
Use of Integrated Core Proteomics, Immuno-Informatics, and In Silico Approaches to Design a Multiepitope Vaccine against Zoonotic Pathogen Edwardsiella tarda

 ,
, 
Abstract
:1. Introduction
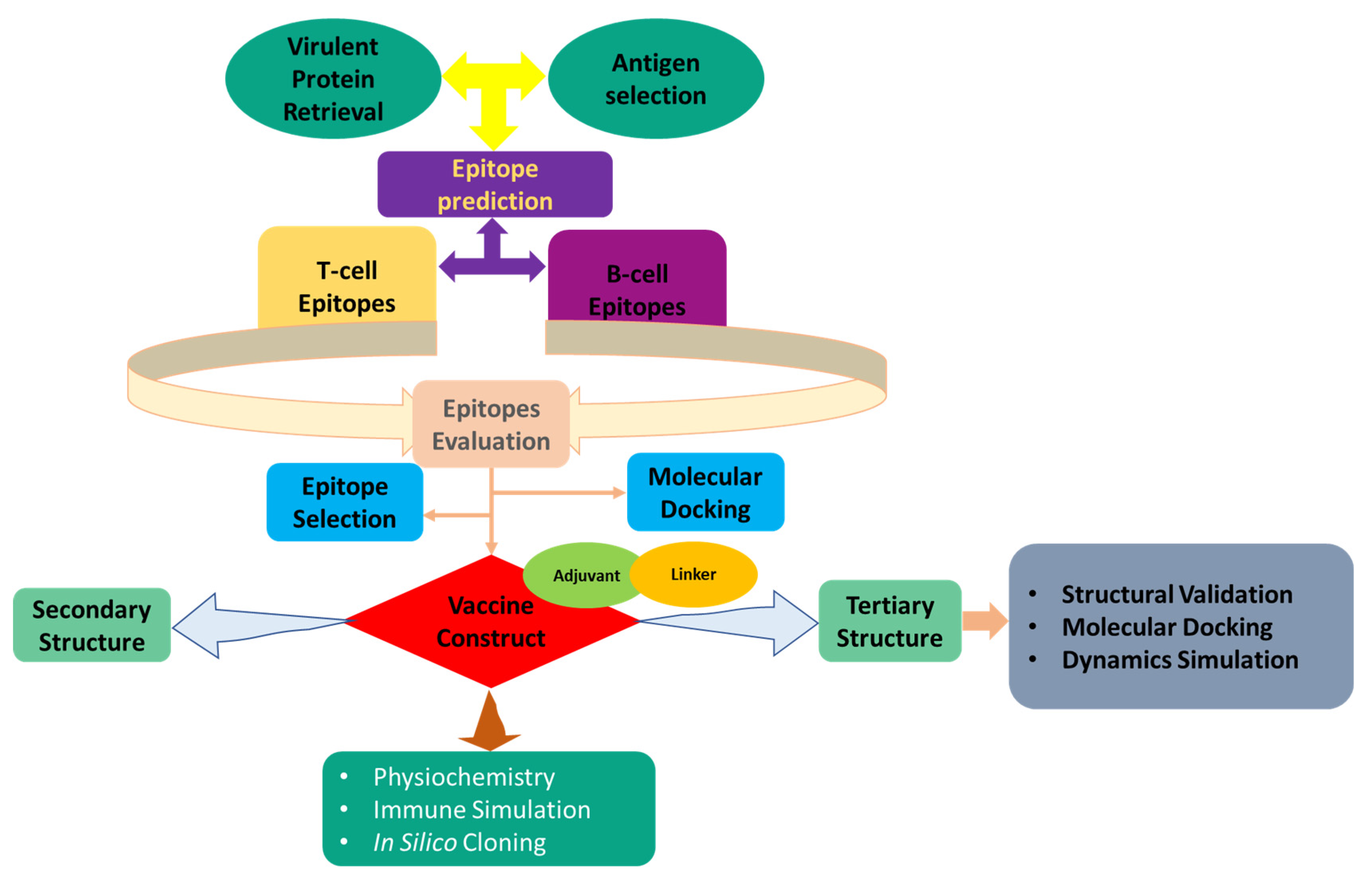
2. Methods and Materials
2.1. E. tarda Core Proteome Identification
2.2. Subtractive Proteomics and Approach
2.3. Epitopes Prediction
2.3.1. T-Cell Epitope Prediction
2.3.2. Epitopes of Linear B-Lymphocytes: Prediction and Evaluation
2.4. Modeling of Peptides and Molecular Docking
2.5. Development of a Multi-Epitope Vaccine
2.6. Evaluation of Design Vaccine Characteristics
2.7. 2D Structural Features Prediction
2.8. Validation and Homology Modeling of Vaccine 3D Structure
2.9. Disulfide Engineering of the Designed Vaccine
2.10. Vaccine-TLR5 Docking
2.11. MD Simulation
2.12. Simulation of Immune Response
2.13. In Silico Cloning and Codon Adaptation
3. Results
3.1. Analysis of Core Proteome
3.2. Identification of Proteins of Interest
3.3. Prediction of Epitopes
3.4. Epitope and Allele Docking Studies
3.5. Construction of Final Vaccine
3.6. Immunological Assessment and Physicochemical Characteristics
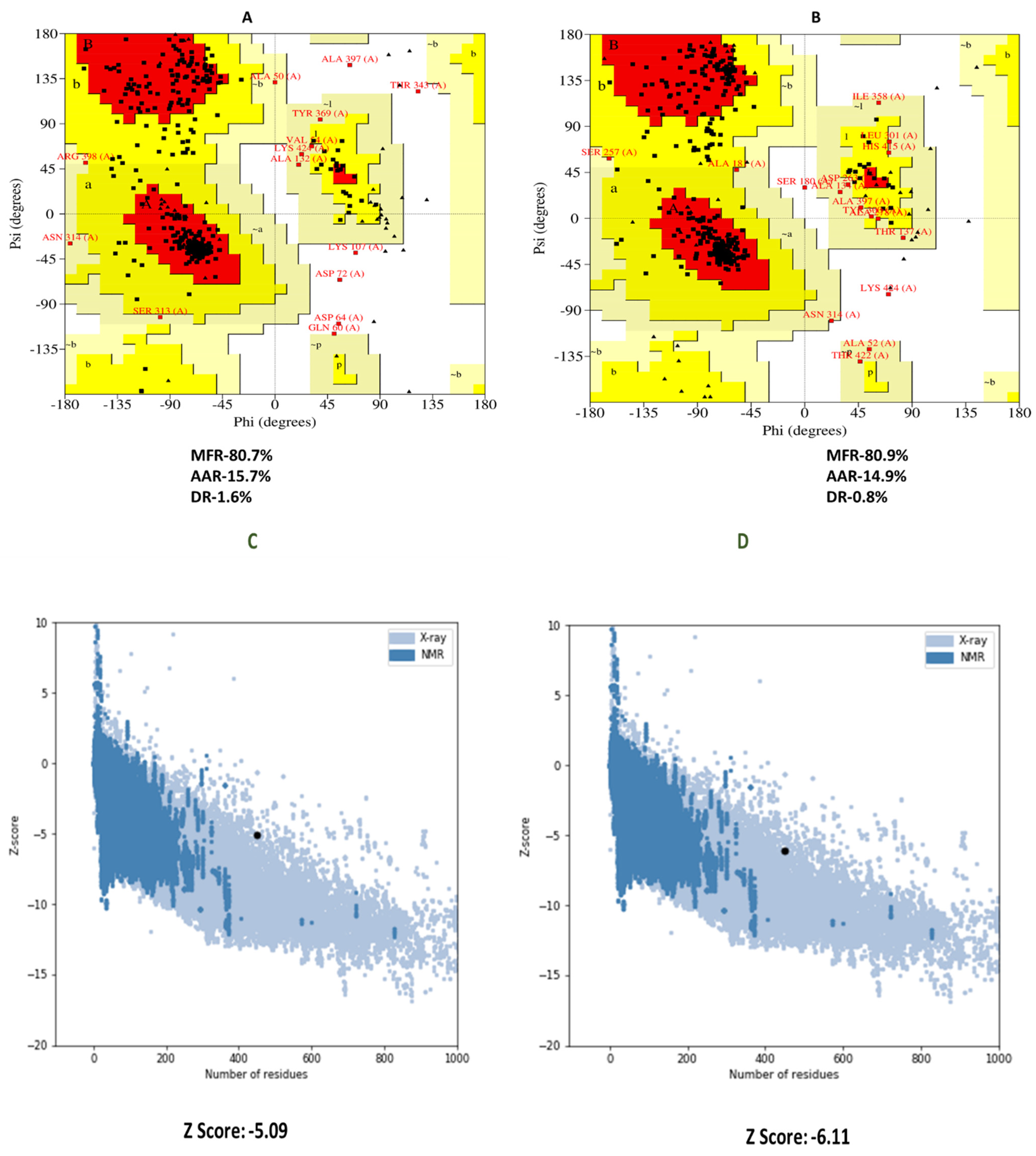
3.7. 3D Structure Refinement and Confirmation
3.8. Vaccine Disulfide Engineering
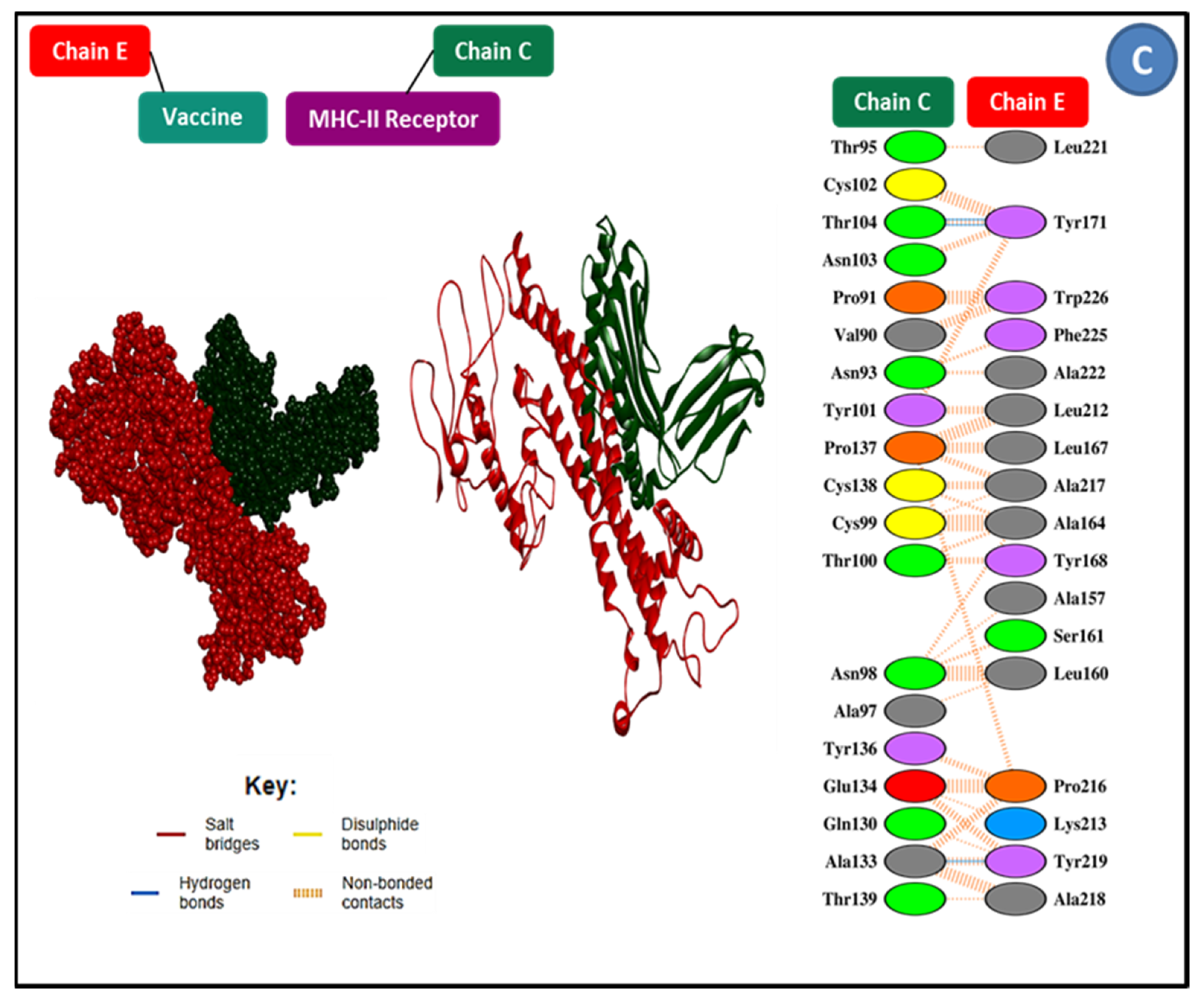
3.9. Molecular Docking Research
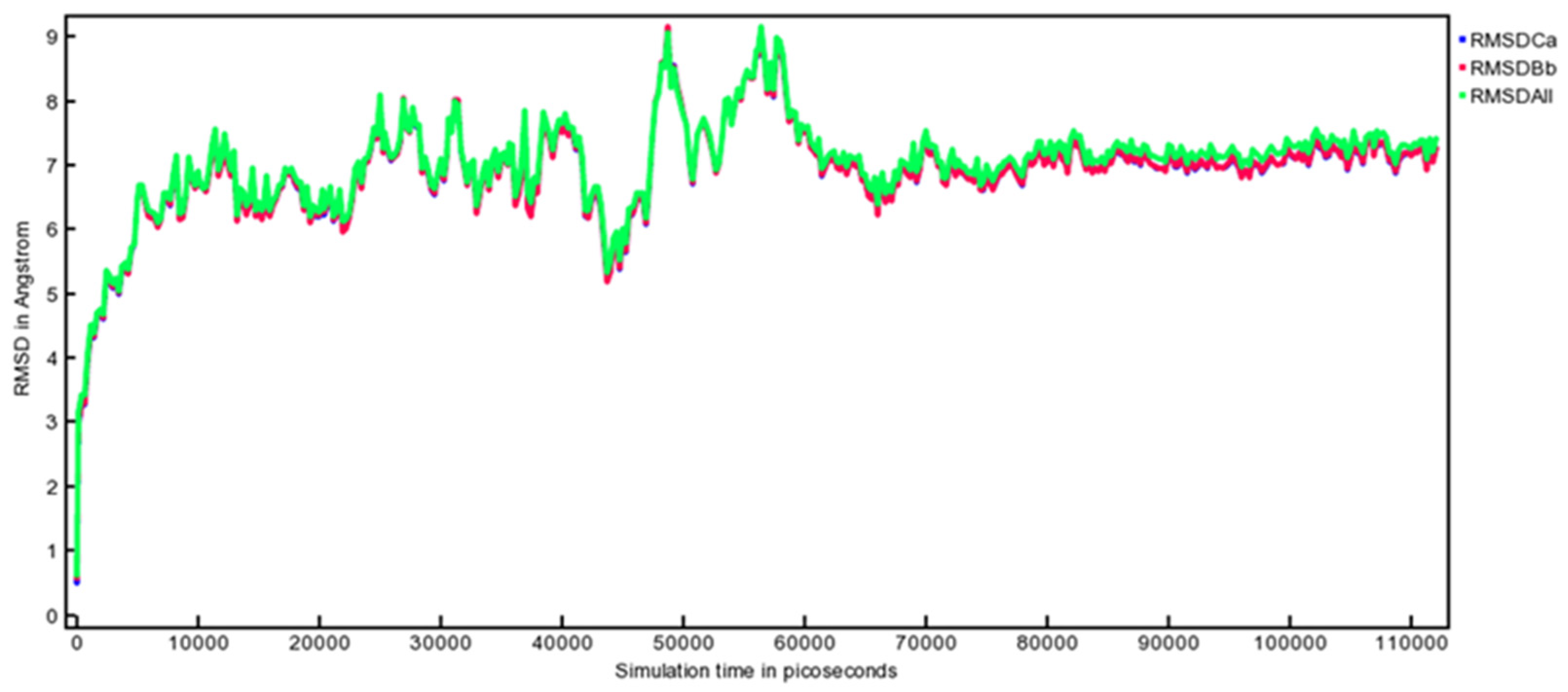
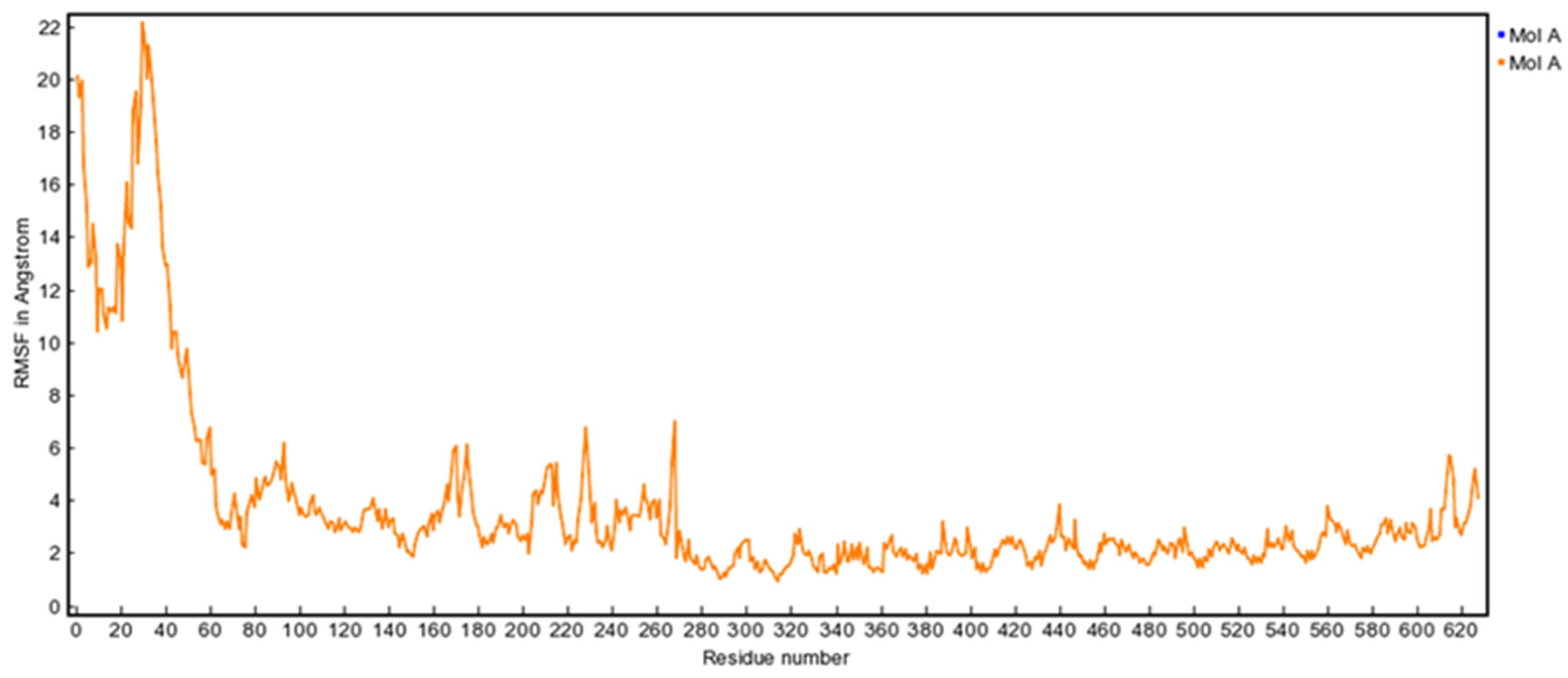
3.10. MD Simulation
3.11. Immune Response Simulation
3.12. In Silico Cloning and Codon Adaptation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassan, H.A.; Ding, X.; Zhang, X.; Zhu, G. Fish borne Edwardsiella tarda eha involved in the bacterial biofilm formation, hemolytic activity, adhesion capability, and pathogenicity. Arch. Microbiol. 2020, 202, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.R.; Sahoo, P.K. Edwardsiellosis in fish: A brief review. J. Biosci. 2007, 32, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.J.; Klesius, P.H.; Plumb, J.; Shoemaker, C.A. Edwardsiella Septicaemias; CABI: Wallingford, UK, 2011; pp. 512–569. [Google Scholar]
- Lima, L.C.; Fernandes, A.A.; Costa, A.A.P.; Velasco, F.O.; Leite, R.C.; Hackett, J.L. Isolation and characterizaton of Edwardsiella tarda from pacu Myleus micans. Arqic. Bras. Med. Vet. Zootec. 2008, 60, 275–277. [Google Scholar] [CrossRef]
