
Computational-Chemistry-Based Prediction of Near-Infrared Rhodamine Fluorescence Peaks with Sub-12 nm Accuracy


Abstract

1. Introduction
2. Computational Methods
3. Results and Discussion
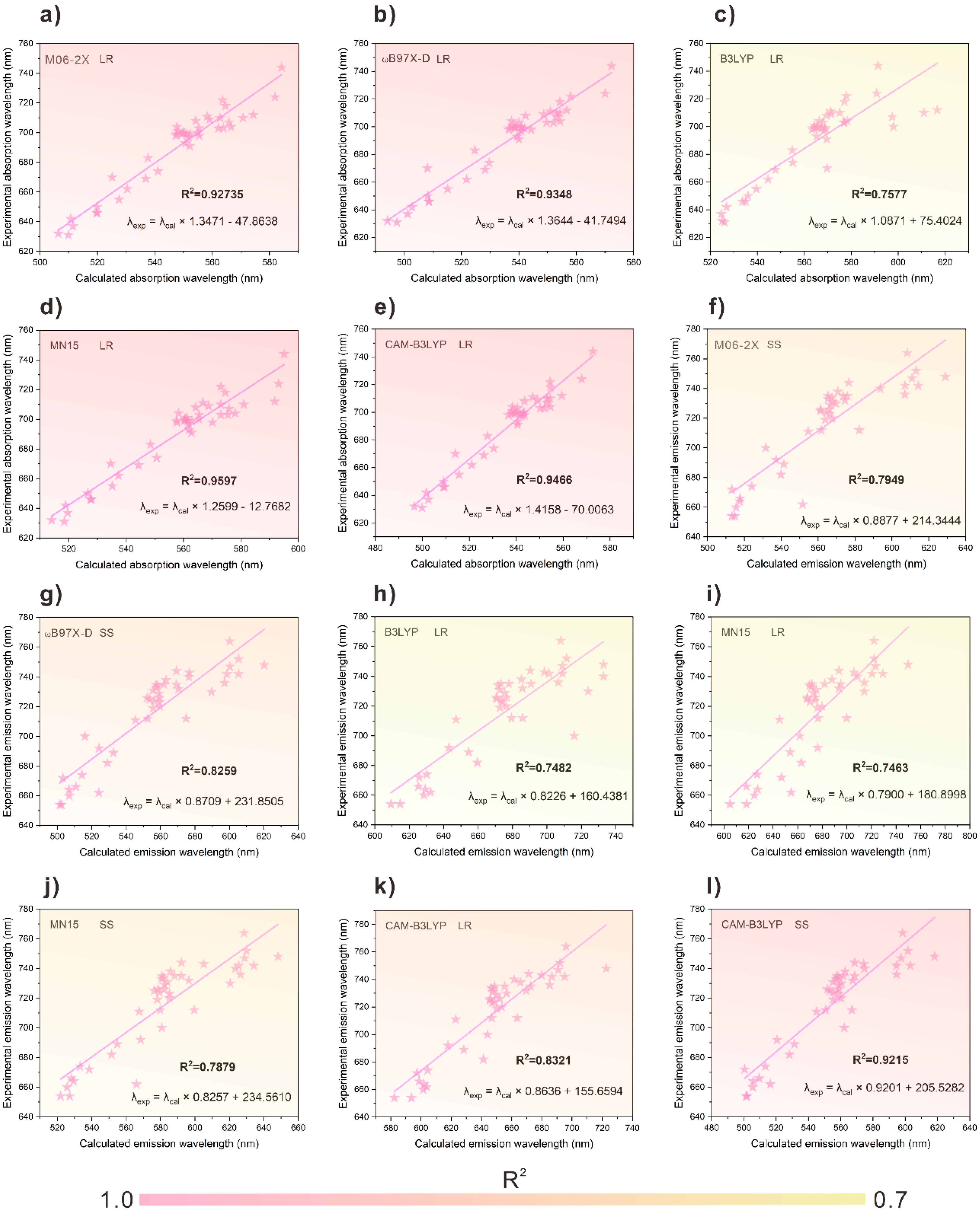
3.1. Calculating the Wavelength of Dyes
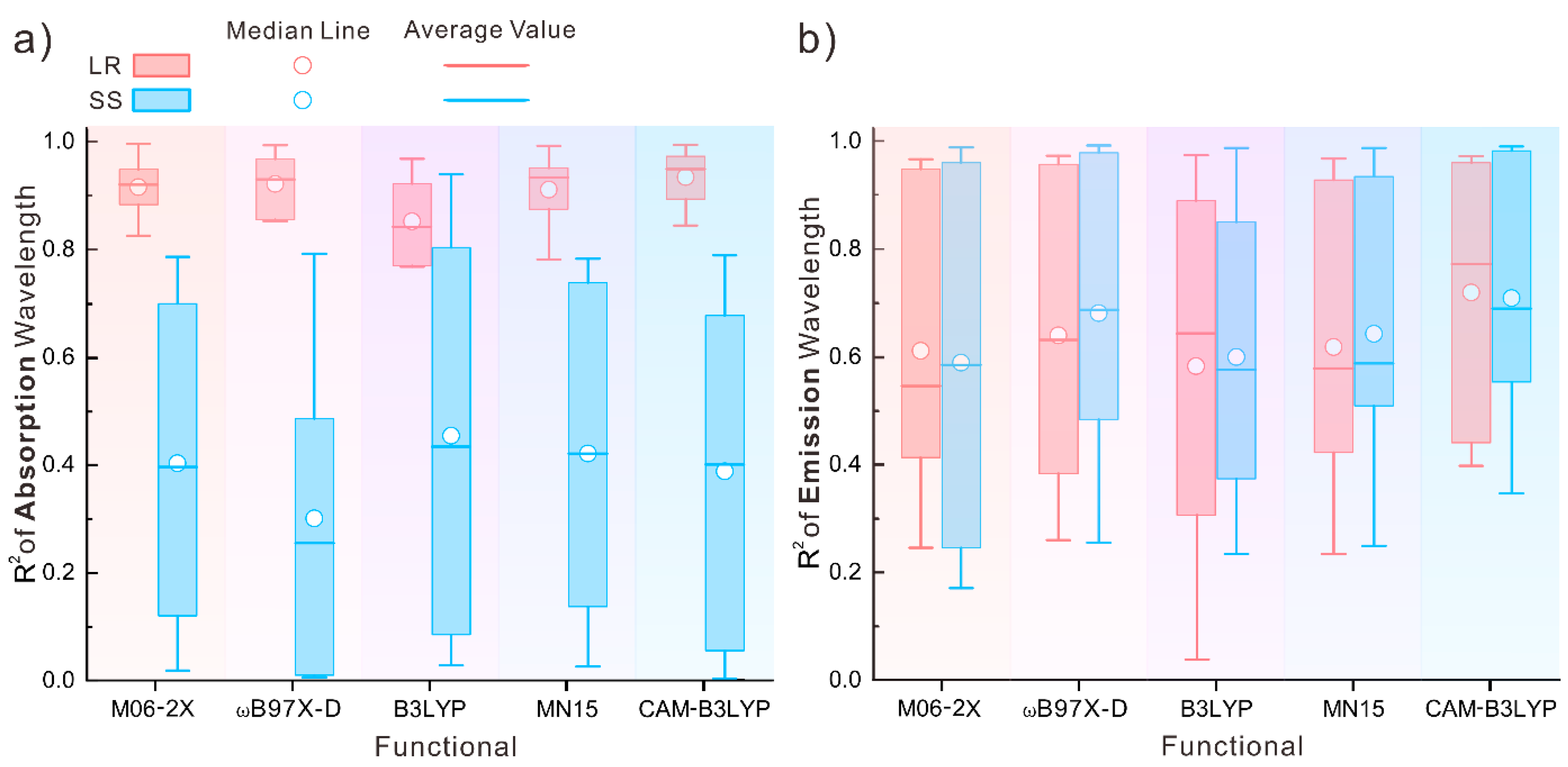
3.2. Establishing a λcal.–λexp. Relationship
3.3. Selection of Wavelength Prediction Model
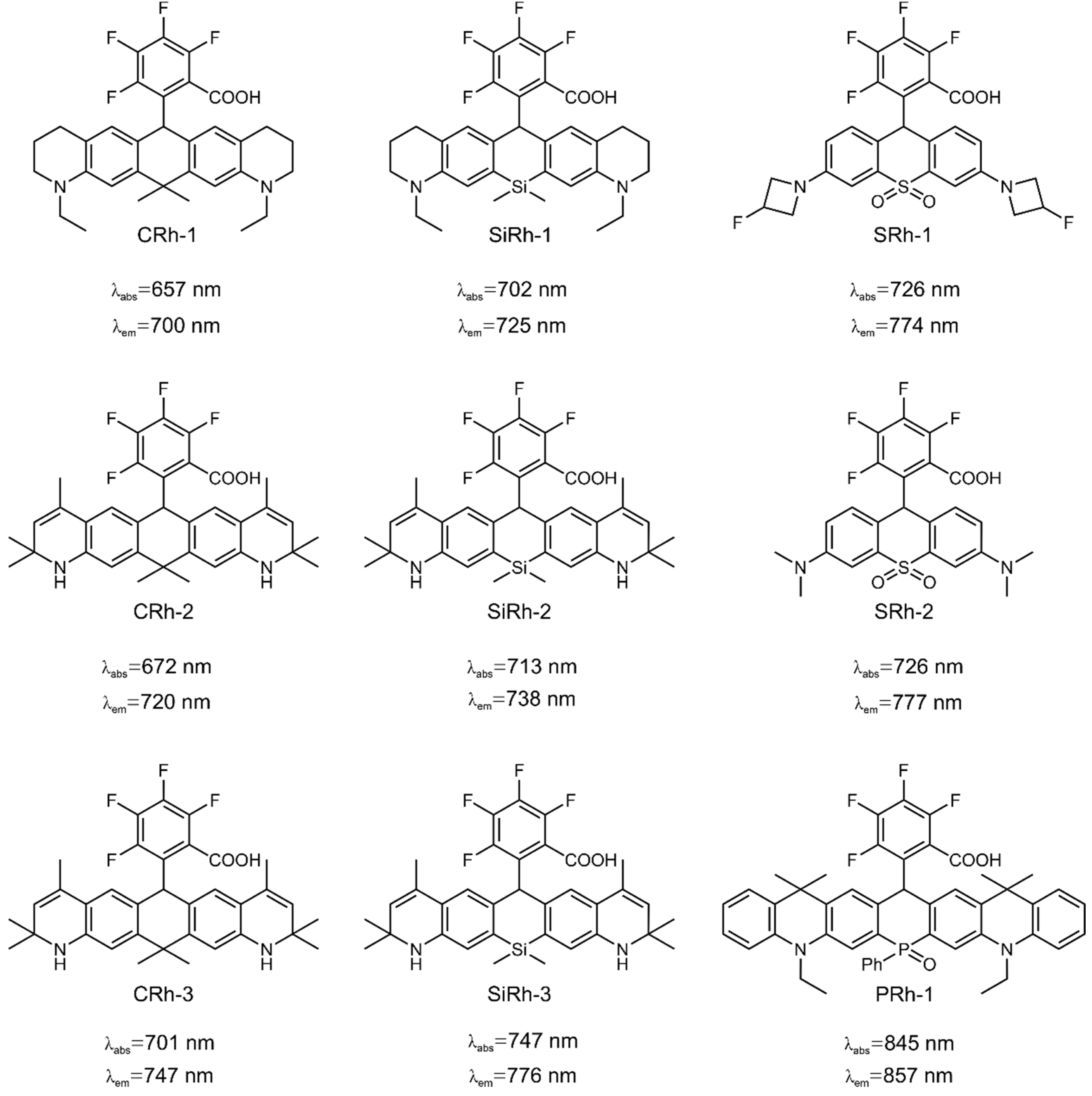
3.4. Computational Design and Screening of Near-Infrared Rhodamines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koide, Y.; Urano, Y.; Hanaoka, K.; Piao, W.; Kusakabe, M.; Saito, N.; Terai, T.; Okabe, T.; Nagano, T. Development of NIR fluorescent dyes based on Si–rhodamine for in vivo imaging. J. Am. Chem. Soc. 2012, 134, 5029–5031. [Google Scholar] [CrossRef]
- Li, H.; Kim, Y.; Jung, H.; Hyun, J.Y.; Shin, I. Near-infrared (NIR) fluorescence-emitting small organic molecules for cancer imaging and therapy. Chem. Soc. Rev. 2022, 51, 8957–9008. [Google Scholar] [CrossRef]
- Feng, Z.; Tang, T.; Wu, T.; Yu, X.; Zhang, Y.; Wang, M.; Zheng, J.; Ying, Y.; Chen, S.; Zhou, J.; et al. Perfecting and extending the near-infrared imaging window. Light Sci. Appl. 2021, 10, 197. [Google Scholar] [CrossRef]
- Usama, S.M.; Inagaki, F.; Kobayashi, H.; Schnermann, M.J. Norcyanine-carbamates are versatile near-infrared fluorogenic probes. J. Am. Chem. Soc. 2021, 143, 5674–5679. [Google Scholar] [CrossRef]
- Dong, M.; Wang, D.; Yang, J.; Sun, P.; Ding, W.; Yang, J.; Yan, J.; Chi, W.J. Computational chemistry-assisted design of hydrazine-based fluorescent molecular rotor for viscosity sensors. Smart Mol. 2023, 1, e20230011. [Google Scholar] [CrossRef]
- Yang, X.Z.; Yao, S.; Wu, J.; Diao, J.; He, W.; Guo, Z.; Chen, Y.J. Recent advances in single fluorescent probes for monitoring dual organelles in two channels. Smart Mol. 2024, 2, e20240040. [Google Scholar] [CrossRef]
