
Competitiveness of Quantitative Polymerase Chain Reaction (qPCR) and Droplet Digital Polymerase Chain Reaction (ddPCR) Technologies, with a Particular Focus on Detection of Antibiotic Resistance Genes (ARGs)


Abstract
:1. Introduction
2. Evolution/Expansion of Nucleic Acid Detection Methods for Molecular Targets
2.1. The Initial State of Polymerase Chain Reaction (PCR)
2.2. The Second-Generation PCR—qPCR
2.3. The Next Generation—ddPCR
2.3.1. The ddPCR Specificity
2.3.2. Multiplex ddPCR
3. Comparison of qPCR and ddPCR Methods and Their Applications

{kind=link}
{kind=link}
| Author (Year) | Gene | Type | LOD | LOQ & Range | Reproducibility | |||
|---|---|---|---|---|---|---|---|---|
| qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | |||
| Laura Cavé et al. (2016) [74] # | sul1, qnrB | ARG | +, 10-fold | + | - | |||
| Cesare, Andrea Di et al. (2018) [75] # | sul2, Intl1 | ARG | - | + | ||||
| Ginn O. et al. (2021) [76] | tetA, qnrB, blaTEM, intl1 | ARG | N/A | + | * | |||
| Kimbell L. et al. (2021) [77] # | blaTEM, blaSHV, sul1, czcD, copA, intl1 | ARG | - | + | * | * | ||
| Sun Y. e al. (2021) [78] | quinolones, tetracyclines, sulfonamides, macrolides | ARG | N/A | * | ||||
| Srisutham S. et al. (2021) [79] | pfmdr1, pfplasmepsin2, pfgch1 | ARG | N/A | * | * | |||
| Xu J. et al. (2021) [80] | mcr-1, blaCTX-M-14, bla CTX-M-55 | ARG | - | + | ||||
| Yang et al. (2014) [53] | Cryptosporidium Oocysts, 18S rRNA | Parasite | N/A | N/A | + | + | ||
| Weerakoon K.G. et al. (2016) [81] # | S. japoricum, SjR2 and nad1 | Parasite | - | 0.05fg, + | - | |||
| Overall | ARG & Parasite | - | + | + | - | * | * | |
| Henrich T.J. et al. (2012) [66] # | HIV-1, human CCR5 DNA | Human Diseases | + | +, HIV-1 | ||||
| Heredia N.J. et al. (2013) [82] | HER2 (= erbB2), CEP17 | Human Diseases | N/A | + | + | |||
| Strain M.C. et al. (2013) [83] # | HIV, episomal 2-LTR | Human Diseases | + | + | - | + | ||
| Bharuthram A. (2014) [84] # | CCL4L, CCL4L1 and CCL4L2 encodes HIV-1 | Human Diseases | - | + | - | + | ||
| Jones M. et al. (2014) [85] | HIV-1 from 8E5/LAV cells | Human Diseases | - | + | + | -, lower target | + | |
| Coudray-Meunier et al. (2015) [86] # | Hepatitis A, Norovirus | Human Diseases | * | * | - | + | - | + |
| Taylor S.C. et al. (2015) [46] # | H275-WT and H275Y-MUT of H1N1 | Human Diseases | +, mutant | + | - | + | ||
| Yan Y. et al. (2016) [87] | H7N9 | Human Diseases | N/A | - | ||||
| Yang Q (2017) [88] # | PRRSV | Human Diseases | - | + | +, false positive | - | ||
| Link-Lenczowska D. et al. (2018) [89]# | JAK2 mutation on V617F | Human Diseases | 0.12% | 0.01%, + | * | * | + | |
| Persson S. et al. (2018) [67] # | norovirus GI (GI.4) and GII (GII.4) | Human Diseases | * | * | + | |||
| Pinheiro T.F. et al. (2018) [45] | foot-and-mouth disease virus RNA | Human Diseases | - | + | ||||
| Baume M. et al. (2019) [90] # | Legionella DNA reference material | Human Disease | + | * | * | |||
| Zhang Y. et al. (2019) [91] # | PCV3 | Human Diseases | - | + | - | + | ||
| Dong L. et al. (2020) [92] | Tumor DNA reference material, BRAF V600E | Human Diseases | N/A | 0.02% | 0.10% | + | ||
| Lin Q. et al. (2020) [50] # | ISKNV | Human Diseases | - | + | - | +, low | ||
| Petiti J. et al. (2020) [64] | BCR-ABL1 disease marker leukemia | Human Diseases | N/A | 0.001% | ||||
| Thwin KKM et al. (2020) [93] # | NB-mRNAs (CRMP1, DBH, DDC, GAP43, ISL1, PHOX2B, and TH mRNA) | Human Diseases | - | + | - | + | ||
| Overall | Human Disease | - | + | * | * | - | + | |
| Milbury C.A. et al. (2014) [94] | EGFR T790M, L858R | Mutation | + | |||||
| Zhao Y. (2019) [95] | MTRNR1-WT | Mutation | N/A | + | + | - | + | |
| Liu Q. et al. (2020) [44] # | CNVs causing somatic mosaicism | Mutation | + | - | * | * | N/A | N/A |
| Overall | Mutation | * | * | + | - | + | ||
| Burns et al. (2010) [96] # | ERM-AD413 carries Mon810 | Plant, Food | - | + | -, lower range | |||
| Coudray-Meunier et al. (2015) [86] # | Hepatitis A, Norovirus | Plant, Food | - | + | + | -, bias | ||
| Porcellato D. et al. (2016) [68] # | gyrB of B. cereus group | Plant, Food | - | + | + | - | * | * |
| Scollo F. et al. (2016) [49] # | 11C Chloroplast locus | Plant, Food | - | + | ||||
| Wang X. et al. (2019) [47] # | transgenic rice line TT51-1 | Plant, Food | - | + | ||||
| Demeke et al. (2020) [97] # | Canola and soybean | Plant, Food | * | * | * | * | + | |
| Overall | Plant & Food | - | + | * | * | + | * | |
| Pinheiro L.B. et al. (2012) [98] | Lambda DNA | Bacteria, Phage | + | + | ||||
| Xi Z. (2018) [48] # | 16S rRNA of Las | Bacteria, Phage | - | + | - | + | ||