- Van Damme, L.R.; Vandepitte, J. Frequent isolation of Edwardsiella tarda and Pleisiomonas shigelloides from healthy Zairese freshwater fish: A possible source of sporadic diarrhea in the tropics. Appl. Environ. Microbiol. 1980, 39, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Wang, H.-L.; Zhang, M.; Xiao, Z.-Z.; Sun, L. Genetic Mechanisms of Multi-Antimicrobial Resistance in a Pathogenic Edwardsiella tarda Strain. Aquaculture 2009, 289, 134–139. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.H.; Austin, B. Comparative analysis of the phenotypic characteristics of high-and low virulent strains of Edwardsiella tarda. J. Fish Dis. 2010, 33, 985–994. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, K.; Sun, L. Regulation of autoinducer 2 production and luxS expression in a pathogenic Edwardsiella tarda strain. Microbiology 2008, 154, 2060–2069. [Google Scholar] [CrossRef] [Green Version]
- Done, H.Y.; Venkatesan, A.K.; Halden, R.U. Does the Recent Growth of Aquaculture Create Antibiotic Resistance Threats Different from those Associated with Land Animal Production in Agriculture? AAPS J. 2015, 17, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.E.M.; Schreier, H.J.; Lanska, L.; Hale, M.S. The Rising Tide of Antimicrobial Resistance in Aquaculture: Sources, Sinks and Solutions. Mar. Drugs 2017, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.E.; Cho, M.Y.; Kim, J.-W.; Kang, H.Y. Large antibiotic-resistance plasmid of Edwardsiella tarda contributes to virulence in fish. Microb. Pathog. 2012, 52, 259–266. [Google Scholar] [CrossRef]
- Roberts, M.C. Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 2005, 245, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; Salamat, M.K.F.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, K.; Morise, M.; Vo, L.K.; Tran, N.T.; Sahashi, D.; Ueda-Wakamatsu, R.; Nishimura, W.; Komatsu, M.; Shiozaki, K. Host lactosylceramide enhances Edwardsiella tarda infection. Cell Microbiol. 2021, 23, e13365. [Google Scholar] [CrossRef] [PubMed]
- Leal, Y.; Velazquez, J.; Hernandez, L.; Swain, J.K.; Rodríguez, A.R.; Martínez, R.; García, C.; Ramos, Y.; Estrada, M.P.; Carpio, Y. Promiscuous T cell epitopes boosts specific IgM immune response against a P0 peptide antigen from sea lice in different teleost species. Fish Shellfish Immunol. 2019, 92, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, H.; Soliman, H.; Fajmann, S.; Sexl, V.; El-Matbouli, M.; Saleh, M. Kinetics of CD4-1+ lymphocytes in brown trout after exposure to viral haemorrhagic septicaemia virus. J. Fish Dis. 2021, 44, 1553–1562. [Google Scholar] [CrossRef]
- Nakanishi, T.; Fischer, U.; Dijkstra, J.; Hasegawa, S.; Somamoto, T.; Okamoto, N.; Ototake, M. Cytotoxic T cell function in fish. Dev. Comp. Immunol. 2002, 26, 131–139. [Google Scholar] [CrossRef]
