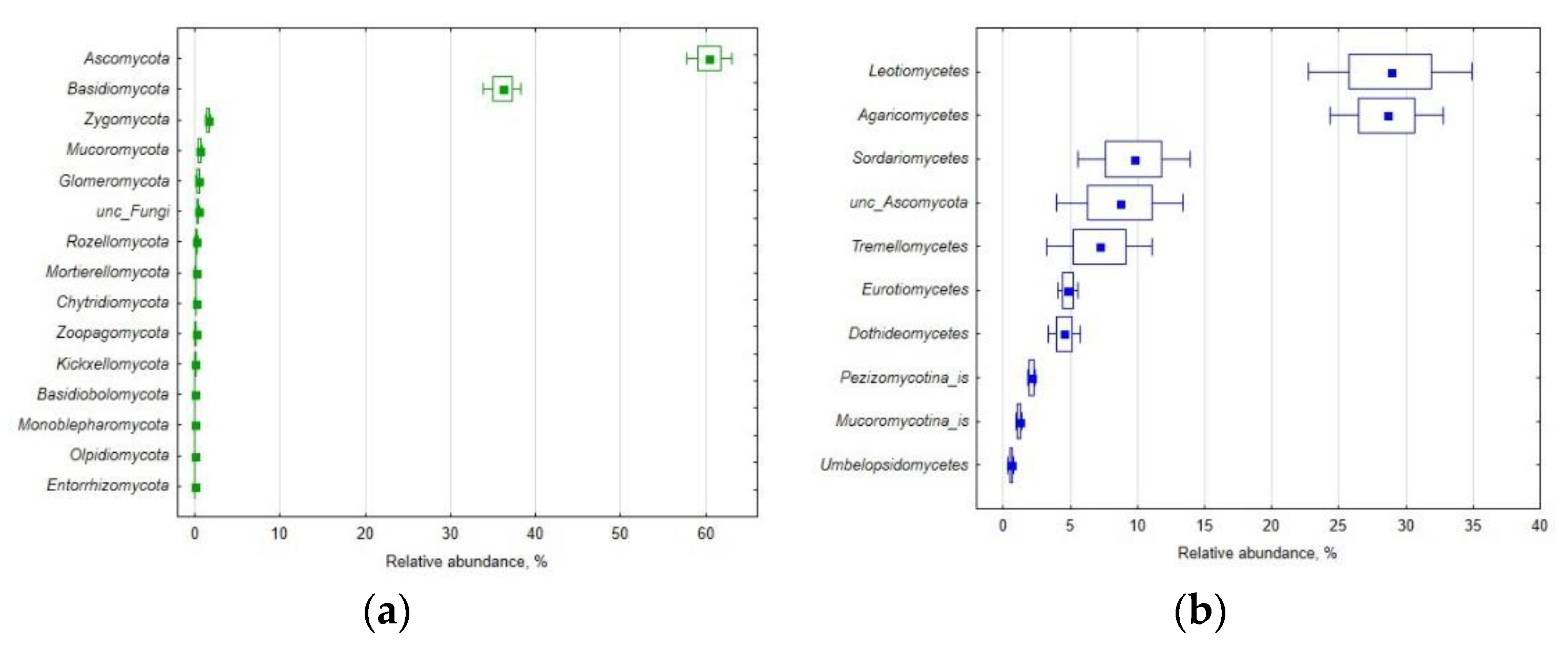
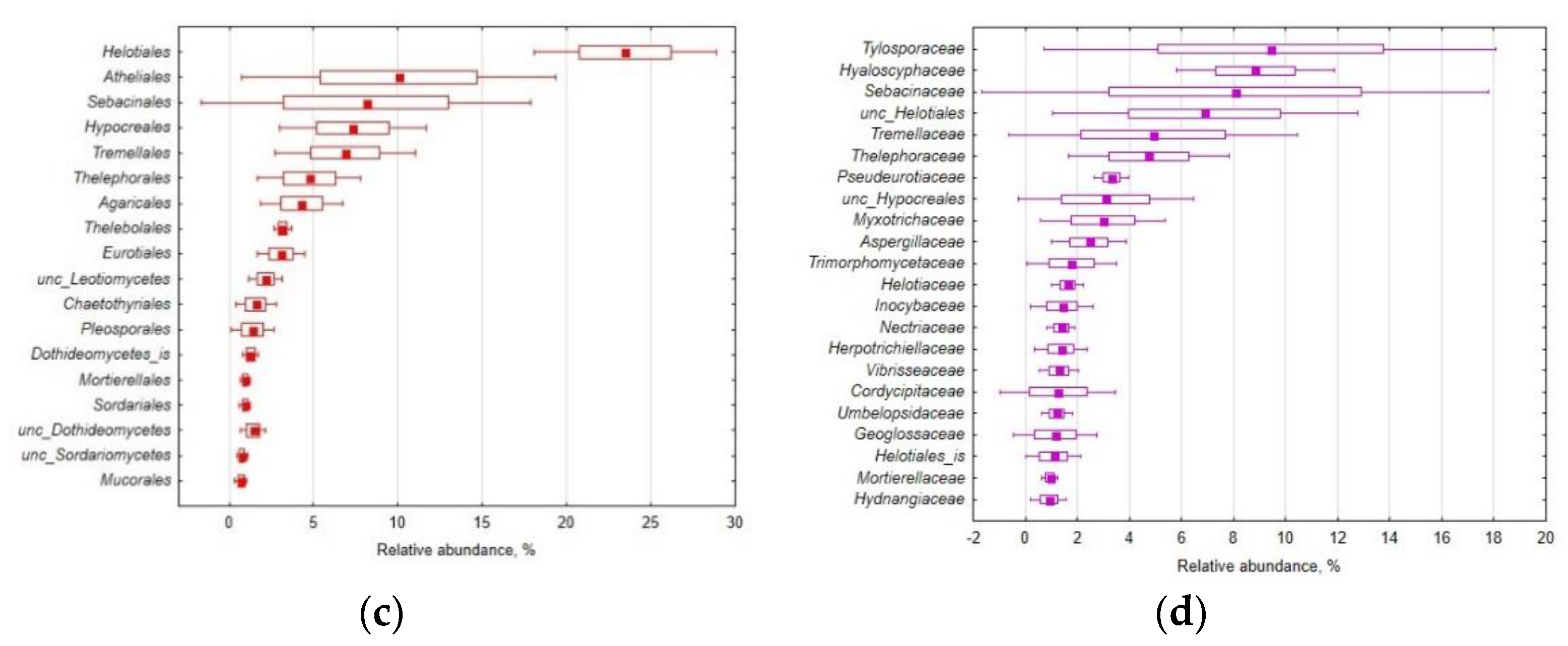
3.1. Mycobiome Diversity
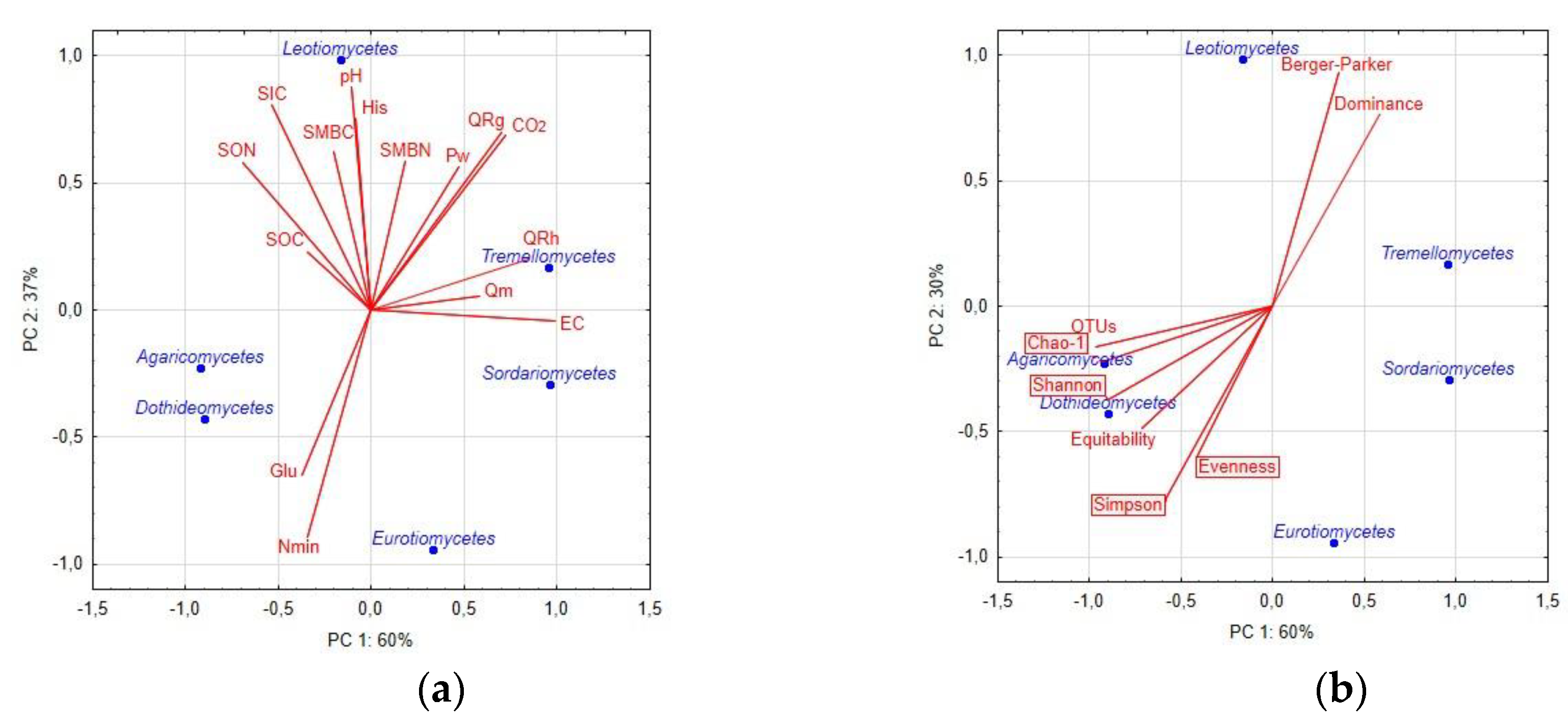
3.2. Mycobiome Diversity and Soil Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Li, F.; Pukkala, T.; Dong, L. Modelling the cone yields of Korean pine. For. Ecol. Manag. 2020, 464, 118086. [Google Scholar] [CrossRef]
- Xu, D.; Yan, H. A study of the impacts of climate change on the geographic distribution of Pinus koraiensis in China. Environ. Int. 2001, 27, 201–205. [Google Scholar] [CrossRef]
- Gao, H.; Liu, Q.; Song, Y.; Jiang, M.; Yin, Y. Modeling Primary Branch Diameter and Length for Planted Pinus koraiensis by Incorporating Neighbor Competition in Northeast China. Forests 2022, 13, 912. [Google Scholar] [CrossRef]
- Vyvodtsev, N.V.; Somov, E.V.; Tutrin, S.A.; Koboyashi, R. Bioecological and morphometric characteristics of the pine forests in Khabarovsk region. For. Tax. Manag. 2012, 40, 27–34. (In Russian) [Google Scholar]
- Liang, Y.; Bai, X.; Xu, X.; Xu, H.; Wang, J.; Pan, P. Direct in vitro organogenesis from sprouted seeds of a highly economical and ecological valued tree, Korean pine. Plant Cell Tissue Organ Cult. 2022, 148, 197–207. [Google Scholar] [CrossRef]
- Qi, D.; Feng, F.; Fu, Y.; Ji, X.; Liu, X. Effects of Soil Microbes on Forest Recovery to Climax Community through theRegulation of Nitrogen Cycling. Forests 2020, 11, 1027. [Google Scholar] [CrossRef]
- Lee, D.; Choi, J. Stocking Diagrams for Silvicultural Guideline in Korean Pines and Japanese Larch. Forests 2020, 11, 833. [Google Scholar] [CrossRef]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Shi, L.L.; Wen, Y.; Yang, Z.J.; Zang, H.D.; Gui, H.; Zou, X.M.; Mortimer, P. Dominant tree species identity effects on soil fungi are context dependent. Mycosphere 2018, 9, 790–802. [Google Scholar] [CrossRef]