- Duo, Y.Y.; Zhao, L.; Wang, Z.G.; Liu, S.L. NIR-II fluorophores: From synthesis to biological applications. J. Anal. Test. 2023, 7, 245–259. [Google Scholar] [CrossRef]
- Wang, L.; Du, W.; Hu, Z.; Uvdal, K.; Li, L.; Huang, W. Hybrid rhodamine fluorophores in the visible/nir region for biological imaging. Angew. Chem. Int. Ed. 2019, 58, 14026–14043. [Google Scholar] [CrossRef]
- Grimm, J.B.; Tkachuk, A.N.; Xie, L.; Choi, H.; Mohar, B.; Falco, N.; Schaefer, K.; Patel, R.; Zheng, Q.; Liu, Z.; et al. A general method to optimize and functionalize red-shifted rhodamine dyes. Nat. Methods 2020, 17, 815–821. [Google Scholar] [CrossRef]
- Ye, Z.; Yu, H.; Yang, W.; Zheng, Y.; Li, N.; Bian, H.; Wang, Z.; Liu, Q.; Song, Y.; Zhang, M.; et al. Strategy to lengthen the on-time of photochromic rhodamine spirolactam for super-resolution photoactivated localization microscopy. J. Am. Chem. Soc. 2019, 141, 6527–6536. [Google Scholar] [CrossRef]
- Zhang, J.; Yue, C.; Ke, Y.; Qu, H.; Zeng, L. Fluorescent probes for the detection of biogenic amines, nitrite and sulfite in food: Progress, challenges and perspective. Adv. Agrochem 2023, 2, 127–141. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Xie, W.; Gu, M.; Mao, G.; Yang, S.J. Recent progress in endoplasmic reticulum-targetable small-molecule probes for fluorescence sensing and phototherapy. J. Anal. Test. 2023, 7, 304–324. [Google Scholar] [CrossRef]
- Fu, M.; Xiao, Y.; Qian, X.; Zhao, D.; Xu, Y. A design concept of long-wavelength fluorescent analogs of rhodamine dyes: Replacement of oxygen with silicon atom. Chem. Commun. 2008, 15, 1780–1782. [Google Scholar] [CrossRef]