- Adams, A. Progress, challenges, and opportunities in fish vaccine development. Fish Shellfish Immunol. 2019, 90, 210–214. [Google Scholar] [CrossRef]
- Grimholt, U. MHC and Evolution in Teleosts. Biology 2016, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, J.M.; Grimholt, U.; Leong, J.; Koop, B.F.; Hashimoto, K. Comprehensive analysis of MHC class II genes in teleost fish genomes reveals dispensability of the peptide-loading DM system in a large part of vertebrates. BMC Evol. Biol. 2013, 13, 260. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Dijkstra, J.M. Major Histocompatibility Complex (MHC) Genes and Disease Resistance in Fish. Cells 2019, 8, 378. [Google Scholar] [CrossRef] [Green Version]
- Stosik, M.; Tokarz-Deptuła, B.; Deptuła, W. Major Histocompatibility Complex in Osteichthyes. J. Vet. Res. 2020, 64, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Snowberg, L.; Stutz, W.; Caporaso, G.; Lauber, C.; Knight, R.; Lauber, C.; Caporaso, J.G. Major Histocompatibility Complex class IIb polymorphism influences gut microbiota composition and diversity. Mol. Ecol. 2014, 23, 4831–4845. [Google Scholar] [CrossRef] [PubMed]
- Marana, M.H.; Jørgensen, L.V.G.; Skov, J.; Chettri, J.K.; Holm Mattsson, A.; Dalsgaard, I.; Kania, W.P.; Buchmann, K. Subunit vaccine candidates against Aeromonas salmonicida in rainbow trout Oncorhynchus mykiss. PLoS ONE 2017, 12, e0171944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahendran, R.; Jeyabaskar, S.; Michael, R.D.; Paul, A.V.; Sitharaman, G. Computer-aided vaccine designing approach against fish pathogens Edwardsiella tarda and Flavobacterium columnare using bioinformatics softwares. Drug Des. Dev. Ther. 2016, 10, 1703–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, U.P.; Soares, S.; Blom, J.; Leal, C.; Ramos, R.; Guimarães, L.C.; Oliveira, L.; Almeida, S.; Hassan, S.; Santos, A.; et al. In silico prediction of conserved vaccine targets in Streptococcus agalactiae strains isolated from fish, cattle, and human samples. Genet. Mol. Res. 2013, 12, 2902–2912. [Google Scholar] [CrossRef] [PubMed]
- Pumchan, A.; Krobthong, S.; Roytrakul, S.; Sawatdichaikul, O.; Kondo, H.; Hirono, I.; Areechon, N.; Unajak, S. Novel Chimeric Multiepitope Vaccine for Streptococcosis Disease in Nile Tilapia (Oreochromis niloticus Linn.). Sci. Rep. 2020, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.I.; Mou, M.J.; Sanjida, S.; Tariq, M.; Nasir, S.; Mahfuj, S. Designing a novel mRNA vaccine against Vibrio harveyi infection in fish: An immunoinformatics approach. Genom. Inform. 2022, 20, e11. [Google Scholar] [CrossRef]
