
Linear and Nonlinear Optical Properties of Quadrupolar Bithiophenes and Cyclopentadithiophenes as Fluorescent Oxygen Photosensitizers
 ,
,  , ,
, ,  ,
,  , ,
, ,  ,
,  and
and 
Abstract
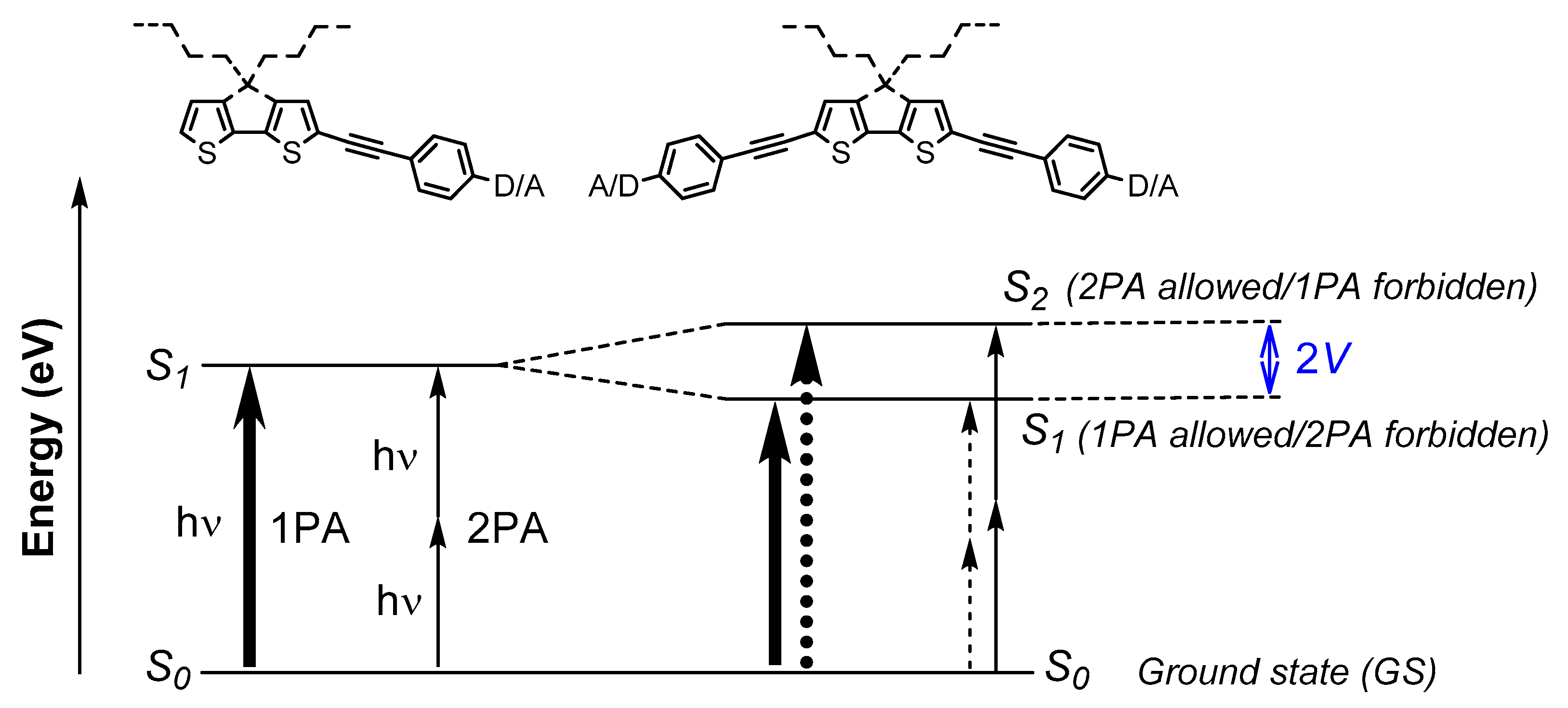
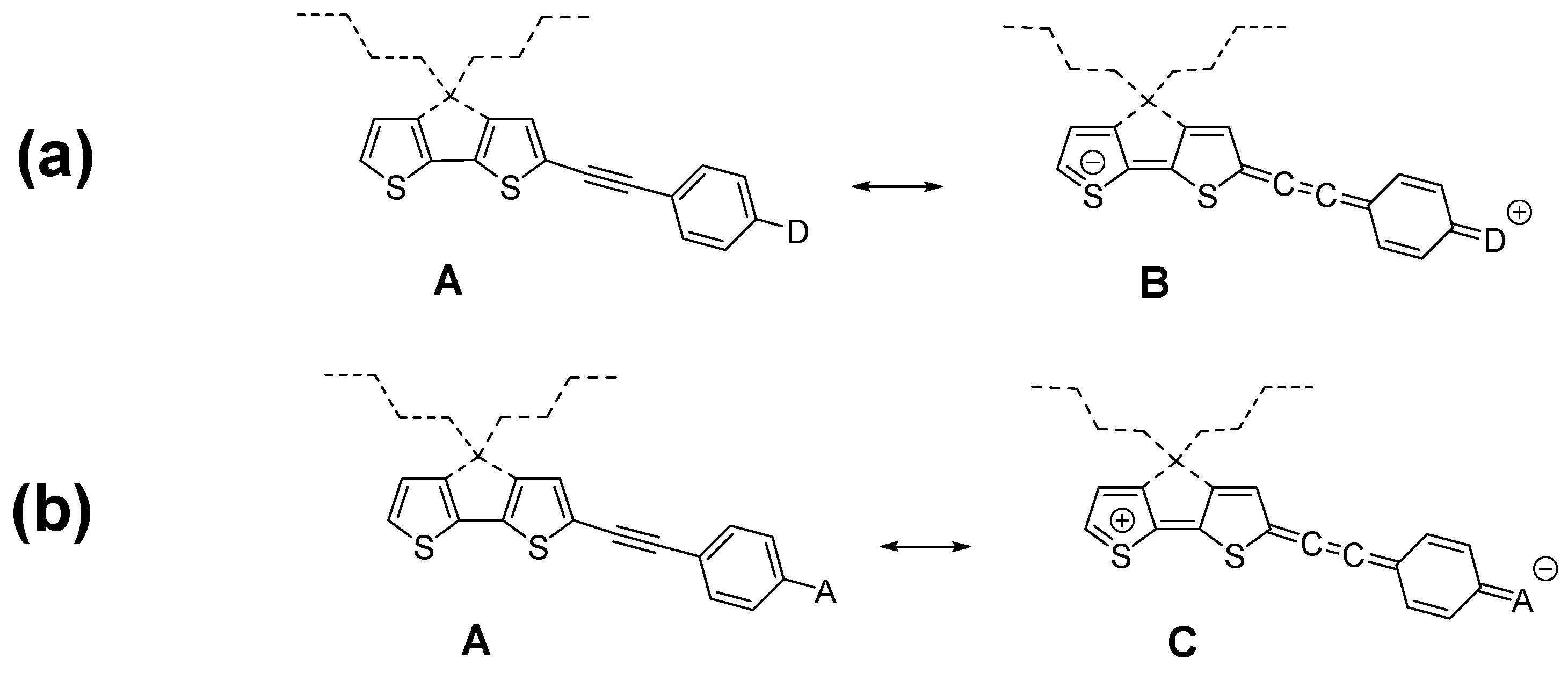
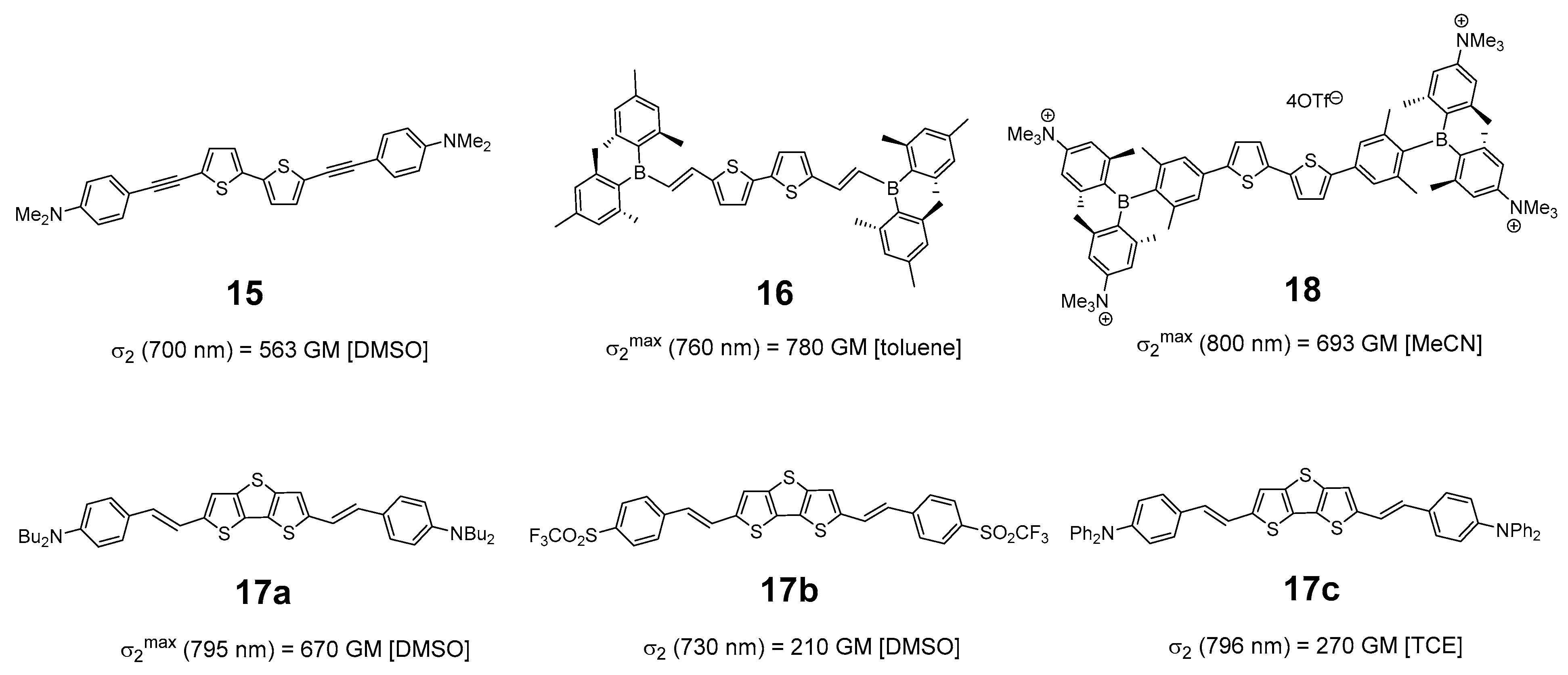
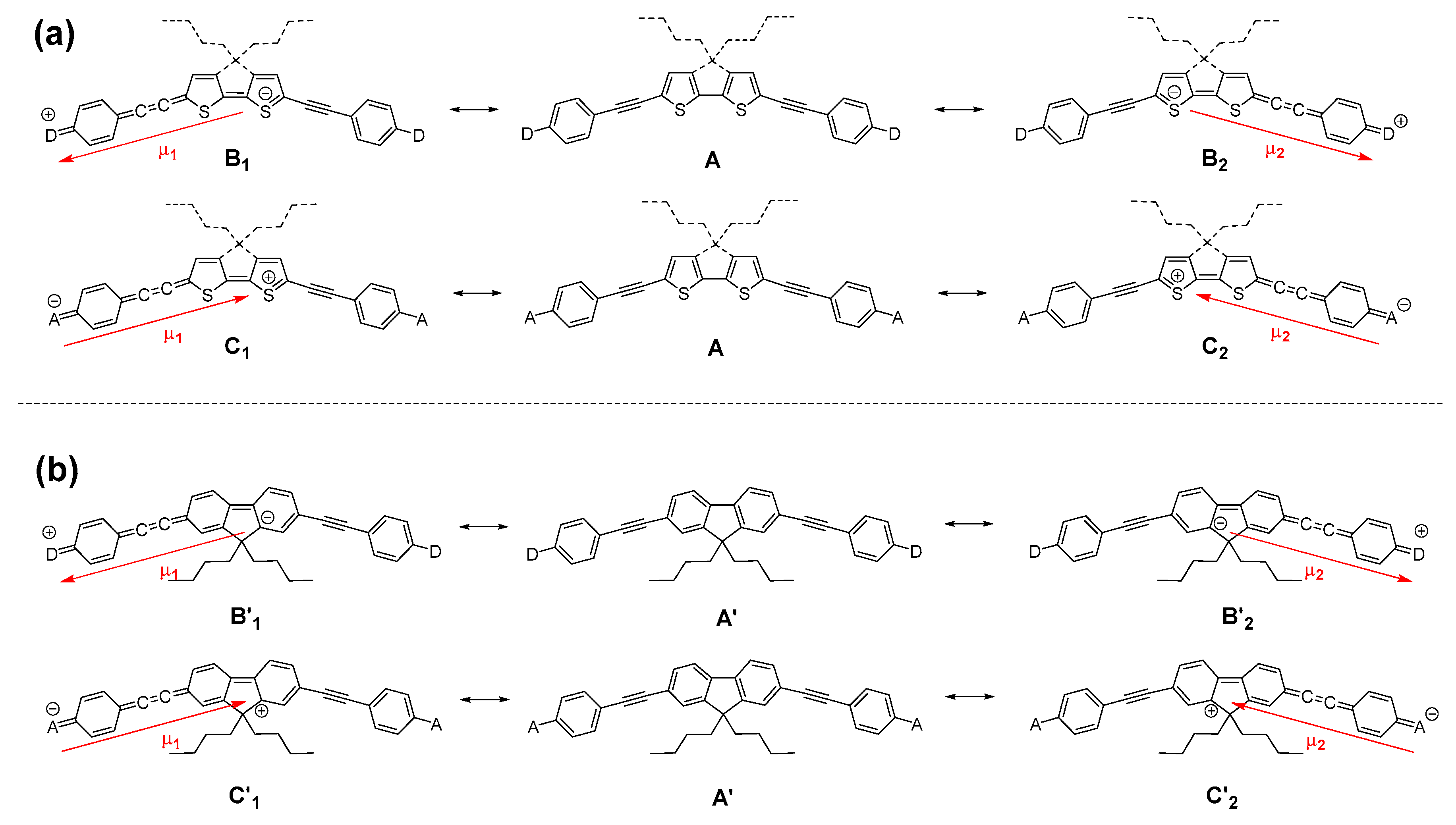
1. Introduction
2. Results
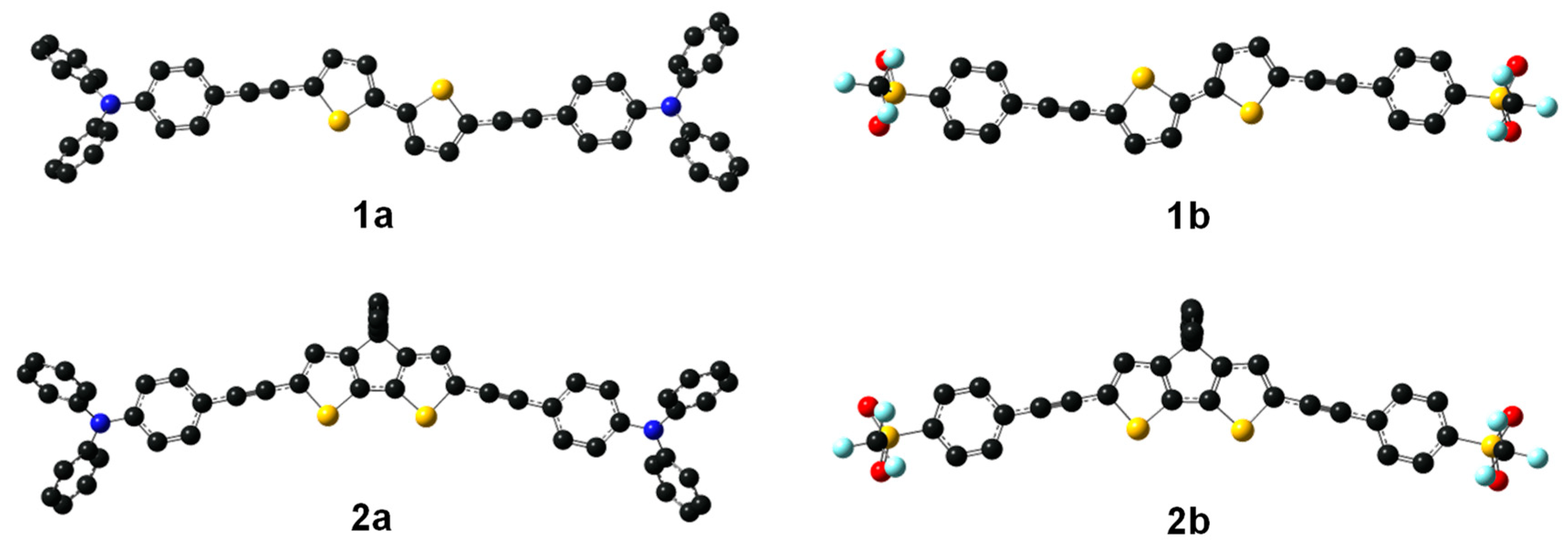
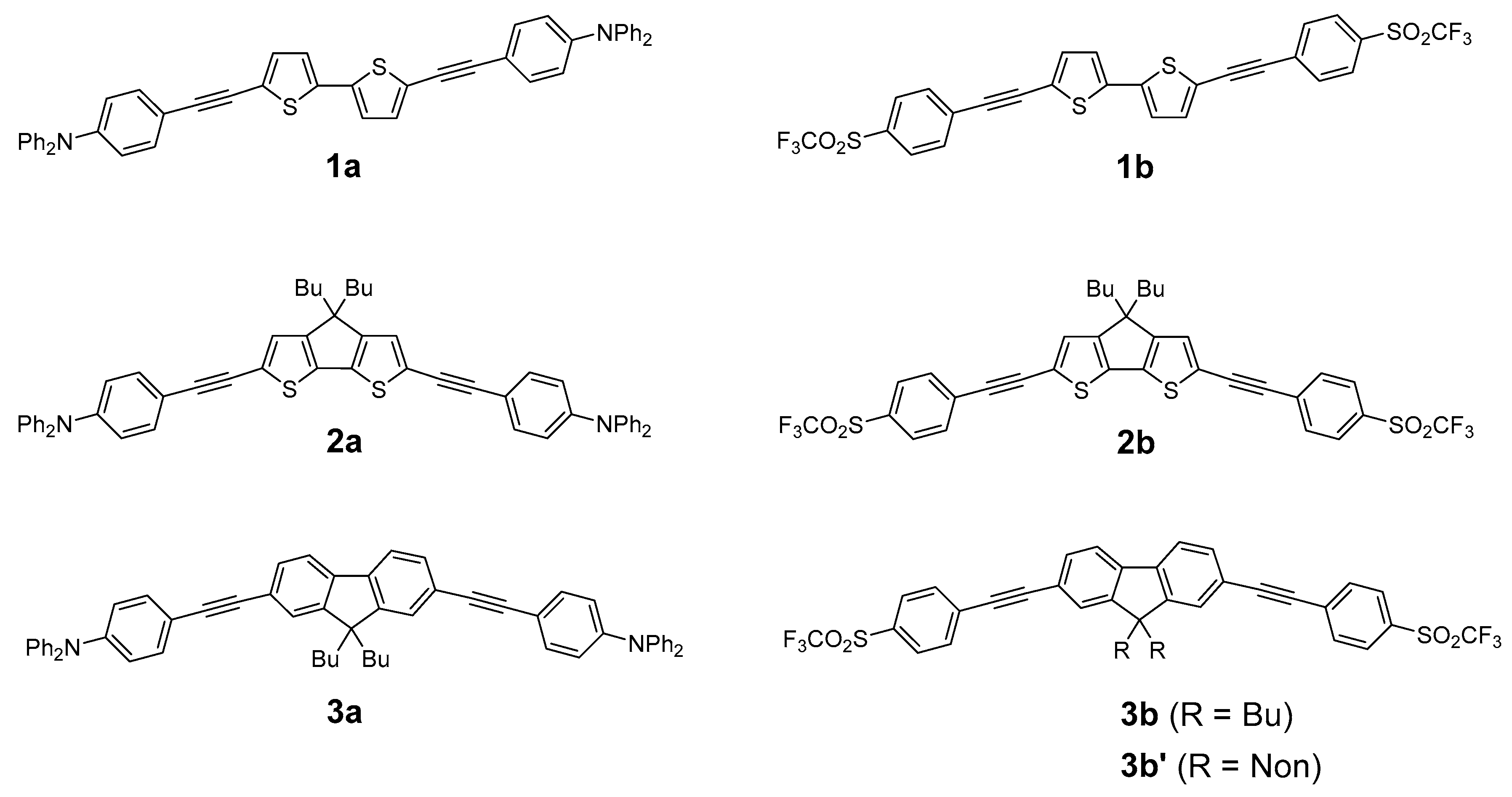
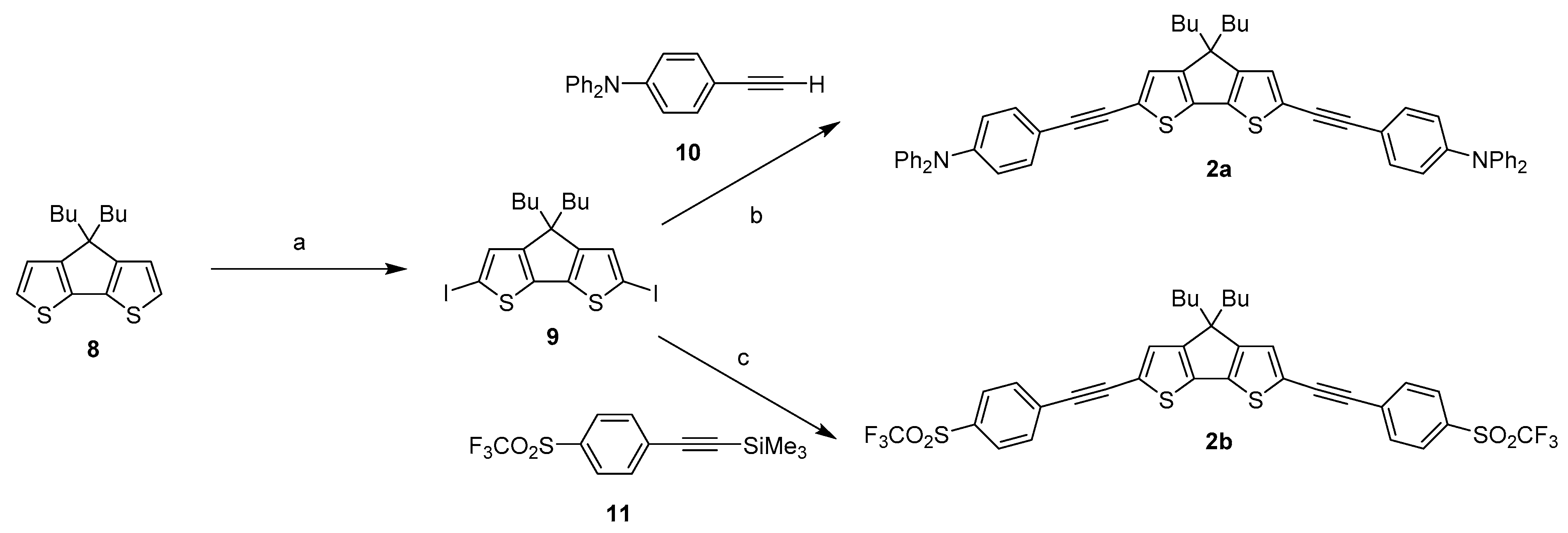
2.1. Syntheses of the New Derivatives
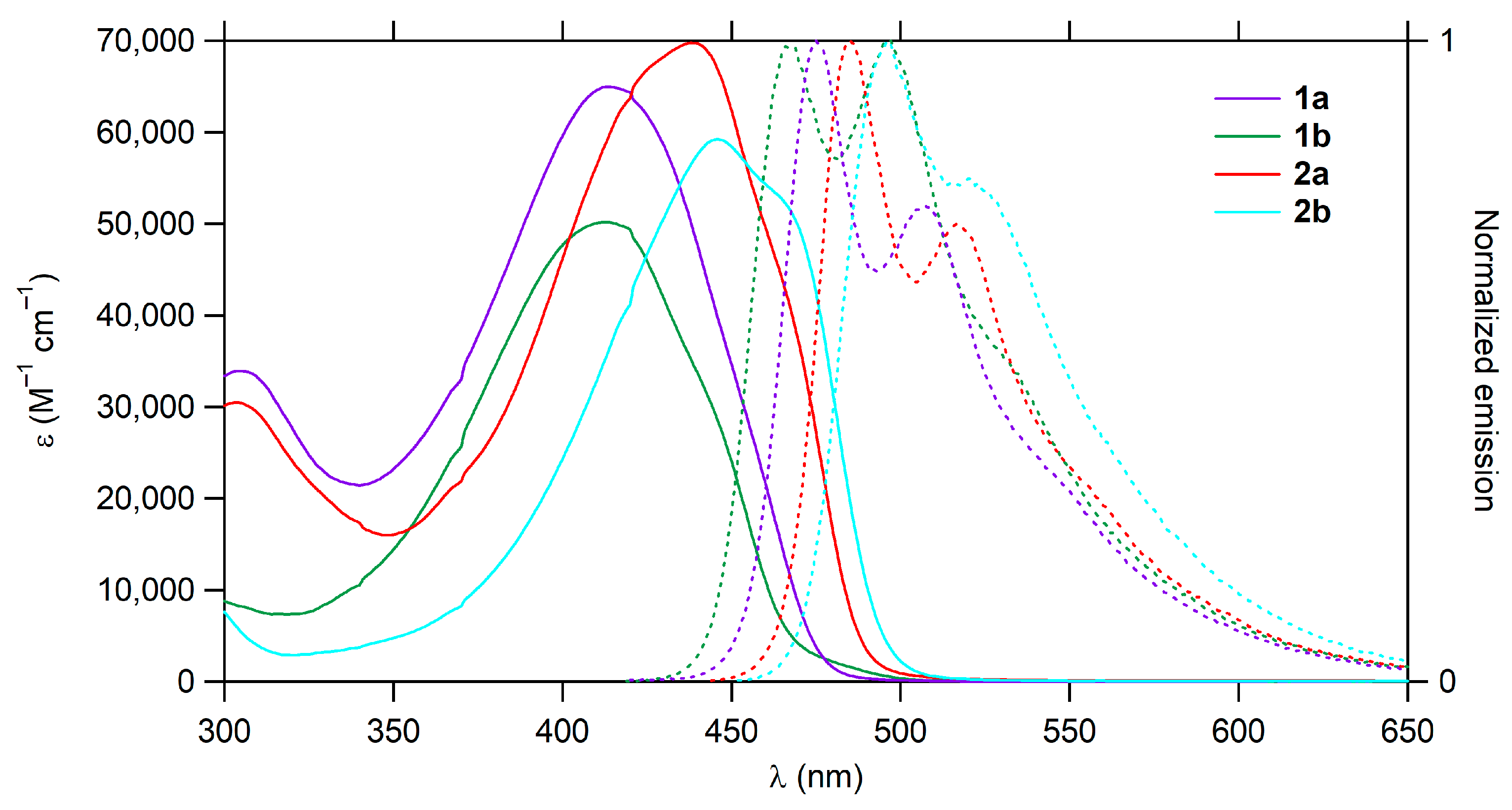
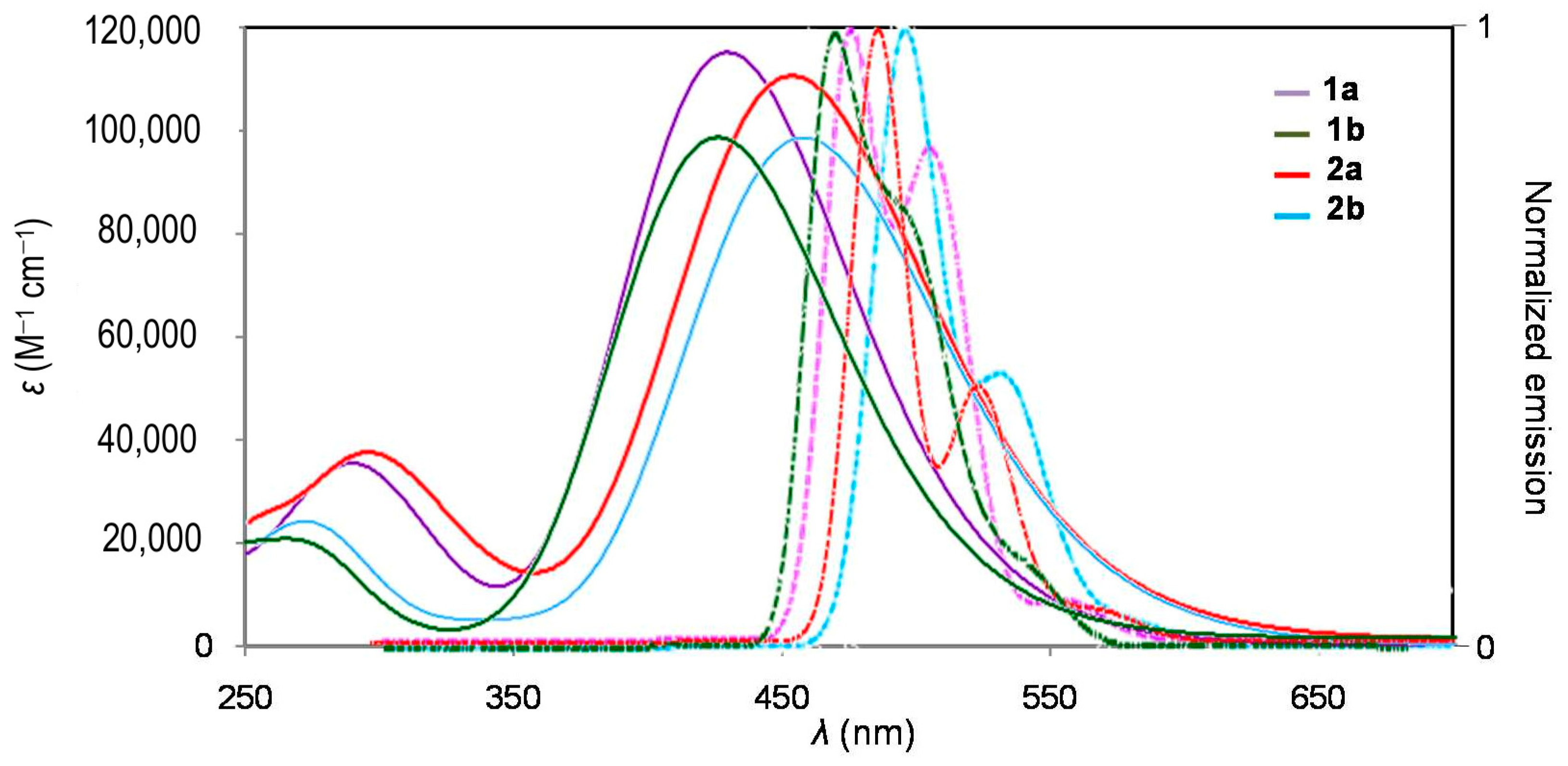
2.2. Photophysical Properties
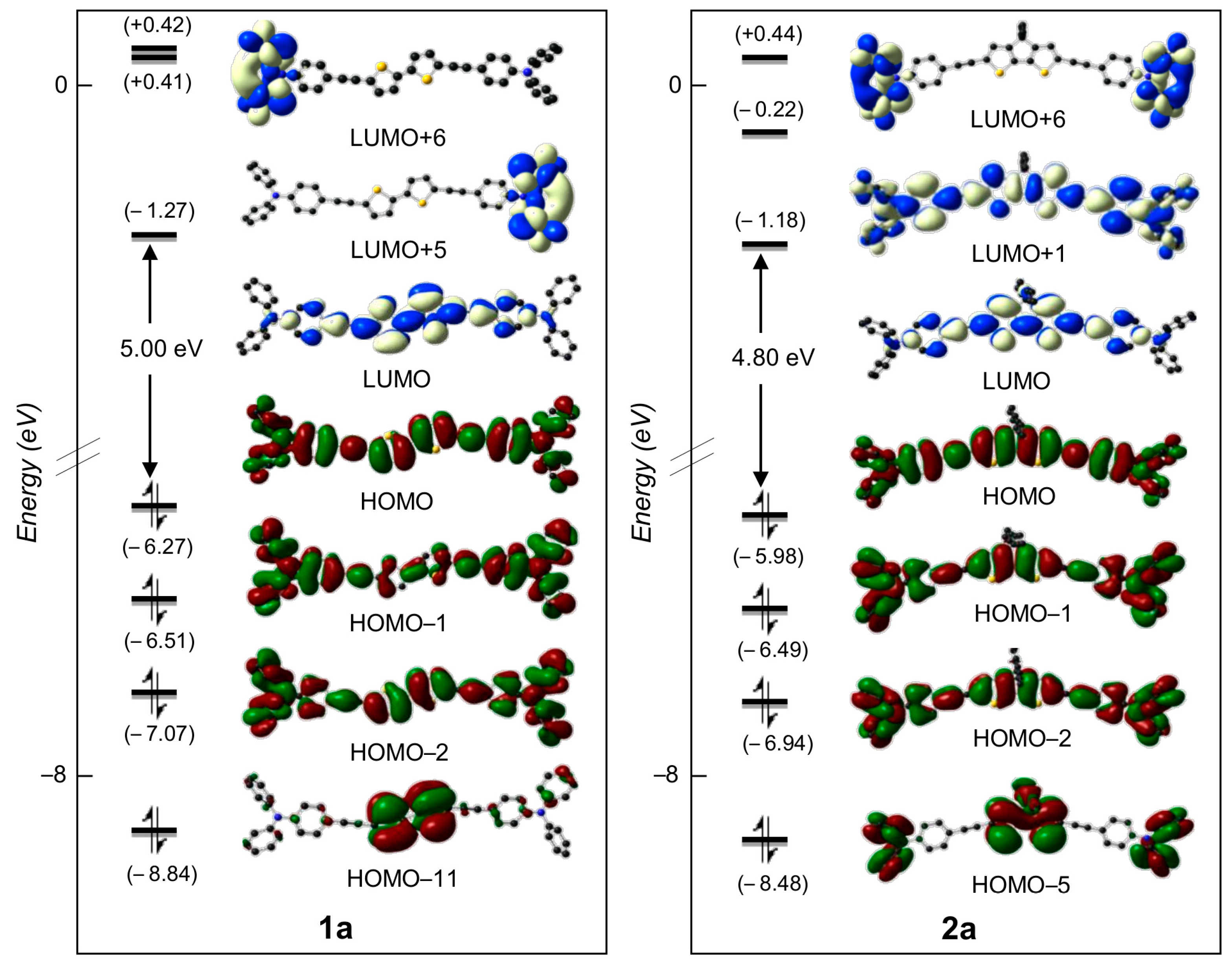
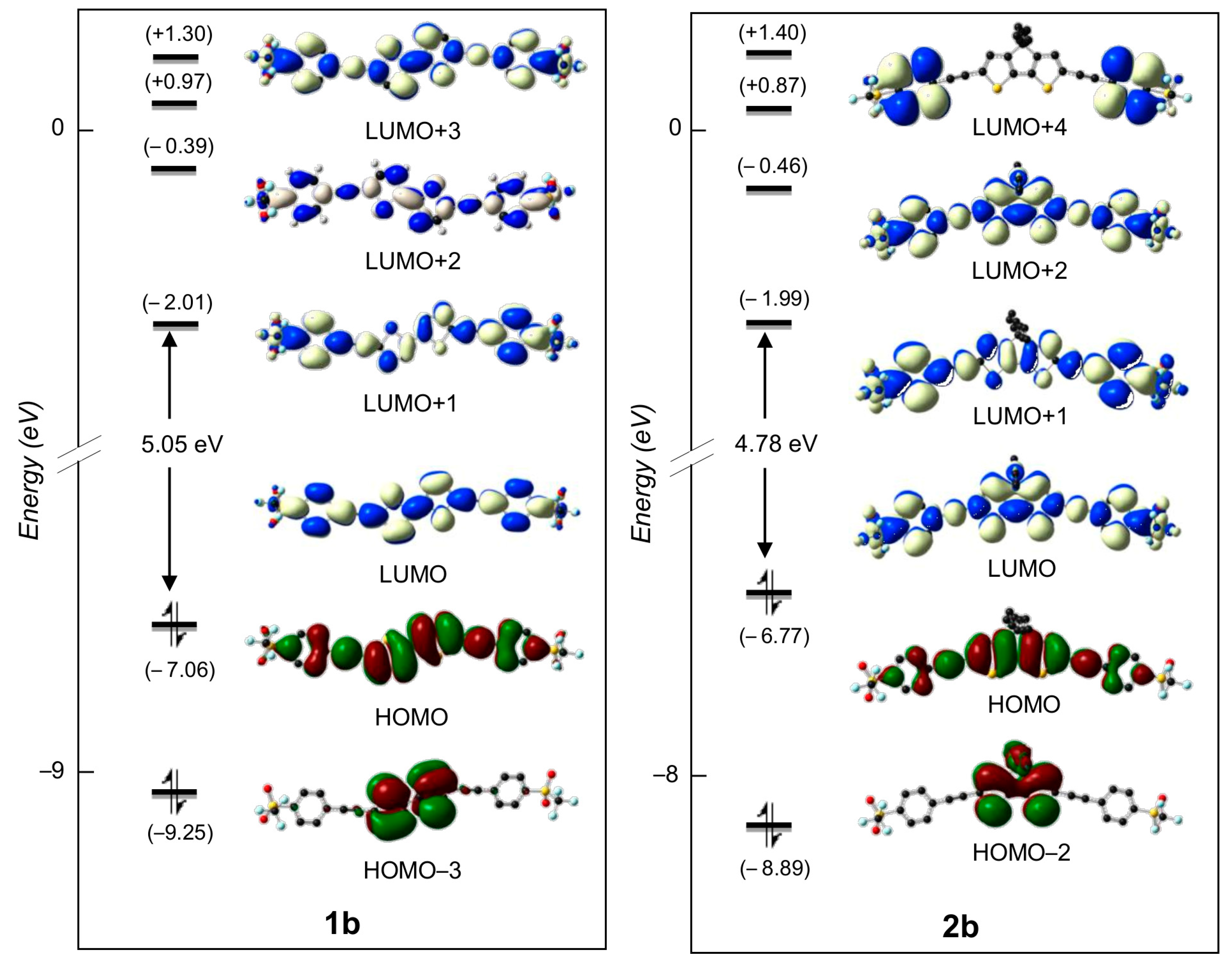
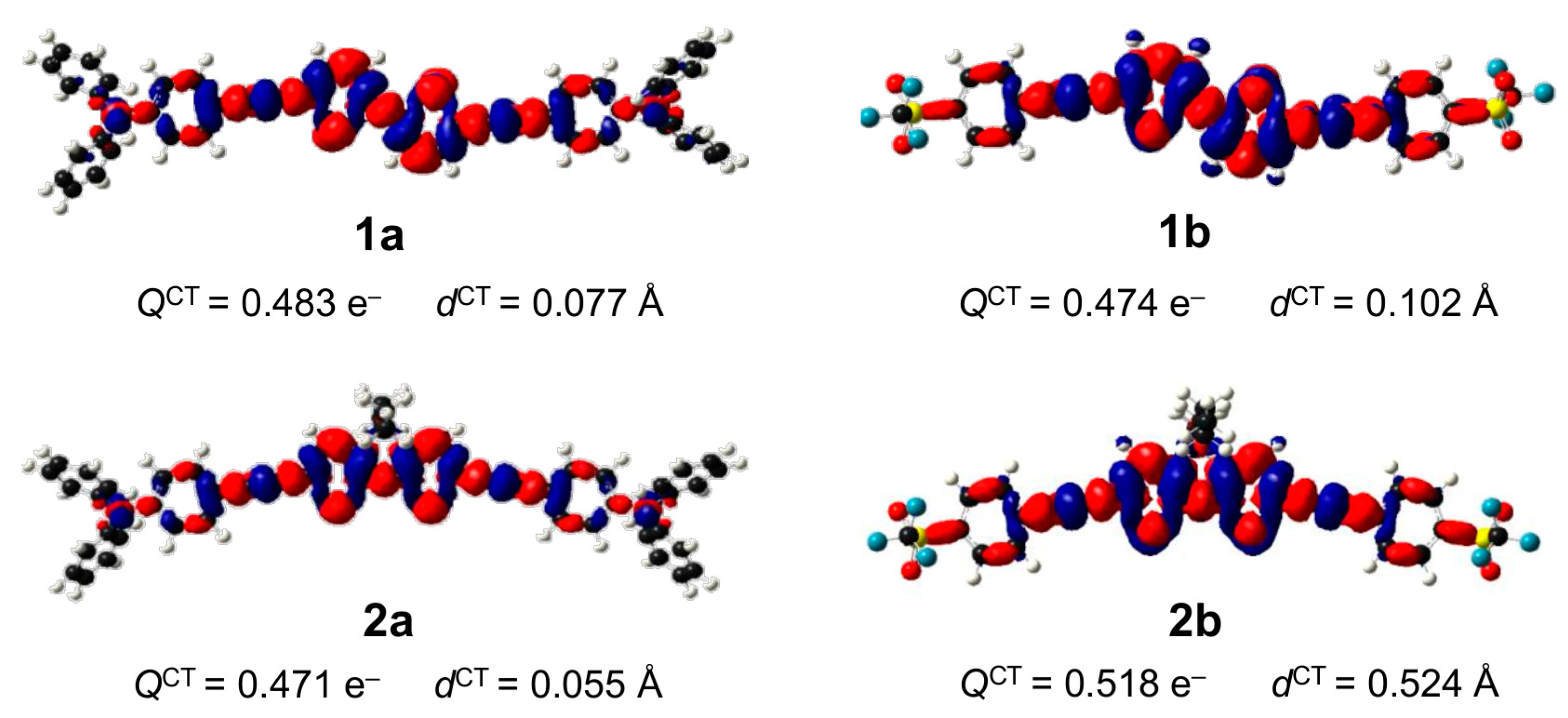