- Zhou, P.; Ning, C.; Alsaedi, A.; Han, K. The effects of heteroatoms Si and S on tuning the optical properties of rhodamine- and fluorescein-based fluorescence probes: A theoretical analysis. ChemPhysChem 2016, 17, 3139–3145. [Google Scholar] [CrossRef]
- Chai, X.; Cui, X.; Wang, B.; Yang, F.; Cai, Y.; Wu, Q.; Wang, T. Near-infrared phosphorus-substituted rhodamine with emission wavelength above 700 nm for bioimaging. Chem. Eur. J. 2015, 21, 16754–16758. [Google Scholar] [CrossRef]
- Gong, Y.J.; Zhang, X.B.; Mao, G.J.; Su, L.; Meng, H.M.; Tan, W.; Feng, S.; Zhang, G. A unique approach toward near-infrared fluorescent probes for bioimaging with remarkably enhanced contrast. Chem. Sci. 2016, 7, 2275–2285. [Google Scholar] [CrossRef]
- McCann, T.E.; Kosaka, N.; Koide, Y.; Mitsunaga, M.; Choyke, P.L.; Nagano, T.; Urano, Y.; Kobayashi, H. Activatable optical imaging with a Silica-rhodamine based Near Infrared (SiR700) fluorophore: A comparison with cyanine based dyes. Bioconjug. Chem. 2011, 22, 2531–2538. [Google Scholar] [CrossRef]
- Grzybowski, M.; Taki, M.; Senda, K.; Sato, Y.; Ariyoshi, T.; Okada, Y.; Kawakami, R.; Imamura, T.; Yamaguchi, S. A highly photostable near-infrared labeling agent based on a phospha-rhodamine for long-term and deep imaging. Angew. Chem. Int. Ed. 2018, 57, 10137–10141. [Google Scholar] [CrossRef]
- Liu, J.; Sun, Y.Q.; Zhang, H.; Shi, H.; Shi, Y.; Guo, W. Sulfone-rhodamines: A new class of near-infrared fluorescent dyes for bioimaging. ACS Appl. Mater. Interfaces 2016, 8, 22953–22962. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; He, Z.; Zhao, Y.; Yang, Y.; Shi, W.; Li, X.; Ma, H. Xanthene-based NIR-II dyes for in vivo dynamic imaging of blood circulation. J. Am. Chem. Soc. 2021, 143, 17136–17143. [Google Scholar] [CrossRef] [PubMed]