- Islam, S.I.; Mou, M.J.; Sanjida, S. Application of reverse vaccinology for designing of an mRNA vaccine against re-emerging marine birnavirus affecting fish species. Inform. Med. Unlocked 2022, 30, 100948. [Google Scholar] [CrossRef]
- Hansson, M.; Nygren, P.-A.K.; Ståhl, S. Design and production of recombinant subunit vaccines. Biotechnol. Appl. Biochem. 2000, 32, 95–107. [Google Scholar] [CrossRef]
- Madonia, A.; Melchiorri, C.; Bonamano, S.; Marcelli, M.; Bulfon, C.; Castiglione, F.; Galeotti, M.; Volpatti, D.; Mosca, F.; Tiscar, P.-G.; et al. Computational modeling of the immune system of the fish for a more effective vaccination in aquaculture. Bioinformatics 2017, 33, 3065–3071. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.; Pathak, D.C.; Mannan, M.A.-U.; Kaushik, V. In-silico designing of an epitope-based vaccine against the seven banded grouper nervous necrosis virus affecting fish species. Netw. Model. Anal. Health Inform. Bioinform. 2021, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, N.; Ajmal, A.; Ali, F.; Rastrelli, L. Core proteome mediated therapeutic target mining and multi-epitope vaccine design for Helicobacter pylori. Genomics 2020, 112, 3473–3483. [Google Scholar] [CrossRef] [PubMed]
- Sanober, G.; Ahmad, S.; Azam, S. Identification of plausible drug targets by investigating the druggable genome of MDR Staphylococcus epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Rédei, G. NCBI (National Center for Biotechnology Information) Encyclopedia of Genetics, Genomics, Proteomics and Informatics. 2008. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 1 June 2022).
- Shenoy, P.; Vin, H. Cello: A Disk Scheduling Framework for Next-Generation Operating Systems *. Real-Time Syst. 2002, 22, 9–48. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Meunier, M.; Guyard-Nicodème, M.; Hirchaud, E.; Parra, A.; Chemaly, M.; Dory, D. Identification of Novel Vaccine Candidates against Campylobacter through Reverse Vaccinology. J. Immunol. Res. 2016, 2016, 5715790. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI-Multi FASTA ProtParam Interface. Bioinformation 2016, 12, 74–77. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8 + cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2018, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Raghava, G.P. Prediction of CTL epitopes using QM, SVM, and ANN techniques. Vaccine 2004, 22, 3195–3204. [Google Scholar] [CrossRef]
