
Tracking Photoinduced Charge Redistribution in a Cu(I) Diimine Donor–Bridge–Acceptor System with Time-Resolved Infrared Spectroscopy

 , , , , and
, , , , and
Abstract

1. Introduction
2. Experimental Methods
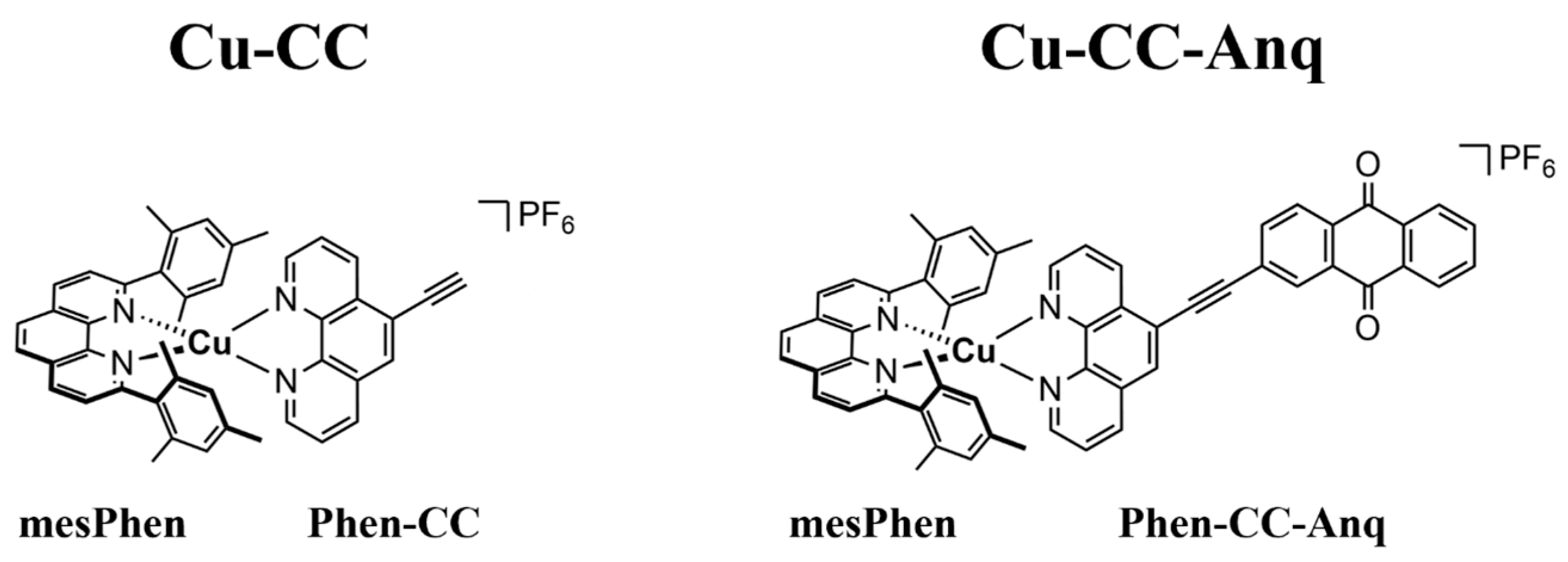
2.1. Sample Preparation
2.2. Spectroscopic Characterization
2.3. Computational Methods
3. Results and Discussion
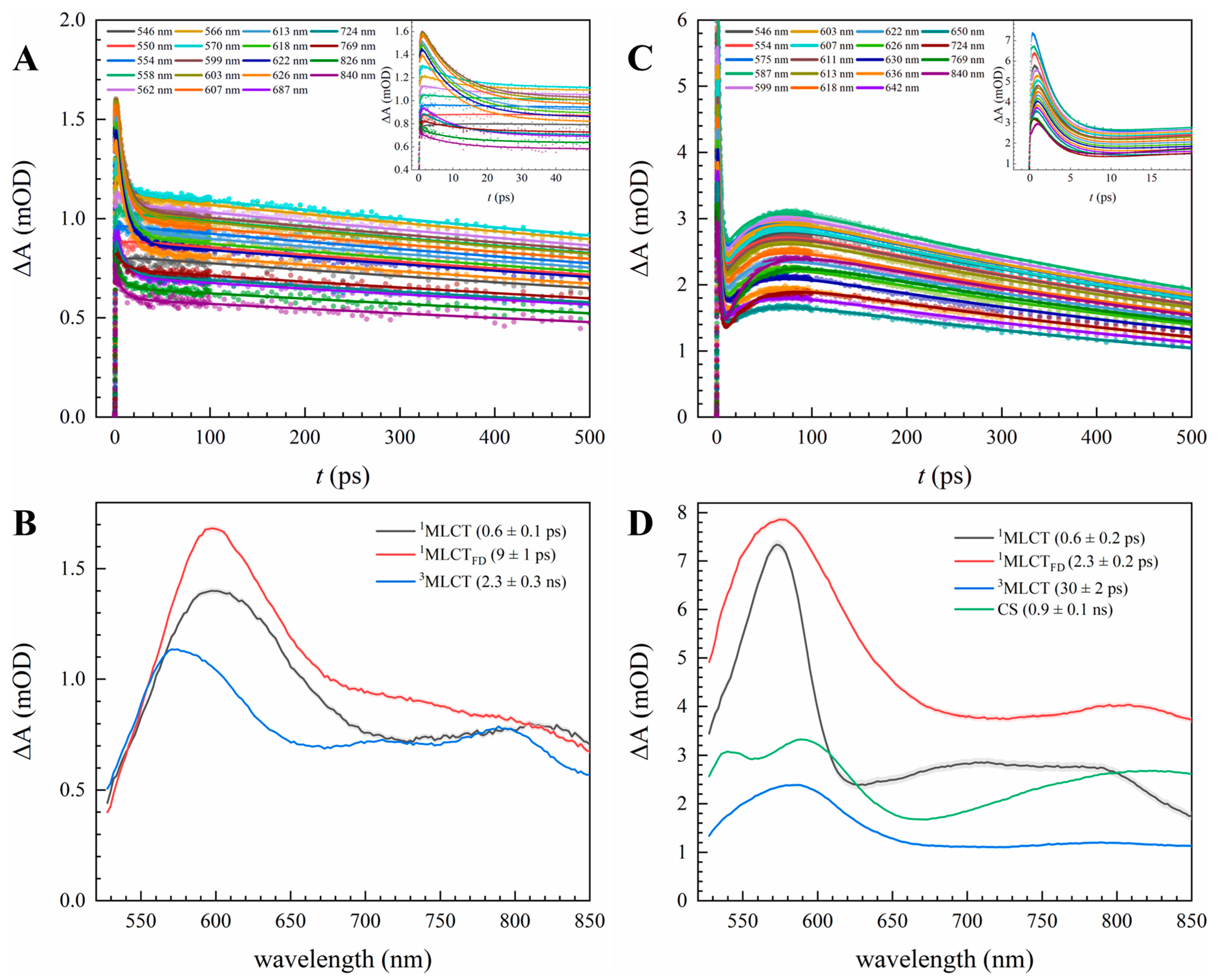
3.1. Optical Characterization of Excited State and PET Dynamics
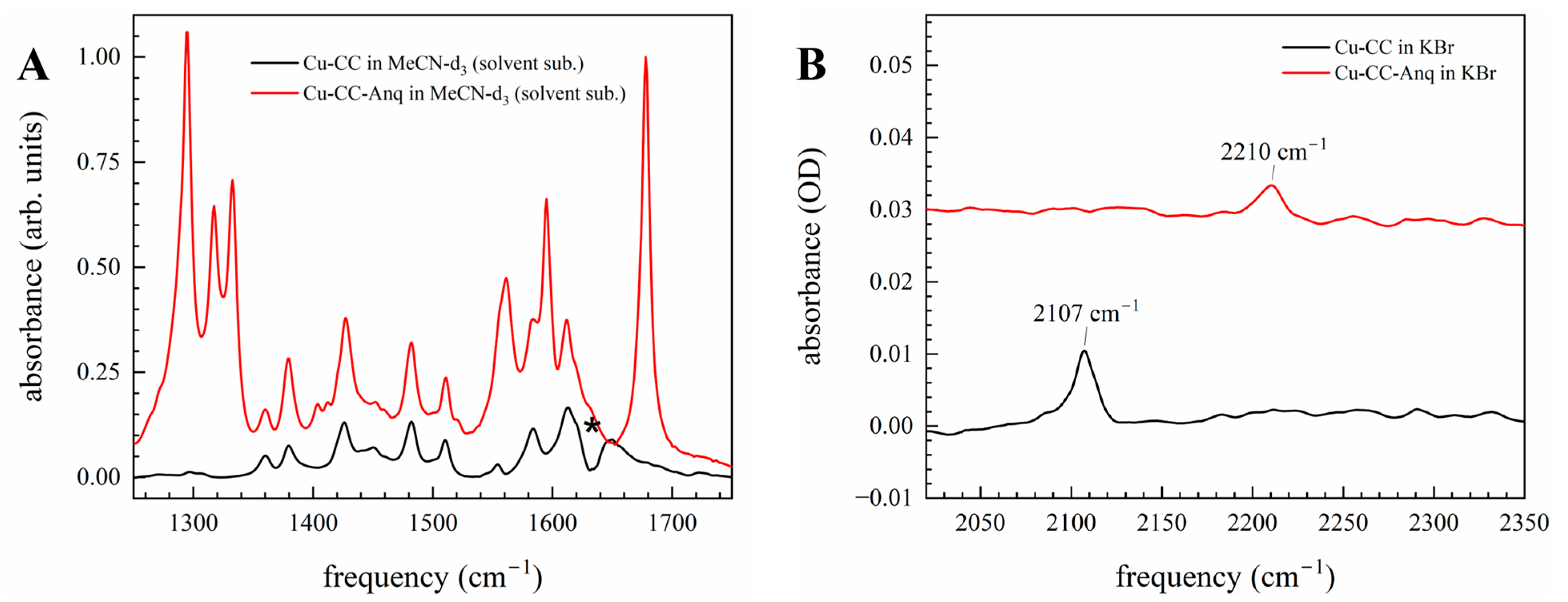
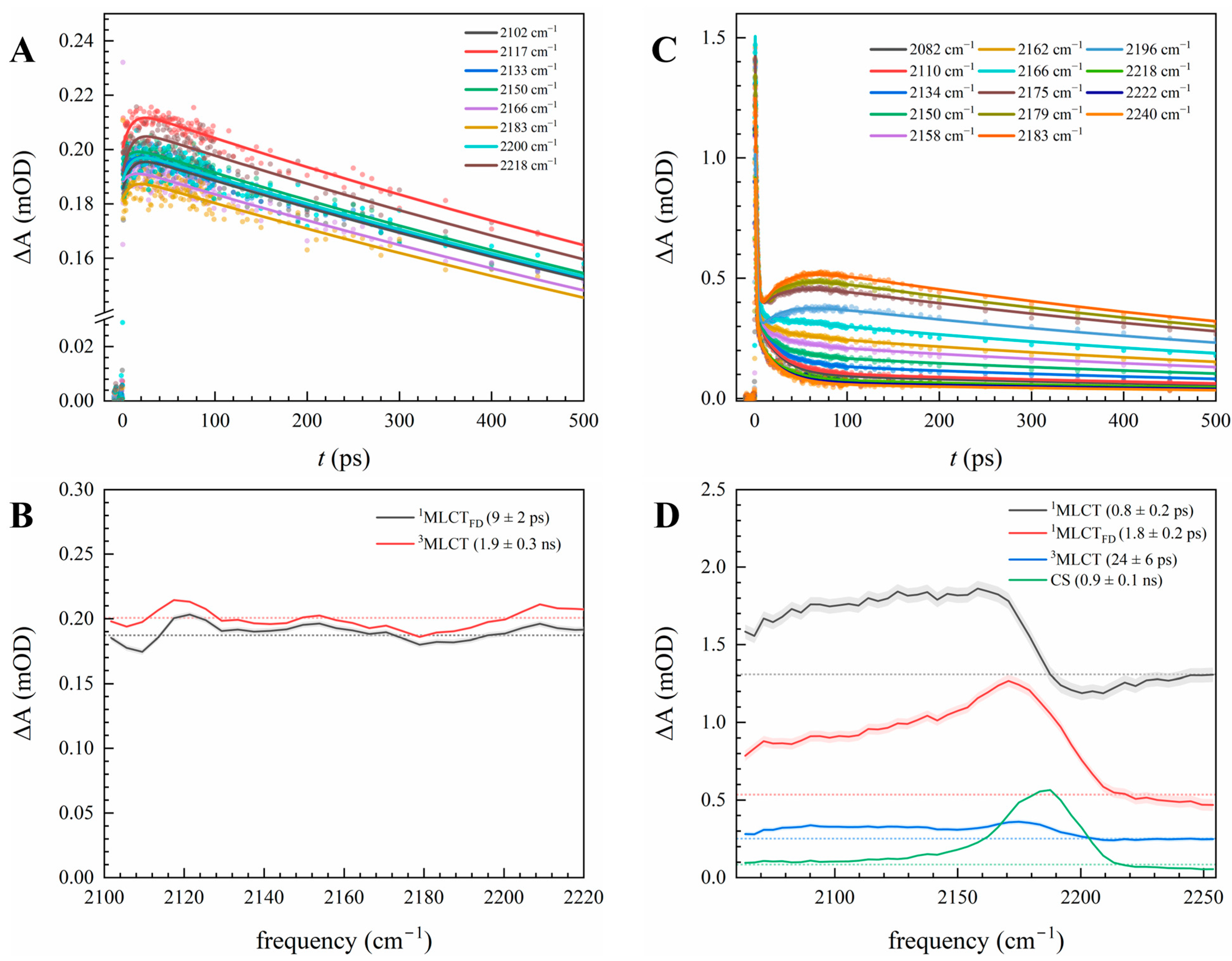
3.2. Electron Delocalization Probed by Infrared Spectroscopy of the Bridge
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Auffèves, A. Quantum Technologies Need a Quantum Energy Initiative. PRX Quantum 2022, 3, 020101. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Wenger, O.S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. [Google Scholar] [CrossRef]
- McCusker, J.K. Electronic structure in the transition metal block and its implications for light harvesting. Science 2019, 363, 484–488. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Constable, E.C. Solar energy conversion using first row d-block metal coordination compound sensitizers and redox mediators. Chem. Sci. 2022, 13, 1225–1262. [Google Scholar] [CrossRef] [PubMed]
- Ruthkosky, M.; Castellano, F.N.; Meyer, G.J. Photodriven Electron and Energy Transfer from Copper Phenanthroline Excited States. Inorg. Chem. 1996, 35, 6406–6412. [Google Scholar] [CrossRef] [PubMed]
- Ruthkosky, M.; Kelly, C.A.; Zaros, M.C.; Meyer, G.J. Long-Lived Charge-Separated States Following Light Excitation of Cu(I) Donor−Acceptor Compounds. J. Am. Chem. Soc. 1997, 119, 12004–12005. [Google Scholar] [CrossRef]
- McMillin, D.R.; Buckner, M.T.; Ahn, B.T. A light-induced redox reaction of bis(2,9-dimethyl-1,10-phenanthroline)copper(I). Inorg. Chem. 1977, 16, 943–945. [Google Scholar] [CrossRef]