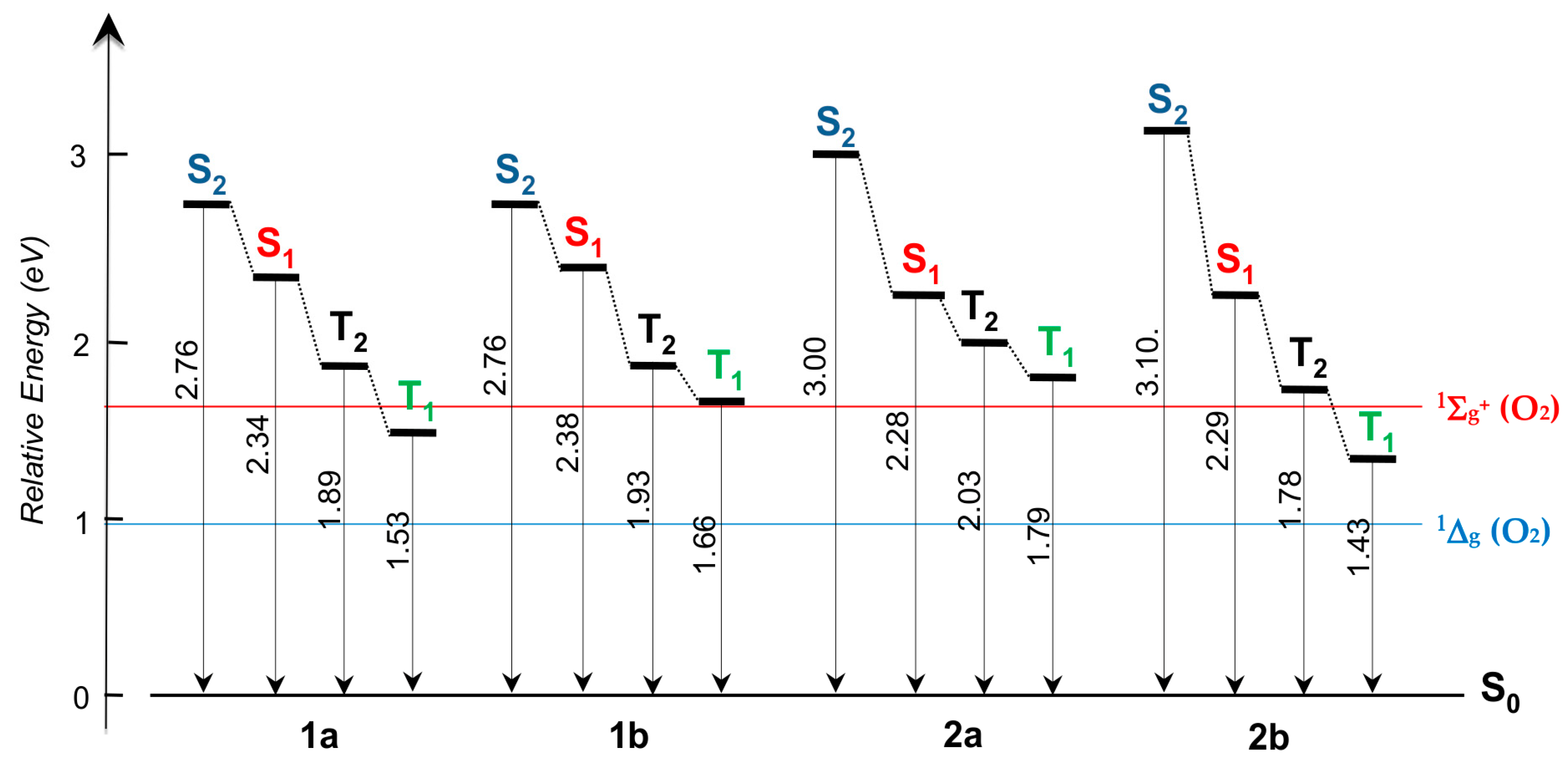
2.3. DFT Studies
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthetic Procedures
4.3. Electronic Absorption and Emission Measurements
4.4. Two-Photon Absorption Experiments
4.5. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References and Notes
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew. Chem. Int. Ed. 2009, 48, 3244–3266. [Google Scholar] [CrossRef]
- He, G.S.; Tan, L.-S.; Zheng, Q.; Prasad, P.N. Multiphoton Absorbing Materials: Molecular Designs, Characterizations, and Applications. Chem. Rev. 2008, 108, 1245–1330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhong, D.; Zhou, S. NIR-I-to-NIR-II fluorescent nanomaterials for biomedical imaging and cancer therapy. J. Mater. Chem. B 2018, 6, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Cho, B.R. Second-order nonlinear optical properties of octupolar molecules structure—Property relationship. J. Mater. Chem. 2009, 19, 7402–7409. [Google Scholar] [CrossRef]
- Prasad, P.N.; Reinhardt, B.A. Is there a role for organic materials chemistry in nonlinear optics and photonics? Chem. Mater. 1990, 2, 660–669. [Google Scholar] [CrossRef]
- Drost, K.J.; Jen, A.K.-Y.; Rao, V.P. Designing organic NLO materials. Chemtech 1995, 25, 16–25. [Google Scholar]
- Nalwa, H.S. Organic Materials for Third-Order Nonlinear Optics. Adv. Mater. 1993, 5, 341–358. [Google Scholar] [CrossRef]
- Ventelon, L.; Moreaux, L.; Mertz, J.; Blanchard-Desce, M. New quadrupolar fluorophores with high two-photon excited fluorescence. Chem. Commun. 1999, 2055–2056. [Google Scholar] [CrossRef]
- Kim, O.-K.; Lee, K.-S.; Woo, H.Y.; Kim, K.-S.; He, G.S.; Swiatkiewicz, J.; Prasad, P.N. New Class of Two-Photon-Absorbing Chromophores Based on Dithienothiophene. Chem. Mater. 2000, 12, 284–286. [Google Scholar] [CrossRef]
- Ventelon, L.; Moreaux, L.; Mertz, J.; Blanchard-Desce, M. Optimization of quadrupolar chromophores for molecular two-photon absorption. Synth. Met. 2002, 127, 17–21. [Google Scholar] [CrossRef]
- Zhang, X.; Abid, S.; Shi, L.; Sun, Z.; Mongin, O.; Blanchard-Desce, M.; Paul, F.; Paul-Roth, C.O. New Conjugated meso-Tetra(thien-2-yl)porphyrin-Cored Derivatives as Two-photon Photosensitizers for Singlet Oxygen Generation. Dye. Pigment 2018, 153, 248–255. [Google Scholar] [CrossRef]