- Kulesza, A.J.; Titov, E.; Daly, S.; Włodarczyk, R.; Megow, J.; Saalfrank, P.; Choi, C.M.; MacAleese, L.; Antoine, R.; Dugourd, P. Excited States of Xanthene Analogues: Photofragmentation and Calculations by CC2 and Time-Dependent Density Functional Theory. ChemPhysChem 2016, 17, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Moore, B., II; Schrader, R.L.; Kowalski, K.; Autschbach, J. Electronic π-to-π* Excitations of Rhodamine Dyes Exhibit a Time-Dependent Kohn–Sham Theory “Cyanine Problem”. ChemistryOpen 2017, 6, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P. Why the lowest electronic excitations of rhodamines are overestimated by time-dependent density functional theory. Int. J. Quantum Chem. 2018, 118, e25780. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, version C; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Grabarz, A.M.; Ośmiałowski, B. Benchmarking Density Functional Approximations for Excited-State Properties of Fluorescent Dyes. Molecules 2021, 26, 7434. [Google Scholar] [CrossRef]
- Chi, W.J.; Qi, Q.L.; Wang, C.; Zheng, J.; Zhou, W.; Xu, N.; Wu, X.; Jiang, X.; Tan, D.; Xu, Z.C.; et al. Descriptor ΔGC-O Enables the Quantitative Design of Spontaneously Blinking Rhodamines for Live-Cell Super-Resolution Imaging. Angew. Chem. Int. Ed. 2020, 59, 20215–20223. [Google Scholar] [CrossRef]
- Chi, W.J.; Qi, Q.L.; Lee, R.; Xu, Z.C.; Liu, X.G. A Unified Push–Pull Model for Understanding the Ring-Opening Mechanism of Rhodamine Dyes. J. Phys. Chem. C 2020, 124, 3793–3801. [Google Scholar] [CrossRef]
- Wang, C.; Qiao, Q.L.; Chi, W.J.; Chen, J.; Liu, W.; Tan, D.; McKechnie, S.; Lyu, D.; Jiang, X.F.; Zhou, W.; et al. Quantitative Design of Bright Fluorophores and AIEgens by the Accurate Prediction of Twisted Intramolecular Charge Transfer (TICT). Angew. Chem. Int. Ed. 2020, 59, 10160–10172. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Casida, M.E.; Huix-Rotllant, M. Progress in time-dependent density-functional theory. Annu. Rev. Phys. Chem. 2012, 63, 287–323. [Google Scholar] [CrossRef]