- Sakaki, S.; Koga, G.; Ohkubo, K. Successful photocatalytic reduction of methylviologen (MV2+) with [Cu(NN)(PPh3)2]+ (NN = 2,9-dimethyl-1,10-phenanthroline or 4,4’,6,6’-tetramethyl-2,2’-bipyridine) upon near-UV-light irradiation and a novel solvent effect on its catalytic activity. Inorg. Chem. 1986, 25, 2330–2333. [Google Scholar] [CrossRef]
- Sakaki, S.; Koga, G.; Hinokuma, S.; Hashimoto, S.; Okubo, K. Improvement of the photochemical reactivity of [Cu(dmp)P2]+ (dmp = 2,9-dimethyl-1,10-phenanthroline; P = tertiary phosphine) by substituting triphenylphosphine with larger phosphines: Application to photoreduction of methylviologen. Inorg. Chem. 1987, 26, 1817–1819. [Google Scholar] [CrossRef]
- Ruthkosky, M.; Kelly, C.A.; Castellano, F.N.; Meyer, G.J. Electron and energy transfer from Cu(I) MLCT excited states. Coord. Chem. Rev. 1998, 171, 309–322. [Google Scholar] [CrossRef]
- Listorti, A.; Accorsi, G.; Rio, Y.; Armaroli, N.; Moudam, O.; Gégout, A.; Delavaux-Nicot, B.; Holler, M.; Nierengarten, J.-F. Heteroleptic Copper(I) Complexes Coupled with Methano[60]fullerene: Synthesis, Electrochemistry, and Photophysics. Inorg. Chem. 2008, 47, 6254–6261. [Google Scholar] [CrossRef]
- Kirner, S.V.; Guldi, D.M.; Megiatto, J.J.D.; Schuster, D.I. Synthesis and photophysical properties of new catenated electron donor–acceptor materials with magnesium and free base porphyrins as donors and C60 as the acceptor. Nanoscale 2015, 7, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Breddels, P.A.; Blasse, G.; Casadonte, D.J.; McMillin, D.R. Photoelectrochemical Studies of Bis(triphenylphosphine)biquinoline Copper(I), Bis(biquinoline) Copper(I) and Bis(2,9-dimethyl-1,10-phenanthroline) Copper(I) on SnO2 Electrodes. Ber. Bunsenges. Phys. Chem. 1984, 88, 572–578. [Google Scholar] [CrossRef]
- Alonso-Vante, N.; Nierengarten, J.-F.; Sauvage, J.-P. Spectral sensitization of large-band-gap semiconductors (thin films and ceramics) by a carboxylated bis(1,10-phenanthroline)copper(I) complex. J. Chem. Soc. Dalton Trans. 1994, 1649–1654. [Google Scholar] [CrossRef]