- IUSS Working Group. WRB, World Reference Base for Soil Resources 2015. In International Soil Classification System for Naming Soils and Creating Legends for Soil Maps; FAO: Rome, Italy, 2015. [Google Scholar]
- Carter, M.R.; Gregorich, E.G. (Eds.) Soil Sampling and Methods of Analysis; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Vance, E.D.; Brookes, P.C.; Jenkinson, D.S. An extraction method for measuring soil microbial biomass-C. Soil Biol. Biochem. 1987, 19, 703–707. [Google Scholar] [CrossRef]
- Brookes, P.C.; Landman, A.; Pruden, G.; Jenkinson, D.S. Chloroform fumigation and the release of soil-nitrogena rapid direct extraction method to measure microbial biomass nitrogen in soil. Soil Biol. Biochem. 1985, 17, 837–842. [Google Scholar] [CrossRef]
- Pell, M.; Stenstrom, L.; Granhall, U. Soil respiration. In Microbiological Methods for Assessing Soil Quality; Bloem, J., Hopkins, D.W., Benedetti, A., Eds.; CABI International: Wallingford, UK, 2005; pp. 117–126. [Google Scholar] [CrossRef]
- Insam, H.; Haselwandter, K. Metabolic quotient of the soil microflora in relation to plant succession. Oecologia 1989, 79, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Igolkina, A.A.; Grekhov, G.A.; Pershina, E.V.; Samosorova, G.G.; Leunova, V.M.; Semenov, A.A.; Baturina, O.A.; Kabilov, M.R.; Andronov, E.E. Identifying components of mixed and contaminated soil samples by detecting specific signatures of control 16S rRNA libraries. Ecol. Ind. 2018, 94, 446–453. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. SINTAX, a Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences. 2016. Available online: http://biorxiv.org/content/early/2016/09/09/074161 (accessed on 12 July 2022).
- Abarenkov, K.; Zirk, A.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.H.; Kõljalg, U. UNITE USEARCH/UTAX Release for Fungi; Version 04.02.2020; UNITE Community: Melbourne, Australia, 2020. [Google Scholar] [CrossRef]