- Dhbaibi, K.; Morgante, P.; Vanthuyne, N.; Autschbach, J.; Favereau, L.; Crassous, J. Low-temperature luminescence in organic helicenes: Singlet versus triplet state circularly polarized emission. J. Phys. Chem. Lett. 2023, 14, 1073–1081. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, W.; Reinholdt, P.; Jensen, H.J.A.; Kongsted, J. Multiconfigurational SCF and Short-Range DFT Combined with Polarizable Density Embedding: Comparison of Linear-Response and State-Specific Solvatochromic Shifts of Acrolein and Para-nitrophenolate in Water. J. Chem. Theory Comput. 2022, 18, 6231–6239. [Google Scholar] [CrossRef] [PubMed]
- Cipolloni, M.; Fresch, B.; Occhiuto, I.; Rukin, P.; Komarova, K.G.; Cecconello, A.; Willner, I.; Levine, R.D.; Remacle, F.; Collini, E. Coherent electronic and nuclear dynamics in a rhodamine heterodimer–DNA supramolecular complex. Phys. Chem. Chem. Phys. 2017, 19, 23043–23051. [Google Scholar] [CrossRef]
- Selenius, E.; Sigurdarson, A.E.; Schmerwitz, Y.L.; Levi, G. Orbital-optimized versus time-dependent density functional calculations of intramolecular charge transfer excited states. J. Chem. Theory Comput. 2024, 20, 3809–3822. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef]
- Paul, B.K.; Rakshit, P.A. Computational assessment of the O−H···O intramolecular hydrogen-bonding in substituted phenalenes: Diverse degrees of covalence. Monatsh. Chem. 2023, 154, 605–613. [Google Scholar] [CrossRef]
- Yuan, Q.L.; Ma, M.Y.; Wang, M.Y.; Sun, P.P.; Xiao, X.Q.; Chi, W.J. Determining the Recruiting Rate of Spontaneously Blinking Rhodamines by Density Functional Calculations. J. Phys. Chem. A 2025, 129, 667–676. [Google Scholar] [CrossRef]