- Castro, M.C.R.; Belsley, M.; Raposo, M.M.M. Synthesis and characterization of push–pull bithienylpyrrole NLOphores with enhanced hyperpolarizabilities. Dye. Pigment 2016, 131, 333–339. [Google Scholar] [CrossRef]
- Fernandes, S.S.M.; Belsley, M.; Pereira, A.I.; Ivanou, D.; Mendes, A.; Justino, L.L.G.; Burrows, H.D.; Raposo, M.M.M. Push–Pull N,N-Diphenylhydrazones Bearing Bithiophene or Thienothiophene Spacers as Nonlinear Optical Second Harmonic Generators and as Photosensitizers for Nanocrystalline TiO2 Dye-Sensitized Solar Cells. ACS Omega 2018, 3, 12893–12904. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.S.M.; Herbivo, C.; Aires-de-Sousa, J.; Comel, A.; Belsley, M.; Raposo, M.M.M. Theoretical and experimental studies of aryl-bithiophene based push-pull π-conjugated heterocyclic systems bearing cyanoacetic or rhodanine-3-acetic acid acceptors for SHG nonlinear optical applications. Dye. Pigment 2018, 149, 566–573. [Google Scholar] [CrossRef]
- Ji, L.; Edkins, R.M.; Sewell, L.J.; Beeby, A.; Batsanov, A.S.; Fucke, K.; Drafz, M.; Howard, J.A.K.; Moutounet, O.; Ibersiene, F.; et al. Experimental and Theoretical Studies of Quadrupolar Oligothiophene-Cored Chromophores Containing Dimesitylboryl Moieties as π-Accepting End-Groups: Syntheses, Structures, Fluorescence, and One- and Two-Photon Absorption. Chem. Eur. J. 2014, 20, 13618–13635. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Yang, Z.; Cao, H.; He, W.; Wang, D.; Zhang, J.; Xing, Y.; Gao, H. Nonlinear optical properties of the novel kind of organic donor-acceptor thiophene derivatives with click chemistry modification. Tetrahedron 2017, 73, 6210–6216. [Google Scholar] [CrossRef]