- He, Y.; Xiang, Z.; Mobley, H.L. Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed. Biotechnol. 2010, 2010, 297505. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P. In silico approach for predicting the toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14 (Suppl. S6), S4. [Google Scholar] [CrossRef] [Green Version]
- Nain, Z.; Abdullah, F.; Rahman, M.M.; Karim, M.M.; Khan, S.A.; Bin Sayed, S.; Mahmud, S.; Rahman, S.M.R.; Sheam, M.; Haque, Z.; et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. [Google Scholar] [CrossRef]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [Green Version]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data-intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design, and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unajak, S.; Pumchan, A.; Roytrakul, S.; Sawatdichaikul, O.; Areechon, N. Novel Vaccine Development for Fish Culture Based on the Multiepitope Concept. Methods Mol. Biol. 2022, 2411, 219–240. [Google Scholar] [PubMed]
- Dorosti, H.; Eslami, M.; Negahdaripour, M.; Ghoshoon, M.B.; Gholami, A.; Heidari, R.; Dehshahri, A.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 2019, 37, 3524–3535. [Google Scholar] [CrossRef]
- Abdellrazeq, G.S.; Fry, L.M.; Elnaggar, M.M.; Bannantine, J.P.; Schneider, D.A.; Chamberlin, W.M.; Mahmoud, A.H.; Park, K.-T.; Hulubei, V.; Davis, W.C. Simultaneous cognate epitope recognition by bovine CD4 and CD8 T cells is essential for the primary expansion of antigen-specific cytotoxic T-cells following ex vivo stimulation with a candidate Mycobacterium avium subsp. paratuberculosis peptide vaccine. Vaccine 2020, 38, 2016–2025. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. In 2-D Proteome Analysis Protocols. Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 1999; Volume 112, pp. 531–552. [Google Scholar]
- Buchan, D.W.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Xu, J.; McPartlon, M.; Li, J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 2021, 3, 601–609. [Google Scholar] [CrossRef]
- Nugent, T.; Cozzetto, D.; Jones, D.T. Evaluation of predictions in the CASP10 model refinement category. Proteins 2014, 82 (Suppl. S2), 98–111. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Cryst. 2002, 40, 82–92. [Google Scholar]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, S.; Bouback, T.A.; Samad, A.; Nur, S.M.; Alam, R.; Abdullah-Al-Mamun, M.; Nain, Z.; Imon, R.R.; Talukder, M.E.K.; Tareq, M.M.I.; et al. Spike protein recognizer receptor ACE2 targeted identification of potential natural antiviral drug candidates against SARS-CoV-2. Int. J. Biol. Macromol. 