- Deshpande, V.; Wang, Q.; Greenfield, P.; Charleston, M.; Porras-Alfaro, A.; Kuske, C.R.; Cole, J.R.; Midgley, D.J.; Tran-Dinh, N. Fungal identification using a Bayesian classifier and the Warcup training set of internal transcribed spacer sequences. Mycologia 2016, 108, 1–5. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Bo-Ra, K.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.-H.; Lee, J.-H.; Kim, H.B.; Isaacson, R.E. Deciphering Diversity Indices for a Better Understanding of Microbial Communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [Green Version]
- Buee, M.; Reich, M.; Murat, C.; Morin, E.; Nilsson, R.H.; Uroz, S.; Martin, F. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytologist 2009, 1842, 449–456. [Google Scholar] [CrossRef]
- Kim, C.S.; Han, S.-K.; Nam, J.W.; Jo, J.W.; Kwag, Y.-N.; Han, J.-G.; Sung, G.-H.; Lim, Y.W.; Oh, S. Fungal communities in a Korean red pine stand, Gwangneung Forest, Korea. J. Asia-Pac. Biodivers. 2017, 10, 559–572. [Google Scholar] [CrossRef]
- Goldmann, K.; Schöning, I.; Buscot, F.; Wubet, T. Forest Management Type Influences Diversity and Community Composition of Soil Fungi across Temperate Forest Ecosystems. Front. Microbiol. 2015, 6, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Santalahti, M.; Pumpanen, J.; Koster, K.; Berninger, F.; Raffaello, T.; Jampponen, A.; Asiegbu, F.O.; Heinonsalo, J. Fungal Community Shifts in Structure and Function across a Boreal Forest Fire Chronosequence. Appl. Environ. Microbiol. 2015, 81, 7869–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siles, J.A.; Margesin, R. Abundance and Diversity of Bacterial, Archaeal, and Fungal Communities Along an Altitudinal Gradient in Alpine Forest Soils, What Are the Driving Factors? Microb. Ecol. 2016, 72, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Ping, Y.; Han, D.; Wang, N.; Hu, Y.; Mu, L.; Feng, F. Vertical zonation of soil fungal community structure in a Korean pine forest on Changbai Mountain, China. World J. Microbiol. Biotechnol. 2017, 33, 1. [Google Scholar] [CrossRef]