- Sakaki, S.; Kuroki, T.; Hamada, T. Synthesis of a new copper(i) complex, [Cu(tmdcbpy)2]+ (tmdcbpy = 4,4′,6,6′-tetramethyl-2,2′-bipyridine-5,5′-dicarboxylic acid), and its application to solar cells. J. Chem. Soc. Dalton Trans. 2002, 840–842. [Google Scholar] [CrossRef]
- Cunningham, C.T.; Moore, J.J.; Cunningham, K.L.H.; Fanwick, P.E.; McMillin, D.R. Structural and Photophysical Studies of Cu(NN)2+ Systems in the Solid State. Emission at Last from Complexes with Simple 1,10-Phenanthroline Ligands. Inorg. Chem. 2000, 39, 3638–3644. [Google Scholar] [CrossRef]
- Mara, M.W.; Fransted, K.A.; Chen, L.X. Interplays of excited state structures and dynamics in copper(I) diimine complexes: Implications and perspectives. Coord. Chem. Rev. 2015, 282–283, 2–18. [Google Scholar] [CrossRef]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Ultrafast Excited-State Dynamics of Copper(I) Complexes. Acc. Chem. Res. 2015, 48, 782–791. [Google Scholar] [CrossRef]
- Siddique, Z.A.; Yamamoto, Y.; Ohno, T.; Nozaki, K. Structure-Dependent Photophysical Properties of Singlet and Triplet Metal-to-Ligand Charge Transfer States in Copper(I) Bis(diimine) Compounds. Inorg. Chem. 2003, 42, 6366–6378. [Google Scholar] [CrossRef]
- McMillin, D.R.; Kirchhoff, J.R.; Goodwin, K.V. Exciplex quenching of photo-excited copper complexes. Coord. Chem. Rev. 1985, 64, 83–92. [Google Scholar] [CrossRef]
- Chen, L.X.; Shaw, G.B.; Novozhilova, I.; Liu, T.; Jennings, G.; Attenkofer, K.; Meyer, G.J.; Coppens, P. MLCT State Structure and Dynamics of a Copper(I) Diimine Complex Characterized by Pump−Probe X-ray and Laser Spectroscopies and DFT Calculations. J. Am. Chem. Soc. 2003, 125, 7022–7034. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.B.; Grant, C.D.; Shirota, H.; Castner, E.W.; Meyer, G.J.; Chen, L.X. Ultrafast Structural Rearrangements in the MLCT Excited State for Copper(I) bis-Phenanthrolines in Solution. J. Am. Chem. Soc. 2007, 129, 2147–2160. [Google Scholar] [CrossRef]
- Eggleston, M.K.; McMillin, D.R.; Koenig, K.S.; Pallenberg, A.J. Steric Effects in the Ground and Excited States of Cu(NN)2+ Systems. Inorg. Chem. 1997, 36, 172–176. [Google Scholar] [CrossRef]