2021, 191, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Desai, V.H.; Kumar, S.P.; Pandya, H.A.; Solanki, H. Receptor-Guided De Novo Design of Dengue Envelope Protein Inhibitors. Appl. Biochem. Biotechnol. 2015, 177, 861–878. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Goldberg, M.F.; Roeske, E.K.; Ward, L.N.; Pengo, T.; Dileepan, T.; Kotov, D.I.; Jenkins, M.K. Salmonella Persist in Activated Macrophages in T Cell-Sparse Granulomas but Are Contained by Surrounding CXCR3 Ligand-Positioned Th1 Cells. Immunity 2018, 49, 1090–1102.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhammad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bol, K.F.; Aarntzen, E.H.J.G.; Pots, J.M.; Nordkamp, M.A.M.O.; Van De Rakt, M.W.M.M.; Scharenborg, N.M.; De Boer, A.J.; Van Oorschot, T.G.M.; Croockewit, S.A.J.; Blokx, W.A.M.; et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol. Immunoth. 2016, 65, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef]
- Bonam, S.R.; Partidos, C.D.; Halmuthur, S.K.M.; Muller, S. An Overview of Novel Adjuvants Designed for Improving Vaccine Efficacy. Trends Pharmacol. Sci. 2017, 38, 771–793. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Protein | Accession No. | Sub-Cellular Localization | Transmembrane Helices | Antigenicity | Molecular Weight (kDa) |
|---|---|---|---|---|---|
| MCP four-helix bundle domain-containing protein | WP_005281935.1 | Inner Membrane | 0 | 0.5040 | 55.87 |
| Extracellular solute-binding protein | WP_035605308.1 | Periplasmic | 0 | 0.5105 | 37.86 |
| Outer membrane protein A | ACY84110.1 | Outer Membrane | 0 | 0.6404 | 37.94 |
| Hypothetical protein | WP_157757873.1 | Extracellular | 0 | 0.5833 | 35.22 |
| Putative transporter protein | WP_005280555.1 | Inner membrane | 0 | 0.6064 | 58.95 |
| Maltoporin protein | WP_005280774.1 | Outer membrane | 0 | 0.7530 | 47.10 |
| Name of Server | Protein Name | Epitopes | Immunogenicity | Allergenicity | Antigenicity | Toxicity |
|---|---|---|---|---|---|---|
| CTLPred | MCP Four helix bundle domain-containing protein | QTNILALNA | Positive | Negative | 0.6679 | Negative |
| MCP Four helix bundle domain-containing protein | LMLILAGLA | Positive | Negative | 0.4431 | Negative | |
| Extracellular solute-binding protein | LSMRARVLY | Positive | Negative | 0.6913 | Negative | |
| Extracellular solute-binding protein | AASLLFGLS | Positive | Negative | 0.4425 | Negative | |
| Outer membrane protein A | YTDRIGSDQ | Positive | Negative | 1.0949 | Negative | |