| Sivagnesan et al. (2018) [99] | Std1_Xhol insert with M13 E coli plasmid DNA | Bacteria, Phage | - | + | * | * | ||
| Furuta-Hanawa B. et al. (2019) [100] # | rAAV2RSM, rAAV8RSM | Bacteria, Phage | * | * | - | + | - | + |
| Nshimyimana J.P. et al. (2019) [69] # | Bacteroidales, BacHum and B. theta | Bacteria, Phage | +, Environmental | +, sensitivity | +, fecal | |||
| Raurich et al. (2019) [101] | Bifidobacterium animalis (BAN) | Bacteria, Phage | + | * | + | - | ||
| Ahn Y. et al. (2020) [102] # | Burkholderia epacian | Bacteria, Phage | - | + | -, recovery | +, recovery | - | + |
| Ibekwe M.A. et al. (2020) [58] # | Shiga toxin-producing E. coli O157:H7 | Bacteria, Phage | + | * | * | - | + | |
| Voegel T.M. et al. (2020) [103] # | amoA, nirS, nirK, nosZI, nosZII | Bacteria, Phage | - | + | + | - | ||
| Overall | Bacteria & Phage | - | + | * | * | - | + | |
4. ddPCR as the Future
5. Current Findings on ddPCR Analysis on Genetic Targets Compared with Other Methods
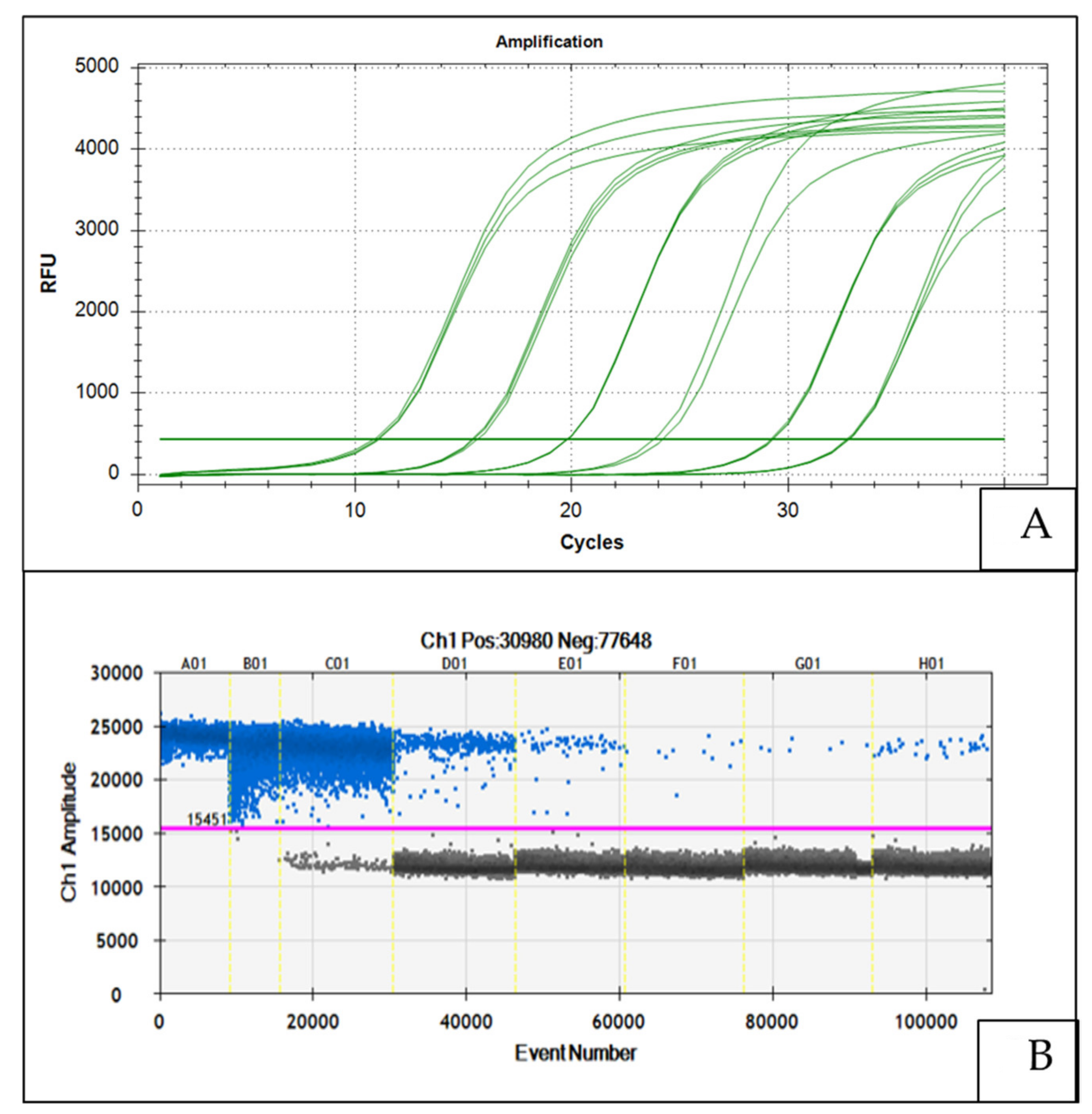
5.1. Sensitivity
5.2. Dynamic Range of Detection and Measurement Variance
5.3. Reproducibility
5.4. Cost
5.5. Risk of Bias in qPCR and ddPCR
5.6. Applicability
6. Current Status in ARG Detection Methods with ddPCR
7. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Knapp, C.; Dolfing, J.; Ehlert, P.A.I.; Graham, D.W. Evidence of Increasing Antibiotic Resistance Gene Abundances in Archived Soils since 1940. Environ. Sci. Technol. 2009, 44, 580–587. [Google Scholar] [CrossRef]
- Bergeron, S.; Brown, R.; Homer, J.; Rehage, S.; Boopathy, R. Presence of antibiotic resistance genes in different salinity gradients of freshwater to saltwater marshes in southeast Louisiana, USA. Int. Biodeterior. Biodegrad. 2016, 113, 80–87. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Genet. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Liu, Y.; Du, P.-P.; Zeng, L.-J.; Mo, C.-H.; Li, Y.-W.; Lü, H.; Cai, Q.-Y. Occurrence and distribution of antibiotics and antibiotic resistant genes in water and sediments of urban rivers with black-odor water in Guangzhou, South China. Sci. Total Environ. 2019, 670, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Eckert, E.; Teruggi, A.; Fontaneto, D.; Bertoni, R.; Callieri, C.; Corno, G. Constitutive presence of antibiotic resistance genes within the bacterial community of a large subalpine lake. Mol. Ecol. 2015, 24, 3888–3900. [Google Scholar] [CrossRef]
- Rizzo, L.; Manaia, C.; Merlin, C.; Schwartz, T.; Dagot, C.; Ploy, M.; Michael, I.; Fatta-Kassinos, D. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: A review. Sci. Total Environ. 2013, 447, 345–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruden, A.; Pei, R.; Storteboom, H.; Carlson, K.H. Antibiotic Resistance Genes as Emerging Contaminants: Studies in Northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Martinez, J.L.; Cantón, R. Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotechnol. 2008, 19, 260–265. [Google Scholar] [CrossRef]
- Morel, C.M.; Lindahl, O.; Harbarth, S.; De Kraker, M.E.A.; Edwards, S.; Hollis, A. Industry incentives and antibiotic resistance: An introduction to the antibiotic susceptibility bonus. J. Antibiot. 2020, 73, 421–428. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Antimicrobial Resistance: Global Report on Surveillance; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- World Health Organization (WHO). Sanitation Safety Planning, Greywater and Excreta; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- World Health Organization (WHO). No Time to Wait: Securing the Future from Drug-Resistant Infections; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Bhattacharyya, A.; Haldar, A.; Bhattacharyya, M.; Ghosh, A. Anthropogenic influence shapes the distribution of antibiotic resistant bacteria (ARB) in the sediment of Sundarban estuary in India. Sci. Total Environ. 2018, 647, 1626–1639. [Google Scholar] [CrossRef]
- Guo, X.-P.; Yang, Y.; Lu, D.-P.; Niu, Z.-S.; Feng, J.-N.; Chen, Y.-R.; Tou, F.-Y.; Garner, E.; Xu, J.; Liu, M.; et al. Biofilms as a sink for antibiotic resistance genes (ARGs) in the Yangtze Estuary. Water Res. 2018, 129, 277–286. [Google Scholar] [CrossRef]
- Reddy, B.; Dubey, S.K.; Reddy, B.; Dubey, S.K. River Ganges water as reservoir of microbes with antibiotic and metal ion resistance genes: High throughput metagenomic approach. Environ. Pollut. 2018, 246, 443–451. [Google Scholar] [CrossRef]
- Fiorentino, A.; De Luca, G.; Rizzo, L.; Viccione, G.; Lofrano, G.; Carotenuto, M. Simulating the fate of indigenous antibiotic resistant bacteria in a mild slope wastewater polluted stream. J. Environ. Sci. 2018, 69, 95–104. [Google Scholar] [CrossRef]