- Castellano, F.N.; Rosko, M.C. Steric and Electronic Influence of Excited-State Decay in Cu(I) MLCT Chromophores. Acc. Chem. Res. 2024, 57, 2872–2886. [Google Scholar] [CrossRef]
- Queffélec, C.; Pati, P.B.; Pellegrin, Y. Fifty Shades of Phenanthroline: Synthesis Strategies to Functionalize 1,10-Phenanthroline in All Positions. Chem. Rev. 2024, 124, 6700–6902. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, Z.-L.; Phelan, B.T.; Lynch, V.M.; Chen, L.X.; Mulfort, K.L. Changing Directions: Influence of Ligand Electronics on the Directionality and Kinetics of Photoinduced Charge Transfer in Cu(I)Diimine Complexes. Inorg. Chem. 2023, 62, 14368–14376. [Google Scholar] [CrossRef]
- Rosko, M.C.; Espinoza, E.M.; Arteta, S.; Kromer, S.; Wheeler, J.P.; Castellano, F.N. Employing Long-Range Inductive Effects to Modulate Metal-to-Ligand Charge Transfer Photoluminescence in Homoleptic Cu(I) Complexes. Inorg. Chem. 2023, 62, 3248–3259. [Google Scholar] [CrossRef]
- Garakyaraghi, S.; McCusker, C.E.; Khan, S.; Koutnik, P.; Bui, A.T.; Castellano, F.N. Enhancing the Visible-Light Absorption and Excited-State Properties of Cu(I) MLCT Excited States. Inorg. Chem. 2018, 57, 2296–2307. [Google Scholar] [CrossRef]
- Rosko, M.C.; Wheeler, J.P.; Alameh, R.; Faulkner, A.P.; Durand, N.; Castellano, F.N. Enhanced Visible Light Absorption in Heteroleptic Cuprous Phenanthrolines. Inorg. Chem. 2024, 63, 1692–1701. [Google Scholar] [CrossRef]
- Schmittel, M.; Ganz, A. Stable mixed phenanthroline copper(I) complexes. Key building blocks for supramolecular coordination chemistry. Chem. Commun. 1997, 999–1000. [Google Scholar] [CrossRef]
- Schmittel, M.; Lüning, U.; Meder, M.; Ganz, A.; Michel, C.; Herderich, M. Synthesis of Sterically Encumbered 2,9-Diaryl Substituted Phenanthrolines. Key Building Blocks for the Preparation of Mixed (Bis-Heteroleptic) Phenanthroline Copper(I) Complexes. Heterocycl. Commun. 1997, 3, 493–498. [Google Scholar] [CrossRef]