- Amma, S.; Toju, H.; Wachrinrat, C.; Sato, H.; Tanabe, A.S.; Artchawakom, T.; Kanzaki, M. Composition and Diversity of Soil Fungi in Dipterocarpaceae-Dominated Seasonal Tropical Forests in Thailand. Microbes Environ. 2018, 33, 135–143. [Google Scholar] [CrossRef]
- Smith, M.L.; Bruhn, J.N.; Anderson, J.B. The fungus Armillaria bulbosa is among the largest and oldest living organisms. Nature 1992, 356, 428–431. [Google Scholar] [CrossRef]
- Lavrinienko, A.; Jernfors, T.; Koskimaki, J.J.; Pirtilla, A.M.; Watts, P.C. Does Intraspecific Variation in rDNA Copy Number Affect Analysis of Microbial Communities? Trends Microbiol. 2020, 29, 19–27. [Google Scholar] [CrossRef]
- Lofgren, L.A.; Uehling, J.K.; Branco, S.; Bruns, T.D.; Martin, F.; Kennedy, P.G. Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol. Ecol. 2019, 28, 721–730. [Google Scholar] [CrossRef]
- Baldrian, P.; Větrovský, T.; Cajthaml, T.; Dobiášová, P.; Petránková, M.; Šnajdr, J.; Eichlerová, I. Estimation of fungal biomass in forest litter and soil. Fungal Ecol. 2013, 6, 1–11. [Google Scholar] [CrossRef]
- Liao, H.L.; Chen, Y.; Vilgalys, R. Metatranscriptomic Study of Common and Host-Specific Patterns of Gene Expression between Pines and Their Symbiotic Ectomycorrhizal Fungi in the Genus Suillus. PLoS Genet. 2016, 12, e1006348. [Google Scholar] [CrossRef] [PubMed]
- Misiak, M.; Goodall-Copestake, W.P.; Sparks, T.H.; Worland, M.R.; Boddy, L.; Magan, N.; Convey, P.; Hopkins, D.W.; Newsham, K.K. Inhibitory effects of climate change on the growth and extracellular enzyme activities of a widespread Antarctic soil fungus. Glob. Chang. Biol. 2020, 27, 1111–1125. [Google Scholar] [CrossRef]