- Pruden, A.; Arabi, M.; Storteboom, H.N. Correlation between Upstream Human Activities and Riverine Antibiotic Resistance Genes. Environ. Sci. Technol. 2012, 46, 11541–11549. [Google Scholar] [CrossRef] [PubMed]
- Christou, A.; Agüera, A.; Bayona, J.M.; Cytryn, E.; Fotopoulos, V.; Lambropoulou, D.; Manaia, C.M.; Michael, C.; Revitt, M.; Schröder, P.; et al. The potential implications of reclaimed wastewater reuse for irrigation on the agricultural environment: The knowns and unknowns of the fate of antibiotics and antibiotic resistant bacteria and resistance genes—A review. Water Res. 2017, 123, 448–467. [Google Scholar] [CrossRef] [Green Version]
- Van Hoek, A.H.A.M.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J.M. Acquired Antibiotic Resistance Genes: An Overview. Front. Microbiol. 2011, 2, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.-G. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2014, 9, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Stedtfeld, R.D.; Guo, X.; Bhalsod, G.D.; Jeon, S.; Tiedje, J.M.; Li, H.; Zhang, W. Pharmaceutical exposure changed antibiotic resistance genes and bacterial communities in soil-surface- and overhead-irrigated greenhouse lettuce. Environ. Int. 2019, 131, 105031. [Google Scholar] [CrossRef]
- Gao, M.; Qiu, T.; Sun, Y.; Wang, X. The abundance and diversity of antibiotic resistance genes in the atmospheric environment of composting plants. Environ. Int. 2018, 116, 229–238. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R.; Da Fonseca, M.M.R. Biotransformations. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017; pp. 574–585. [Google Scholar]
- Garibyan, L.; Avashia, N. Polymerase Chain Reaction. J. Investig. Dermatol. 2013, 133, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Clewley, J.P. The polymerae chain reaction, a review of the practical limitations for human immunodeficiency virus diagnosis. J. Virol. Methods 1989, 25, 179–188. [Google Scholar] [CrossRef]
- Bustin, S.; Huggett, J. qPCR primer design revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef]
- Chandler, D.P.; Wagnon, C.A.; Bolton, H. Reverse Transcriptase (RT) Inhibition of PCR at Low Concentrations of Template and Its Implications for Quantitative RT-PCR. Appl. Environ. Microbiol. 1998, 64, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romsos, E.L.; Vallone, P. Estimation of extraction efficiency by droplet digital PCR. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 515–517. [Google Scholar] [CrossRef]
- Perry, D.J.; Harper, P.L.; Fairham, S.; Daly, M.; Carrell, R.W. Antithrombin Cambridge, 384 Ala to Pro: A new variant identified using the polymerase chain reaction. FEBS Lett. 1989, 254, 174–176. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Li, Y.; Chen, X.; Chang, Y.; Li, L.; Shi, L.; Bai, W.; Ye, L. Species identification and quantification of silver pomfret using the droplet digital PCR assay. Food Chem. 2019, 302, 125331. [Google Scholar] [CrossRef] [PubMed]
- Vishnuraj, M.; Devatkal, S.; Vaithiyanathan, S.; Kumar, R.U.; Srinivas, C.; Mendiratta, S. Detection of giblets in chicken meat products using microRNA markers and droplet digital PCR assay. LWT 2020, 140, 110798. [Google Scholar] [CrossRef]
- Ekman, S. Pcr Optimization and Troubleshooting, with Special Reference to the Amplification of Ribosomal DNA in Lichenized Fungi. Lichenologist 1999, 31, 517–531. [Google Scholar] [CrossRef]
- Butler, J.M.; Ruitberg, C.M.; Vallone, P. Capillary electrophoresis as a tool for optimization of multiplex PCR reactions. Anal. Bioanal. Chem. 2001, 369, 200–205. [Google Scholar] [CrossRef]
- Wong, W.H.; Tay, Y.C.; Puniamoorthy, J.; Balke, M.; Cranston, P.S.; Meier, R. ‘Direct PCR’ optimization yields a rapid, cost-effective, nondestructive and efficient method for obtaining DNA barcodes without DNA extraction. Mol. Ecol. Resour. 2014, 14, 1271–1280. [Google Scholar] [CrossRef]
- Ponchel, F.; Toomes, C.; Bransfield, K.; Leong, F.T.; Douglas, S.H.; Field, S.L.; Bell, S.M.; Combaret, V.; Puisieux, A.; Mighell, A.J.; et al. Real-time PCR based on SYBR-Green I fluorescence: An alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alía, A.; Andrade, M.J.; Córdoba, J.J.; Martín, I.; Rodríguez, A. Development of a multiplex real-time PCR to differentiate the four major Listeria monocytogenes serotypes in isolates from meat processing plants. Food Microbiol. 2019, 87, 103367. [Google Scholar] [CrossRef]
- Huggett, J.F.; Novak, T.; Garson, J.; Green, C.; Morris-Jones, S.D.; Miller, R.F.; Zumla, A. Differential susceptibility of PCR reactions to inhibitors: An important and unrecognised phenomenon. BMC Res. Notes 2008, 1, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suslov, O. PCR inhibition by reverse transcriptase leads to an overestimation of amplification efficiency. Nucleic Acids Res. 2005, 33, e181. [Google Scholar] [CrossRef] [PubMed]