- Kohler, L.; Hayes, D.; Hong, J.; Carter, T.J.; Shelby, M.L.; Fransted, K.A.; Chen, L.X.; Mulfort, K.L. Synthesis, structure, ultrafast kinetics, and light-induced dynamics of CuHETPHEN chromophores. Dalton Trans. 2016, 45, 9871–9883. [Google Scholar] [CrossRef]
- Hayes, D.; Kohler, L.; Chen, L.X.; Mulfort, K.L. Ligand Mediation of Vectorial Charge Transfer in Cu(I)diimine Chromophore–Acceptor Dyads. J. Phys. Chem. Lett. 2018, 9, 2070–2076. [Google Scholar] [CrossRef]
- Phelan, B.T.; Xie, Z.-L.; Liu, X.; Li, X.; Mulfort, K.L.; Chen, L.X. Photodriven electron-transfer dynamics in a series of heteroleptic Cu(I)–anthraquinone dyads. J. Chem. Phys. 2024, 160, 144905. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Lawrence, C.M.; Xiao, D.; Kireev, V.V.; Skourtis, S.S.; Sessler, J.L.; Beratan, D.N.; Rubtsov, I.V. Modulating Unimolecular Charge Transfer by Exciting Bridge Vibrations. J. Am. Chem. Soc. 2009, 131, 18060–18062. [Google Scholar] [CrossRef]
- Delor, M.; Keane, T.; Scattergood, P.A.; Sazanovich, I.V.; Greetham, G.M.; Towrie, M.; Meijer, A.J.H.M.; Weinstein, J.A. On the mechanism of vibrational control of light-induced charge transfer in donor–bridge–acceptor assemblies. Nat. Chem. 2015, 7, 689–695. [Google Scholar] [CrossRef]
- Yue, Y.; Grusenmeyer, T.; Ma, Z.; Zhang, P.; Schmehl, R.H.; Beratan, D.N.; Rubtsov, I.V. Electron transfer rate modulation in a compact Re(i) donor–acceptor complex. Dalton Trans. 2015, 44, 8609–8616. [Google Scholar] [CrossRef]
- Delor, M.; Archer, S.A.; Keane, T.; Meijer, A.J.H.M.; Sazanovich, I.V.; Greetham, G.M.; Towrie, M.; Weinstein, J.A. Directing the path of light-induced electron transfer at a molecular fork using vibrational excitation. Nat. Chem. 2017, 9, 1099–1104. [Google Scholar] [CrossRef]
- Wintergerst, P.; Mengele, A.K.; Nauroozi, D.; Tschierlei, S.; Rau, S. Impact of Alkyne Functionalization on Photophysical and Electrochemical Properties of 1,10-Phenanthrolines and Their RuII Complexes. Eur. J. Inorg. Chem. 2019, 2019, 1988–1992. [Google Scholar] [CrossRef]