| Outer membrane protein A | PLAAIGVEY | Positive | Negative | 0.7107 | Negative | |
| Hypothetical protein | MSLVLKIIP | Positive | Negative | 1.2527 | Negative | |
| Putative transporter protein | LLALLFWSV | Positive | Negative | 2.0790 | Negative | |
| Putative transporter protein | PQLLALLFW | Positive | Negative | 2.1342 | Negative | |
| Maltoporin protein | MIDFYYWDI | Positive | Negative | 2.5781 | Negative | |
| Maltoporin protein | GSLELGFDY | Positive | Negative | 1.2685 | Negative | |
| Vaxitop Server | Extracellular solute-binding protein | INTWLRLGAASLLFG | Positive | Negative | 1.0671 | Negative |
| Outer membrane protein A | GAFFGYQANPYLGFE | Positive | Negative | 2.2112 | Negative | |
| Maltoporin protein | MTASNSGHSGGSSVN | Positive | Negative | 2.0832 | Negative |
| Protein Name | Sequence | Position | Score | Antigenicity | Allergenicity |
|---|---|---|---|---|---|
| MCP four-helix bundle domain-containing protein | FLMLILAGLAAA | 200 | 0.52 | 0.8265 | Negative |
| MCP four-helix bundle domain-containing protein | GLTSGSGELAAR | 291 | 0.55 | 1.5861 | Negative |
| Outer membrane protein A | PAPIPAPAPAPV | 203 | 0.74 | 0.9919 | Negative |
| Outer membrane protein A | YQYVNKVGTRSE | 166 | 0.57 | 1.0305 | Negative |
| Hypothetical protein | SVSAVDIEKRQA | 221 | 0.53 | 0.9263 | Negative |
| Putative transporter | ARLVIGEQVDTS | 270 | 0.75 | 0.7535 | Negative |
| Putative transporter | SRNGHHHELLQT | 195 | 0.71 | 1.0494 | Negative |
| Maltoporin | TNPGGSLELGFD | 206 | 0.74 | 1.3095 | Negative |
| Maltoporin | GAGSKYRLGNEC | 51 | 0.73 | 1.9150 | Negative |
| T-Cell Epitope | HLA Allele | Epitope Affinity (kcal/mol) | Control Affinity (kcal/mol) | Number of Hydrogens Bonds (CHB) | Residues Involved in CHB Networks (n) |
|---|---|---|---|---|---|
| INTWLRLGAASLLFG | DAB1*07:01 | −7.2 | −6.9 | 9 (7) | Met69, Ala149, Thr7, Ile8, Gln19, Ile1, Ala2, Trp7, Tyr74 (9) |
| GAFFGYQANPYLGFE | DAB1*15:01 | −7.2 | −7.1 | 8 (7) | Tyr80, Lys84, Val146, Thr7, Lys9, Val66, Thr77, Asn143 (8) |
| MTASNSGHSGGSSVN | DAA2*01:01 | −7.1 | −7.0 | 16 (14) | Tyr7, Asp2, Asp9, Glu63, Lys66, Arg69, Asn77, Asn77, Lys80, Tyr84, Tyr99, Thr143, Lys146, Trp147, Glu15, Glu152 (16) |
| INTWLRLGAASLLFG | HLA-A*0201 | −6.7 | −7.3 | 9 (7) | Arg71, Ala12, Asn82, Val1, Glu6, Ser4, Thr77, Thr13, Val14 (9) |
| GAFFGYQANPYLGFE | HLA-B*3501 | −7.1 | −7.9 | 12 (10) | Tyr7, Asp9, Asp9, Ser24, Glu63, Lys66, Arg69, Arg69, Tyr99, Glu152, Glu152, Gln155 (12) |
| MTASNSGHSGGSSVN | HLA-B*3508 | −6.9 | −7.3 | 9 (8) | Ser63, Glu85, Asn72, Trp326, His7, Glu45, Phe17, Phe8, Ile17 (9) |