- Karlen, Y.; McNair, A.; Perseguers, S.; Mazza, C.; Mermod, N. Statistical significance of quantitative PCR. BMC Bioinform. 2007, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Nyaruaba, R.; Mwaliko, C.; Kering, K.K.; Wei, H. Droplet digital PCR applications in the tuberculosis world. Tuberculosis 2019, 117, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Biorad. Droplet Digital TM PCR Droplet Digital TM PCR Applications Guide; Biorad: Hercules, CA, USA, 2018; p. 145. [Google Scholar]
- Tone, M.; Torunn, K. Presence and Levels of Antibiotic Resistance Genes in Saliva from Dental Students in Tromsø. Master’s Thesis, The Arctic University of Norway, Tromsø, Norway, May 2016. [Google Scholar]
- Liu, Q.; Karolak, J.; Grochowski, C.M.; Wilson, T.A.; Rosenfeld, J.A.; Bacino, C.A.; Lalani, S.R.; Patel, A.; Breman, A.; Smith, J.L.; et al. Parental somatic mosaicism for CNV deletions—A need for more sensitive and precise detection methods in clinical diagnostics settings. Genomics 2020, 112, 2937–2941. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.; Fonseca, A.A.; Camargos, M.F.; Laguardia-Nascimento, M.; de Oliveira, A.M.; Cottorello, A.C.; Goes-Neto, A.; Barbosa-Stancioli, E.F. Development of a droplet digital RT-PCR for the quantification of foot-and-mouth virus RNA. J. Virol. Methods 2018, 259, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Carbonneau, J.; Shelton, D.N.; Boivin, G. Optimization of Droplet Digital PCR from RNA and DNA extracts with direct comparison to RT-qPCR: Clinical implications for quantification of Oseltamivir-resistant subpopulations. J. Virol. Methods 2015, 224, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tang, T.; Miao, Q.; Xie, S.; Chen, X.; Tang, J.; Peng, C.; Xu, X.; Wei, W.; You, Z.; et al. Detection of transgenic rice line TT51-1 in processed foods using conventional PCR, real-time PCR, and droplet digital PCR. Food Control. 2018, 98, 380–388. [Google Scholar] [CrossRef]
- Zhong, X.; Liu, X.-L.; Lou, B.-H.; Zhou, C.-Y.; Wang, X.-F. Development of a sensitive and reliable droplet digital PCR assay for the detection of ‘Candidatus Liberibacter asiaticus’. J. Integr. Agric. 2018, 17, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Scollo, F.; Egea, L.A.; Gentile, A.; La Malfa, S.; Dorado, G.; Hernandez, P. Absolute quantification of olive oil DNA by droplet digital-PCR (ddPCR): Comparison of isolation and amplification methodologies. Food Chem. 2016, 213, 388–394. [Google Scholar] [CrossRef]
- Lin, Q.; Fu, X.; Liu, L.; Liang, H.; Niu, Y.; Wen, Y.; Huang, Z.; Li, N. Development and application of a sensitive droplet digital PCR (ddPCR) for the detection of infectious spleen and kidney necrosis virus. Aquaculture 2020, 529, 735697. [Google Scholar] [CrossRef]
- Naaum, A.M.; Shehata, H.R.; Chen, S.; Li, J.; Tabujara, N.; Awmack, D.; Lutze-Wallace, C.; Hanner, R. Complementary molecular methods detect undeclared species in sausage products at retail markets in Canada. Food Control 2018, 84, 339–344. [Google Scholar] [CrossRef]
- Floren, C.; Wiedemann, I.; Brenig, B.; Schütz, E.; Beck, J. Species identification and quantification in meat and meat products using droplet digital PCR (ddPCR). Food Chem. 2015, 173, 1054–1058. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Paparini, A.; Monis, P.; Ryan, U. Comparison of next-generation droplet digital PCR (ddPCR) with quantitative PCR (qPCR) for enumeration of Cryptosporidium oocysts in faecal samples. Int. J. Parasitol. 2014, 44, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrberg, C.D.; Lee, J.M.; Chung, B.G. Microwell Array-based Digital PCR for Influenza Virus Detection. BioChip J. 2019, 13, 269–276. [Google Scholar] [CrossRef]
- Heyries, K.; Tropini, C.; VanInsberghe, M.; Doolin, C.; Petriv, O.; Singhal, A.; Leung, K.; Hughesman, C.B.; Hansen, C.L. Megapixel digital PCR. Nat. Methods 2011, 8, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. dPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef] [Green Version]
- Huggett, J.F.; Cowen, S.; Foy, C.A. Considerations for Digital PCR as an Accurate Molecular Diagnostic Tool. Clin. Chem. 2015, 61, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Ibekwe, A.M.; Murinda, S.E.; Park, S.; Obayiuwana, A.; Murry, M.A.; Schwartz, G.; Lundquist, T. Comparative Use of Quantitative PCR (qPCR), Droplet Digital PCR (ddPCR), and Recombinase Polymerase Amplification (RPA) in the Detection of Shiga Toxin-Producing E. coli (STEC) in Environmental Samples. Water 2020, 12, 3507. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, N.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Maeda, R.; Kami, D.; Maeda, H.; Shikuma, A.; Gojo, S. High throughput single cell analysis of mitochondrial heteroplasmy in mitochondrial diseases. Sci. Rep. 2020, 10, 10821. [Google Scholar] [CrossRef]