- Vohník, M.; Figura, T.; Réblová, M. Hyaloscypha gabretae and Hyaloscypha gryndleri spp. nov. (Hyaloscyphaceae, Helotiales), two new mycobionts colonizing conifer, ericaceous and orchid roots. Mycorrhiza 2022, 32, 105–122. [Google Scholar] [CrossRef]
- Six, J.; Bossuyt, H.; Degryze, S.; Denef, K. A history of research on the link between (micro) aggregates, soil biota, and soil organic matter dynamics. Soil Tillage Res. 2004, 79, 7–31. [Google Scholar] [CrossRef]
- Ranjard, L.; Lejon, D.P.; Mougel, C.; Schehrer, L.; Merdinoglu, D.; Chaussod, R. Sampling strategy in molecular microbial ecology, influence of soil sample size on DNA fingerprinting analysis of fungal and bacterial communities. Environ. Microbiol. 2003, 5, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.M.; Williams, R.J.; Hargreaves, S.K.; Yang, F.; Hofmockel, K.S. Greatest soil microbial diversity found in micro-habitats. Soil Biol. Biochem. 2018, 118, 217–226. [Google Scholar] [CrossRef]
- Meiser, A.; Bálint, M.; Schmitt, I. Meta-analysis of deep-sequenced fungal communities indicates limited taxon sharing between studies and the presence of biogeographic patterns. New Phytol. 2014, 201, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, M.; Hagedorn, F.; Wipf, S.; Donhauser, J.; Vittoz, P.; Rixen, C.; Frossard, A.; Theurillat, J.P.; Frey, B. The Soil Microbiome of GLORIA Mountain Summits in the Swiss Alps. Front. Microbiol. 2019, 10, 1080. [Google Scholar] [CrossRef]
- Wang, N.N.; Yang, X.; Li, S.L.; Sui, X.; Han, S.J.; Feng, F.J. Effects of precipitation variation on the distribution pattern of soil fungal diversity in broad-leaved Korean pine mixed forest. Ying Yong Sheng Tai Xue Bao 2013, 24, 1985–1990. (In Chinese) [Google Scholar]
- He, L.; Rodrigues, J.L.M.; Soudzilovskaia, N.A.; Barceló, M.; Olsson, P.A.; Song, C.; Tedersoo, L.; Yuan, F.; Yuan, F.; Lipson, D.A.; et al. Global biogeography of fungal and bacterial biomass carbon in topsoil. Soil Biol. Biochem. 2020, 151, 108024. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Property | Mean | S.E.M. | C.V. |