- Ren, X.; Wang, C.; Wu, X.; Rong, M.; Huang, R.; Liang, Q.; Shen, T.; Sun, H.; Zhang, R.; Zhang, Z.; et al. Auxochrome Dimethyl-Dihydroacridine Improves Fluorophores for Prolonged Live-Cell Super-Resolution Imaging. J. Am. Chem. Soc. 2024, 146, 6566–6579. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M062X | WB97XD | B3LYP | MN15 | CAM-B3LYP | |||
|---|---|---|---|---|---|---|---|
| MAE | Abs | LR | 142.17 | 153.59 | 124.61 | 132.10 | 153.07 |
| SS | 250.04 | 254.93 | 241.15 | 241.15 | 256.27 | ||
| Em | LR | 50.83 | 67.63 | 40.80 | 38.74 | 67.24 | |
| SS | 150.98 | 160.13 | 128.17 | 132.99 | 161.24 |
| M062X | WB97XD | B3LYP | MN15 | CAM-B3LYP | |||
|---|---|---|---|---|---|---|---|
| MAE | Abs | LR | 7.38 | 6.75 | 11.90 | 7.25 | 6.26 |
| SS | 17.72 | 18.92 | 18.23 | 17.05 | 17.18 | ||
| Em | LR | 14.33 | 14.23 | 15.23 | 14.85 | 11.96 | |
| SS | 13.25 | 12.79 | 15.56 | 13.74 | 11.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Q.; Wang, H.; Sun, P.; Zeng, C.; Chi, W. Computational-Chemistry-Based Prediction of Near-Infrared Rhodamine Fluorescence Peaks with Sub-12 nm Accuracy. Photochem 2025, 5, 15. https://doi.org/10.3390/photochem5020015
Yuan Q, Wang H, Sun P, Zeng C, Chi W. Computational-Chemistry-Based Prediction of Near-Infrared Rhodamine Fluorescence Peaks with Sub-12 nm Accuracy. Photochem. 2025; 5(2):15. https://doi.org/10.3390/photochem5020015
Chicago/Turabian StyleYuan, Qinlin, Hanwei Wang, Pingping Sun, Chaoyuan Zeng, and Weijie Chi. 2025. "Computational-Chemistry-Based Prediction of Near-Infrared Rhodamine Fluorescence Peaks with Sub-12 nm Accuracy" Photochem 5, no. 2: 15. https://doi.org/10.3390/photochem5020015
APA StyleYuan, Q., Wang, H., Sun, P., Zeng, C., & Chi, W. (2025). Computational-Chemistry-Based Prediction of Near-Infrared Rhodamine Fluorescence Peaks with Sub-12 nm Accuracy. Photochem, 5(2), 15. https://doi.org/10.3390/photochem5020015