- Takahashi, M.; Wu, X.; Ho, M.; Chomchan, P.; Rossi, J.J.; Burnett, J.C.; Zhou, J. High throughput sequencing analysis of RNA libraries reveals the influences of initial library and PCR methods on SELEX efficiency. Sci. Rep. 2016, 6, 33697. [Google Scholar] [CrossRef] [Green Version]
- Marangi, M.; Giangaspero, A.; Lacasella, V.; Lonigro, A.; Gasser, R.B. Multiplex PCR for the detection and quantification of zoonotic taxa of Giardia, Cryptosporidium and Toxoplasma in wastewater and mussels. Mol. Cell. Probes 2015, 29, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Giantsis, I.; Chaskopoulou, A. Broadening the tools for studying sand fly breeding habitats: A novel molecular approach for the detection of phlebotomine larval DNA in soil substrates. Acta Trop. 2018, 190, 123–128. [Google Scholar] [CrossRef]
- Petiti, J.; Iacono, M.L.; Dragani, M.; Pironi, L.; Fantino, C.; Rapanotti, M.C.; Quarantelli, F.; Izzo, B.; Divona, M.; Rege-Cambrin, G.; et al. Novel Multiplex Droplet Digital PCR Assays to Monitor Minimal Residual Disease in Chronic Myeloid Leukemia Patients Showing Atypical BCR-ABL1 Transcripts. J. Clin. Med. 2020, 9, 1457. [Google Scholar] [CrossRef]
- Malic, L.; Daoud, J.; Geissler, M.; Boutin, A.; Lukic, L.; Janta, M.; Elmanzalawy, A.; Veres, T. Epigenetic subtyping of white blood cells using a thermoplastic elastomer-based microfluidic emulsification device for multiplexed, methylation-specific digital droplet PCR. Analyst 2019, 144, 6541–6553. [Google Scholar] [CrossRef]
- Henrich, T.J.; Gallien, S.; Li, J.Z.; Pereyra, F.; Kuritzkes, D.R. Low-level detection and quantitation of cellular HIV-1 DNA and 2-LTR circles using droplet digital PCR. J. Virol. Methods 2012, 186, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Persson, S.; Eriksson, R.; Lowther, J.; Ellström, P.; Simonsson, M. Comparison between RT droplet digital PCR and RT real-time PCR for quantification of noroviruses in oysters. Int. J. Food Microbiol. 2018, 284, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Porcellato, D.; Narvhus, J.; Skeie, S.B. Detection and quantification of Bacillus cereus group in milk by droplet digital PCR. J. Microbiol. Methods 2016, 127, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nshimyimana, J.P.; Cruz, M.C.; Wuertz, S.; Thompson, J.R. Variably improved microbial source tracking with digital droplet PCR. Water Res. 2019, 159, 192–202. [Google Scholar] [CrossRef]
- NejcRački, N.; Dreo, T.; Gutierrez-Aguirre, I.; Blejec, A.; Ravnikar, M. Reverse transcriptase droplet digital PCR shows high resilience to PCR inhibitors from plant, soil and water samples. Plant Methods 2014, 10, 1–10. [Google Scholar] [CrossRef]
- Rački, N.; Morisset, D.; Gutierrez-Aguirre, I.; Ravnikar, M. One-step RT-droplet digital PCR: A breakthrough in the quantification of waterborne RNA viruses. Anal. Bioanal. Chem. 2013, 406, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Aguirre, I.; Rački, N.; Dreo, T.; Ravnikar, M. Droplet Digital PCR for Absolute Quantification of Pathogens. Methods Mol. Biol. 2015, 1302, 331–347. [Google Scholar] [CrossRef]
- Deprez, L.; Corbisier, P.; Kortekaas, A.-M.; Mazoua, S.; Hidalgo, R.B.; Trapmann, S.; Emons, H. Validation of a digital PCR method for quantification of DNA copy number concentrations by using a certified reference material. Biomol. Detect. Quantif. 2016, 9, 29–39. [Google Scholar] [CrossRef]
- Cavé, L.; Brothier, E.; Abrouk, D.; Bouda, P.S.; Hien, E.; Nazaret, S. Efficiency and sensitivity of the digital droplet PCR for the quantification of antibiotic resistance genes in soils and organic residues. Appl. Microbiol. Biotechnol. 2016, 100, 10597–10608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare, A.; Petrin, S.; Fontaneto, D.; LoSasso, C.; Eckert, E.M.; Tassistro, G.; Borello, A.; Ricci, A.; Wilson, W.H.; Pruzzo, C.; et al. ddPCR applied on archived Continuous Plankton Recorder samples reveals long-term occurrence of class 1 integrons and a sulphonamide resistance gene in marine plankton communities. Environ. Microbiol. Rep. 2018, 10, 458–464. [Google Scholar] [CrossRef]
- Ginn, O.; Nichols, D.; Rocha-Melogno, L.; Bivins, A.; Berendes, D.; Soria, F.; Andrade, M.; Deshusses, M.A.; Bergin, M.; Brown, J. Antimicrobial resistance genes are enriched in aerosols near impacted urban surface waters in La Paz, Bolivia. Environ. Res. 2021, 194, 110730. [Google Scholar] [CrossRef]