|---|---|---|---|
| pH | 6.08 | 0.05 | 1 |
| EC, dS∙m−1 | 0.06 | 0.0 | 20 |
| SOC,% | 8.9 | 2.7 | 30 |
| SIC,% | 0.33 | 0.09 | 26 |
| SMBC, mg/g soil | 2.0 | 0.8 | 39 |
| SMBC/SOC,% | 2.1 | 0.3 | 15 |
| SMBN, mg/g soil | 0.2 | 0.1 | 57 |
| DOC, mg/kg soil | 30 | 30 | 79 |
| Pw, mg/kg soil | 0.5 | 0.2 | 47 |
| NO3, mg N/kg soil | 6.9 | 2.7 | 40 |
| NH4, mg N/kg soil | 2.0 | 1.2 | 59 |
| SON, % | 0.45 | 0.1 | 23 |
| SO(C/N) | 19.6 | 4.5 | 23 |
| CO2, µgC-CO2/g soil·h | 5.0 | 1.6 | 31 |
| Glu, µgC-CO2/g soil·h | 14.6 | 0.6 | 4 |
| His, µgC-CO2/g soil·h | 6.1 | 1.3 | 22 |
| QRg | 0.34 | 0.12 | 33 |
| QRh | 0.83 | 0.29 | 35 |
| Qm, µgC-CO2/mg SMBC | 2.8 | 1.5 | 5.2 |
| Taxon | Mean | S.E.M. | C.V. |
|---|---|---|---|
| Amphinema | 10.0 | 9.4 | 94 |
| Sebacina | 8.1 | 9.8 | 120 |
| un.1 Hyaloscyphaceae | 7.9 | 2.9 | 36 |
| un. Helotiales | 6.9 | 5.8 | 84 |
| Cryptococcus | 4.9 | 5.5 | 113 |
| Tomentella | 4.7 | 3.1 | 65 |
| un. Hypocreales | 3.1 | 3.4 | 109 |
| Pseudogymnoascus | 3.1 | 0.5 | 15 |
| Oidiodendron | 2.7 | 2.5 | 92 |
| Penicillium | 2.3 | 1.3 | 57 |
| Chalara | 2.3 | 0.5 | 20 |
| un. Leotiomycetes | 2.2 | 1.0 | 45 |
| Saitozyma | 1.8 | 1.7 | 96 |
| Inocybe | 1.4 | 1.2 | 84 |
| un. Dothideomycetes | 1.4 | 0.7 | 51 |
| Lecanicillium | 1.3 | 2.2 | 176 |
| Umbelopsis | 1.2 | 0.6 | 48 |
| Phialocephala | 1.2 | 0.8 | 65 |
| Trichoglossum | 1.1 | 1.5 | 142 |
| Tetracladium | 1.0 | 1.0 | 104 |
| Mortierella | 1.0 | 0.3 | 32 |
| OTU’s No. | Taxon | Mean | S.E.M. | C.V. |
|---|---|---|---|---|
| 4 | un.1 Amphinema | 8.2 | 9.5 | 116 |
| 3 | un. Sebacina | 7.6 | 9.4 | 123 |
| 5 2 | un. Hyaloscyphaceae | 7.0 | 2.1 | 30 |
| 7 | un. Cryptococcus | 4.9 | 5.5 | 113 |
| 6 | un. Helotiales | 3.6 | 5.9 | 162 |
| 8 | un. Pseudogymnoascus | 3.1 | 0.5 | 15 |
| 17 | un. Hypocreales | 3.0 | 3.4 | 111 |