- Xie, Z.-L.; Liu, X.; Valentine, A.J.S.; Lynch, V.M.; Tiede, D.M.; Li, X.; Mulfort, K.L. Bimetallic Copper/Ruthenium/Osmium Complexes: Observation of Conformational Differences Between the Solution Phase and Solid State by Atomic Pair Distribution Function Analysis. Angew. Chem. Int. Ed. 2022, 61, e202111764. [Google Scholar] [CrossRef]
- Pellegrin, Y.; Sandroni, M.; Blart, E.; Planchat, A.; Evain, M.; Bera, N.C.; Kayanuma, M.; Sliwa, M.; Rebarz, M.; Poizat, O.; et al. New Heteroleptic Bis-Phenanthroline Copper(I) Complexes with Dipyridophenazine or Imidazole Fused Phenanthroline Ligands: Spectral, Electrochemical, and Quantum Chemical Studies. Inorg. Chem. 2011, 50, 11309–11322. [Google Scholar] [CrossRef] [PubMed]
- Sandroni, M.; Kayanuma, M.; Planchat, A.; Szuwarski, N.; Blart, E.; Pellegrin, Y.; Daniel, C.; Boujtita, M.; Odobel, F. First application of the HETPHEN concept to new heteroleptic bis(diimine) copper(i) complexes as sensitizers in dye sensitized solar cells. Dalton Trans. 2013, 42, 10818–10827. [Google Scholar] [CrossRef] [PubMed]
- Singh, Z.; Kamal, S.; Majewski, M.B. Copper(I) Donor–Chromophore–Acceptor Assembly for Light-Driven Oxidation on a Zinc Oxide Nanowire Electrode. J. Phys. Chem. C 2022, 126, 16732–16743. [Google Scholar] [CrossRef]
- Gaussian Developmental, Version Rev. J.14+; Gaussian, Inc.: Wallingford, CT, USA, 2020.
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Boettger, J.C. Approximate two-electron spin-orbit coupling term for density-functional-theory DFT calculations using the Douglas-Kroll-Hess transformation. Phys. Rev. B 2000, 62, 7809–7815. [Google Scholar] [CrossRef]
- Liao, C.; Kasper, J.M.; Jenkins, A.J.; Yang, P.; Batista, E.R.; Frisch, M.J.; Li, X. State Interaction Linear Response Time-Dependent Density Functional Theory with Perturbative Spin–Orbit Coupling: Benchmark and Perspectives. JACS Au 2023, 3, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Ehrman, J.; Martinez-Baez, E.; Jenkins, A.J.; Li, X. Improving One-Electron Exact-Two-Component Relativistic Methods with the Dirac–Coulomb–Breit-Parameterized Effective Spin–Orbit Coupling. J. Chem. Theory Comput. 2023, 19, 5785–5790. [Google Scholar] [CrossRef]