- Kimbell, L.K.; LaMartina, E.L.; Kappell, A.D.; Huo, J.; Wang, Y.; Newton, R.J.; McNamara, P.J. Cast iron drinking water pipe biofilms support diverse microbial communities containing antibiotic resistance genes, metal resistance genes, and class 1 integrons. Environ. Sci. Water Res. Technol. 2021, 7, 584–598. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, Y.; Shi, M.; Qiu, T.; Gao, M.; Tian, S.; Wang, X. Effect of antibiotic type and vegetable species on antibiotic accumulation in soil-vegetable system, soil microbiota, and resistance genes. Chemosphere 2020, 263, 128099. [Google Scholar] [CrossRef] [PubMed]
- Srisutham, S.; Suwannasin, K.; Sugaram, R.; Dondorp, A.M.; Imwong, M. Measurement of gene amplifications related to drug resistance in Plasmodium falciparum using droplet digital PCR. Malar. J. 2021, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, N.; Luo, M.; Wang, M.; Wang, L.; Li, J.; Li, Z.; Zhao, H.; Li, Z.; Kan, B.; et al. Rapid Identification of Plasmid Replicon Type and Coexisting Plasmid-Borne Antimicrobial Resistance Genes by S1-Pulsed-Field Gel Electrophoresis-Droplet Digital Polymerase Chain Reaction. Foodborne Pathog. Dis. 2021, 18, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Weerakoon, K.G.; Gordon, C.; Gobert, G.; Cai, P.; McManus, D.P. Optimisation of a droplet digital PCR assay for the diagnosis of Schistosoma japonicum infection: A duplex approach with DNA binding dye chemistry. J. Microbiol. Methods 2016, 125, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Heredia, N.J.; Belgrader, P.; Wang, S.; Koehler, R.; Regan, J.; Cosman, A.M.; Saxonov, S.; Hindson, B.; Tanner, S.C.; Brown, A.S.; et al. Droplet Digital™ PCR quantitation of HER2 expression in FFPE breast cancer samples. Methods 2012, 59, S20–S23. [Google Scholar] [CrossRef]
- Strain, M.C.; Lada, S.M.; Luong, T.; Rought, S.E.; Gianella, S.; Terry, V.H.; Spina, C.A.; Woelk, C.H.; Richman, D.D. Highly Precise Measurement of HIV DNA by Droplet Digital PCR. PLoS ONE 2013, 8, e55943. [Google Scholar] [CrossRef]
- Bharuthram, A.; Paximadis, M.; Picton, A.C.; Tiemessen, C. Comparison of a quantitative Real-Time PCR assay and droplet digital PCR for copy number analysis of the CCL4L genes. Infect. Genet. Evol. 2014, 25, 28–35. [Google Scholar] [CrossRef]
- Jones, M.; Williams, J.; Gärtner, K.; Phillips, R.; Hurst, J.; Frater, J. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, ‘definetherain’. J. Virol. Methods 2014, 202, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Coudray-Meunier, C.; Fraisse, A.; Martin-Latil, S.; Guillier, L.; Delannoy, S.; Fach, P.; Perelle, S. A comparative study of digital RT-PCR and RT-qPCR for quantification of Hepatitis A virus and Norovirus in lettuce and water samples. Int. J. Food Microbiol. 2015, 201, 17–26. [Google Scholar] [CrossRef]
- Yan, Y.; Jia, X.-J.; Wang, H.-H.; Fu, X.-F.; Ji, J.-M.; He, P.-Y.; Chen, L.-X.; Luo, J.-Y.; Chen, Z.-W. Dynamic quantification of avian influenza H7N9(A) virus in a human infection during clinical treatment using droplet digital PCR. J. Virol. Methods 2016, 234, 22–27. [Google Scholar] [CrossRef]
- Yang, Q.; Xi, J.; Chen, X.; Hu, S.; Chen, N.; Qiao, S.; Wan, S.; Bao, D. The development of a sensitive droplet digital PCR for quantitative detection of porcine reproductive and respiratory syndrome virus. Int. J. Biol. Macromol. 2017, 104, 1223–1228. [Google Scholar] [CrossRef]
- Link-Lenczowska, D.; Pallisgaard, N.; Cordua, S.; Zawada, M.; Czekalska, S.; Krochmalczyk, D.; Kanduła, Z.; Sacha, T. A comparison of qPCR and ddPCR used for quantification of the JAK2 V617F allele burden in Ph negative MPNs. Ann. Hematol. 2018, 97, 2299–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baume, M.; Cariou, A.; Leveau, A.; Fessy, N.; Pastori, F.; Jarraud, S.; Pierre, S. Quantification of Legionella DNA certified reference material by digital droplet PCR. J. Microbiol. Methods 2018, 157, 50–53. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Wang, Z.; Wang, Z.; Wang, C.; Feng, C.; Yuan, W.; Lin, X.; Wu, S. Development of a droplet digital PCR assay for sensitive detection of porcine circovirus 3. Mol. Cell. Probes 2018, 43, 50–57. [Google Scholar] [CrossRef]
- Dong, L.; Wang, X.; Wang, S.; Du, M.; Niu, C.; Yang, J.; Li, L.; Zhang, G.; Fu, B.; Gao, Y.; et al. Interlaboratory assessment of droplet digital PCR for quantification of BRAF V600E mutation using a novel DNA reference material. Talanta 2019, 207, 120293. [Google Scholar] [CrossRef] [PubMed]