| 10 | un. Ascomycota | 2.9 | 4.1 | 141 |
| 14 | un. Ascomycota | 2.1 | 3.5 | 163 |
| 32 | un. Tomentella | 1.8 | 3.0 | 166 |
| 16 | Saitozyma podzolica | 1.8 | 1.7 | 96 |
| 26 | un. Penicillium | 1.6 | 1.7 | 110 |
| 47 | un. Tomentella | 1.4 | 1.1 | 78 |
| 24 | Lecanicillium primulinum | 1.2 | 2.2 | 179 |
| 38 | un. Amphinema | 1.2 | 1.8 | 152 |
| 19 | un. Chalara | 1.1 | 1.3 | 116 |
| 27 | Phialocephala fortinii | 1.1 | 0.8 | 71 |
| 617 | un. Oidiodendron | 1.1 | 1.1 | 106 |
| 23 | un. Leotiomycetes | 1.0 | 0.3 | 31 |
| Index | Mean | S.E.M. | C.V. |
|---|---|---|---|
| Richness | 419 | 109 | 26 |
| Chao-1 | 437 | 99 | 23 |
| Simpson (1-D) | 0.95 | 0.01 | 2 |
| Shannon | 4.0 | 0.4 | 9 |
| Evenness | 0.14 | 0.03 | 18 |
| Equitability | 0.67 | 0.04 | 6 |
| Berger-Parker | 0.16 | 0.03 | 22 |
| Dominance (D) | 0.05 | 0.01 | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naumova, N.; Kuznetsova, G.; Alikina, T.; Kabilov, M. Cambisol Mycobiome in a Long-Term Field Experiment with Korean Pine as a Sole Edificator: A Case Study. Appl. Microbiol. 2022, 2, 470-480. https://doi.org/10.3390/applmicrobiol2030036
Naumova N, Kuznetsova G, Alikina T, Kabilov M. Cambisol Mycobiome in a Long-Term Field Experiment with Korean Pine as a Sole Edificator: A Case Study. Applied Microbiology. 2022; 2(3):470-480. https://doi.org/10.3390/applmicrobiol2030036
Chicago/Turabian StyleNaumova, Natalia, Galina Kuznetsova, Tatiana Alikina, and Marsel Kabilov. 2022. "Cambisol Mycobiome in a Long-Term Field Experiment with Korean Pine as a Sole Edificator: A Case Study" Applied Microbiology 2, no. 3: 470-480. https://doi.org/10.3390/applmicrobiol2030036
APA StyleNaumova, N., Kuznetsova, G., Alikina, T., & Kabilov, M. (2022). Cambisol Mycobiome in a Long-Term Field Experiment with Korean Pine as a Sole Edificator: A Case Study. Applied Microbiology, 2(3), 470-480. https://doi.org/10.3390/applmicrobiol2030036