| Characteristics | Finding | Remark |
|---|---|---|
| Number of amino acids | 450 | Suitable |
| Molecular weight | 47,284.67 | Average |
| Theoretical pI | 8.82 | Base |
| Chemical formula | C2157H3410N552O620S9 | - |
| Instability index of vaccine | 28.48 | Stable |
| Aliphatic index of vaccine | 96.29 | Thermostable |
| Grand average of hydropathicity (GRAVY) | 0.122 | Hydrophobic |
| Antigenicity | 0.7818 | Antigenic |
| Immunogenicity | 1.47082 | Immunogenic |
| Allergenicity | No | Non-allergen |
| Solubility | 0.891343 | Soluble |
| Features | MEBV-MHCI | MEBV-MHCII | MHC-TLR5 |
|---|---|---|---|
| HADDOCK Score | 211.3 ± 12.2 | 169.4 ± 22.3 | 207.6 ± 14.6 |
| Cluster Size | 6 | 5 | 7 |
| Van der Waals energy | −41.8 ± 3.6 | −69.1 ± 2.2 | −41.1 ± 2.3 |
| Desolvation energy | −1.4 ± 0.7 | −12.7 ± 4.8 | −0.5 ± 3.3 |
| Electrostatic energy | −60.8 ± 9.0 | −261.1 ± 24.8 | −67.1 ± 27.8 |
| RMSD from the overall lowest-energy structure | 37.6 ± 0.3 | 9.3 ± 0.5 | 49.4 ± 0.1 |
| Buried surface area | 2219.9 ± 110.2 | 2977.6 ± 63.1 | 2101.9 ± 120.9 |
| Z-Score | −1.1 | −0.9 | −1.9 |
| Restraint violation energy | 2746.2 ± 114.9 | 3124.6 ± 172.4 | 2529.9 ± 182.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, S.I.; Mahfuj, S.; Islam, M.J.; Mou, M.J.; Sanjida, S. Use of Integrated Core Proteomics, Immuno-Informatics, and In Silico Approaches to Design a Multiepitope Vaccine against Zoonotic Pathogen Edwardsiella tarda. Appl. Microbiol. 2022, 2, 414-437. https://doi.org/10.3390/applmicrobiol2020031
Islam SI, Mahfuj S, Islam MJ, Mou MJ, Sanjida S. Use of Integrated Core Proteomics, Immuno-Informatics, and In Silico Approaches to Design a Multiepitope Vaccine against Zoonotic Pathogen Edwardsiella tarda. Applied Microbiology. 2022; 2(2):414-437. https://doi.org/10.3390/applmicrobiol2020031
Chicago/Turabian StyleIslam, Sk Injamamul, Sarower Mahfuj, Md Jakiul Islam, Moslema Jahan Mou, and Saloa Sanjida. 2022. "Use of Integrated Core Proteomics, Immuno-Informatics, and In Silico Approaches to Design a Multiepitope Vaccine against Zoonotic Pathogen Edwardsiella tarda" Applied Microbiology 2, no. 2: 414-437. https://doi.org/10.3390/applmicrobiol2020031
APA StyleIslam, S. I., Mahfuj, S., Islam, M. J., Mou, M. J., & Sanjida, S. (2022). Use of Integrated Core Proteomics, Immuno-Informatics, and In Silico Approaches to Design a Multiepitope Vaccine against Zoonotic Pathogen Edwardsiella tarda. Applied Microbiology, 2(2), 414-437. https://doi.org/10.3390/applmicrobiol2020031