- Thwin, K.K.; Ishida, T.; Uemura, S.; Yamamoto, N.; Lin, K.S.; Tamura, A.; Kozaki, A.; Saito, A.; Kishimoto, K.; Mori, T.; et al. Level of Seven Neuroblastoma-Associated mRNAs Detected by Droplet Digital PCR Is Associated with Tumor Relapse/Regrowth of High-Risk Neuroblastoma Patients. J. Mol. Diagn. 2020, 22, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Milbury, C.A.; Zhong, Q.; Lin, J.; Williams, M.; Olson, J.; Link, D.R.; Hutchison, B. Determining lower limits of detection of digital PCR assays for cancer-related gene mutations. Biomol. Detect. Quantif. 2014, 1, 8–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y. A comparative study of ddPCR and sanger sequencing for quantitative detection of low-frequency mutation rate. IOP Conf. Ser. Earth Environ. Sci. 2019, 332, 032023. [Google Scholar] [CrossRef]
- Burns, M.J.; Burrell, A.M.; Foy, C.A. The applicability of digital PCR for the assessment of detection limits in GMO analysis. Eur. Food Res. Technol. 2010, 231, 353–362. [Google Scholar] [CrossRef]
- Demeke, T.; Beecher, B.; Eng, M. Assessment of genetically engineered events in heat-treated and non-treated samples using droplet digital PCR and real-time quantitative PCR. Food Control 2020, 115, 107291. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a Droplet Digital Polymerase Chain Reaction Format for DNA Copy Number Quantification. Anal. Chem. 2011, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Sivaganesan, M.; Varma, M.; Siefring, S.; Haugland, R. Quantification of plasmid DNA standards for U.S. EPA fecal indicator bacteria qPCR methods by droplet digital PCR analysis. J. Microbiol. Methods 2018, 152, 135–142. [Google Scholar] [CrossRef]
- Furuta-Hanawa, B.; Yamaguchi, T.; Uchida, E. Two-Dimensional Droplet Digital PCR as a Tool for Titration and Integrity Evaluation of Recombinant Adeno-Associated Viral Vectors. Hum. Gene Ther. Methods 2019, 30, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raurich, S.; Weber, B.; Klose, V.; Mohnl, M.; Petri, D.; Fibi-Smetana, S. Optimisation of a droplet digital PCR for strain specific quantification of a probiotic Bifidobacterium animalis strain in poultry feed. J. Microbiol. Methods 2019, 163, 105646. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Gibson, B.; Williams, A.; Alusta, P.; Buzatu, D.A.; Lee, Y.-J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Cerniglia, C.E. A comparison of culture-based, real-time PCR, droplet digital PCR and flow cytometric methods for the detection of Burkholderia cepacia complex in nuclease-free water and antiseptics. J. Ind. Microbiol. Biotechnol. 2020, 47, 475–484. [Google Scholar] [CrossRef]
- Voegel, T.M.; Larrabee, M.M.; Nelson, L.M. Development of droplet digital PCR assays to quantify genes involved in nitrification and denitrification, comparison with quantitative real-time PCR and validation of assays in vineyard soil. Can. J. Microbiol. 2021, 67, 174–187. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kline, M.C.; Romsos, E.L.; Duewer, D.L. Evaluating Digital PCR for the Quantification of Human Genomic DNA: Accessible Amplifiable Targets. Anal. Chem. 2016, 88, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, J.D.; Herrera, G.; Muskus, C.; Mendez, C.; Duque, M.C.; Butcher, R. Development of a Digital Droplet Polymerase Chain Reaction (ddPCR) assay to detect Leishmania DNA in samples from Cutaneous Leishmaniasis patients. Int. J. Infect. Dis. 2019, 79, 1–3. [Google Scholar] [CrossRef]
- Galluzzi, L.; Ceccarelli, M.; Diotallevi, A.; Menotta, M.; Magnani, M. Real-time PCR applications for diagnosis of leishmaniasis. Parasites Vectors 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.S. Digital Assays Part I: Partitioning Statistics and Digital PCR. SLAS Technol. Transl. Life Sci. Innov. 2017, 22, 369–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhao, Z.; Avillan, J.J.; Call, D.R.; Davis, M.; Sischo, W.M.; Zhang, A. Dairy farm soil presents distinct microbiota and varied prevalence of antibiotic resistance across housing areas. Environ. Pollut. 2019, 254, 113058. [Google Scholar] [CrossRef] [PubMed]
- Arvia, R.; Sollai, M.; Pierucci, F.; Urso, C.; Massi, D.; Zakrzewska, K. Droplet digital PCR (ddPCR) vs quantitative real-time PCR (qPCR) approach for detection and quantification of Merkel cell polyomavirus (MCPyV) DNA in formalin fixed paraffin embedded (FFPE) cutaneous biopsies. J. Virol. Methods 2017, 246, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Koepfli, C.; Nguitragool, W.; Hofmann, N.E.; Robinson, L.J.; Ome-Kaius, M.; Sattabongkot, J.; Felger, I.; Mueller, I. Sensitive and accurate quantification of human malaria parasites using droplet digital PCR (ddPCR). Sci. Rep. 2016, 6, 39183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-R.; Guo, X.-P.; Feng, J.-N.; Lu, D.-P.; Niu, Z.-S.; Tou, F.-Y.; Hou, L.-J.; Liu, M.; Yang, Y. Impact of ZnO nanoparticles on the antibiotic resistance genes (ARGs) in estuarine water: ARG variations and their association with the microbial community. Environ. Sci. Nano 2019, 6, 2405–2419. [Google Scholar] [CrossRef]