- Englman, R.; Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar] [CrossRef]
- Jortner, J.; Rice, S.A.; Hochstrasser, R.M. Radiationless Transitions in Photochemistry. In Advances in Photochemistry; Wiley: Hoboken, NJ, USA, 1969; pp. 149–309. [Google Scholar]
- Gutmann, V. Principles of Coordination Chemistry in Non-Aqueous Solutions; Springer: New York, NY, USA, 1968. [Google Scholar]
- Büschel, M.; Stadler, C.; Lambert, C.; Beck, M.; Daub, J. Heterocyclic quinones as core units for redox switches: UV–vis/NIR, FTIR spectroelectrochemistry and DFT calculations on the vibrational and electronic structure of the radical anions. J. Electroanal. Chem. 2000, 484, 24–32. [Google Scholar] [CrossRef]
- Larsen, C.B.; Farrow, G.A.; Smith, L.D.; Appleby, M.V.; Chekulaev, D.; Weinstein, J.A.; Wenger, O.S. Solvent-Mediated Activation/Deactivation of Photoinduced Electron-Transfer in a Molecular Dyad. Inorg. Chem. 2020, 59, 10430–10438. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Smith, B.C. An IR spectral interpretation potpourri: Carbohydrates and alkynes. Spectroscopy 2017, 32, 18–24. [Google Scholar]
- Vauthey, E. Watching Excited-State Symmetry Breaking in Multibranched Push–Pull Molecules. J. Phys. Chem. Lett. 2022, 13, 2064–2071. [Google Scholar] [CrossRef] [PubMed]
- Dereka, B.; Svechkarev, D.; Rosspeintner, A.; Aster, A.; Lunzer, M.; Liska, R.; Mohs, A.M.; Vauthey, E. Solvent tuning of photochemistry upon excited-state symmetry breaking. Nat. Commun. 2020, 11, 1925. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Hedström, S.; Méndez-Hernández, D.D.; Hendrickson, H.P.; Jung, K.A.; Ho, J.; Batista, V.S. Electron Transfer Assisted by Vibronic Coupling from Multiple Modes. J. Chem. Theory Comput. 2017, 13, 6000–6009. [Google Scholar] [CrossRef]
- Ulstrup, J.; Jortner, J. The effect of intramolecular quantum modes on free energy relationships for electron transfer reactions. J. Chem. Phys. 1975, 63, 4358–4368. [Google Scholar] [CrossRef]
- Jortner, J.; Bixon, M. Intramolecular vibrational excitations accompanying solvent-controlled electron transfer reactions. J. Chem. Phys. 1988, 88, 167–170. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Frequency (cm−1) | IR Intensity (arb. Units) | Bond Length (Å) |
|---|---|---|---|
| DFT | |||
| S0 | 2230 | 1.0 | 1.214 |
| S1 (531 nm) | 2233 | 0.9 | 1.214 |
| Experiment | |||
| S0 | 2107.1 ± 0.1 | — | — |
| S1 | 2107.1 ± 0.1 1 | — | — |
| State | Frequency (cm−1) | IR Intensity (arb. Units) | Bond Length (Å) |
|---|---|---|---|
| DFT | |||
| S0 | 2332 | 34.4 | 1.219 |
| S1 (590 nm) | 2257 | 12,830.0 | 1.226 |
| S2 (531 nm) | 2335 | 0.2 | 1.218 |
| Experiment | |||
| S0 | 2211.5 ± 0.1 | — | — |
| 1MLCT | 2170.2 ± 0.4 | — | — |
| 3MLCT | 2173.6 ± 0.4 | — | — |
| CS | 2183.7 ± 0.3 | — | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roget, S.A.; Henke, W.C.; Taub, M.; Kim, P.; Yarranton, J.T.; Li, X.; Mulfort, K.L.; Chen, L.X. Tracking Photoinduced Charge Redistribution in a Cu(I) Diimine Donor–Bridge–Acceptor System with Time-Resolved Infrared Spectroscopy. Photochem 2025, 5, 16. https://doi.org/10.3390/photochem5020016
Roget SA, Henke WC, Taub M, Kim P, Yarranton JT, Li X, Mulfort KL, Chen LX. Tracking Photoinduced Charge Redistribution in a Cu(I) Diimine Donor–Bridge–Acceptor System with Time-Resolved Infrared Spectroscopy. Photochem. 2025; 5(2):16. https://doi.org/10.3390/photochem5020016
Chicago/Turabian StyleRoget, Sean A., Wade C. Henke, Maxwell Taub, Pyosang Kim, Jonathan T. Yarranton, Xiaosong Li, Karen L. Mulfort, and Lin X. Chen. 2025. "Tracking Photoinduced Charge Redistribution in a Cu(I) Diimine Donor–Bridge–Acceptor System with Time-Resolved Infrared Spectroscopy" Photochem 5, no. 2: 16. https://doi.org/10.3390/photochem5020016
APA StyleRoget, S. A., Henke, W. C., Taub, M., Kim, P., Yarranton, J. T., Li, X., Mulfort, K. L., & Chen, L. X. (2025). Tracking Photoinduced Charge Redistribution in a Cu(I) Diimine Donor–Bridge–Acceptor System with Time-Resolved Infrared Spectroscopy. Photochem, 5(2), 16. https://doi.org/10.3390/photochem5020016