- Fujimoto, M.; Carey, D.E.; McNamara, P.J. Metagenomics reveal triclosan-induced changes in the antibiotic resistome of anaerobic digesters. Environ. Pollut. 2018, 241, 1182–1190. [Google Scholar] [CrossRef]
- Gaviria-Figueroa, A.; Preisner, E.C.; Hoque, S.; Feigley, C.E.; Norman, R.S. Emission and dispersal of antibiotic resistance genes through bioaerosols generated during the treatment of municipal sewage. Sci. Total Environ. 2019, 686, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Yu, S.; Rysz, M.; Luo, Y.; Yang, F.; Li, F.; Hou, J.; Mu, Q.; Alvarez, P. Prevalence and proliferation of antibiotic resistance genes in two municipal wastewater treatment plants. Water Res. 2015, 85, 458–466. [Google Scholar] [CrossRef]
- Gerdes, L.; Iwobi, A.; Busch, U.; Pecoraro, S. Optimization of digital droplet polymerase chain reaction for quantification of genetically modified organisms. Biomol. Detect. Quantif. 2016, 7, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Noh, E.S.; Park, Y.J.; Kim, E.-M.; Park, J.Y.; Shim, K.B.; Choi, T.-J.; Kim, K.-H.; Kang, J.-H. Quantitative analysis of Alaska pollock in seafood products by droplet digital PCR. Food Chem. 2018, 275, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Talarico, S.; Korson, A.S.; Leverich, C.K.; Park, S.; Jalikis, F.G.; Upton, M.P.; Broussard, E.; Salama, N.R. High prevalence of Helicobacter pylori clarithromycin resistance mutations among Seattle patients measured by droplet digital PCR. Helicobacter 2018, 23, e12472. [Google Scholar] [CrossRef]
- Liao, Y.; Chen, Y.; Kou, X.; Xiao, Y.; Ye, J.; Wu, A. Diagnostic test accuracy of droplet digital PCR for the detection of EGFR mutation (T790M) in plasma: Systematic review and meta-analysis. Clin. Chim. Acta 2019, 503, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Überbacher, C.; Obergasteiger, J.; Volta, M.; Venezia, S.; Müller, S.; Pesce, I.; Pizzi, S.; Lamonaca, G.; Picard, A.; Cattelan, G.; et al. Application of CRISPR/Cas9 editing and digital droplet PCR in human iPSCs to generate novel knock-in reporter lines to visualize dopaminergic neurons. Stem Cell Res. 2019, 41, 101656. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J. Recent advances in the detection of methicillin resistant Staphylococcus aureus (MRSA). BioChip J. 2017, 11, 89–100. [Google Scholar] [CrossRef]
- Koch, H.; Jeschke, A.; Becks, L. Use of dd PCR in experimental evolution studies. Methods Ecol. Evol. 2015, 7, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Ram, M.R.; Teh, X.; Rajakumar, T.; Goh, K.L.; Leow, A.H.R.; Poh, B.H.; Mariappan, V.; Shankar, E.M.; Loke, M.F.; Vadivelu, J. Polymorphisms in the host CYP2C19 gene and antibiotic-resistance attributes of Helicobacter pyloriisolates influence the outcome of triple therapy. J. Antimicrob. Chemother. 2018, 74, 11–16. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Rana, A.; Sung, W.; Munir, M. Competitiveness of Quantitative Polymerase Chain Reaction (qPCR) and Droplet Digital Polymerase Chain Reaction (ddPCR) Technologies, with a Particular Focus on Detection of Antibiotic Resistance Genes (ARGs). Appl. Microbiol. 2021, 1, 426-444. https://doi.org/10.3390/applmicrobiol1030028
Park S, Rana A, Sung W, Munir M. Competitiveness of Quantitative Polymerase Chain Reaction (qPCR) and Droplet Digital Polymerase Chain Reaction (ddPCR) Technologies, with a Particular Focus on Detection of Antibiotic Resistance Genes (ARGs). Applied Microbiology. 2021; 1(3):426-444. https://doi.org/10.3390/applmicrobiol1030028
Chicago/Turabian StylePark, Sol, Anita Rana, Way Sung, and Mariya Munir. 2021. "Competitiveness of Quantitative Polymerase Chain Reaction (qPCR) and Droplet Digital Polymerase Chain Reaction (ddPCR) Technologies, with a Particular Focus on Detection of Antibiotic Resistance Genes (ARGs)" Applied Microbiology 1, no. 3: 426-444. https://doi.org/10.3390/applmicrobiol1030028
APA StylePark, S., Rana, A., Sung, W., & Munir, M. (2021). Competitiveness of Quantitative Polymerase Chain Reaction (qPCR) and Droplet Digital Polymerase Chain Reaction (ddPCR) Technologies, with a Particular Focus on Detection of Antibiotic Resistance Genes (ARGs). Applied Microbiology, 1(3), 426-444. https://doi.org/10.3390/applmicrobiol1030028