- Wang, C.-K.; Macak, P.; Luo, Y.; Ågren, H. Effects of π centers and symmetry on two-photon absorption cross sections of organic chromophores. J. Chem. Phys. 2001, 114, 9813–9820. [Google Scholar] [CrossRef]
- Chow, C.-F. Two-photon induced emissive thiophene donor–acceptor systems as molecular probes for in vitro bio-imaging: Synthesis, crystal structure, and spectroscopic properties. RSC Adv. 2013, 3, 18835–18843. [Google Scholar] [CrossRef]
- Griesbeck, S.; Zhang, Z.; Gutmann, M.; Lühmann, T.; Edkins, R.M.; Clermont, G.; Lazar, A.N.; Haehnel, M.; Edkins, K.; Eichhorn, A.; et al. Water-Soluble Triarylborane Chromophores for One- and Two-Photon Excited Fluorescence Imaging of Mitochondria in Cells. Chem. Eur. J. 2016, 22, 14701–14706. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, S.; Ferger, M.; Czernetzi, C.; Wang, C.; Bertermann, R.; Friedrich, A.; Haehnel, M.; Sieh, D.; Taki, M.; Yamaguchi, S.; et al. Optimization of Aqueous Stability versus π-Conjugation in Tetracationic Bis(triarylborane) Chromophores: Applications in Live-Cell Fluorescence Imaging. Chem. Eur. J. 2019, 25, 7679–7688. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.M.; Rühe, J.; Schwarzmann, J.; Phillipps, A.; Richard, A.-K.; Ferger, M.; Krummenacher, I.; Tumir, L.-M.; Ban, Ž.; Crnolatac, I.; et al. Bithiophene-Cored, mono-, bis-, and tris-(Trimethylammonium)-Substituted, bis-Triarylborane Chromophores: Effect of the Number and Position of Charges on Cell Imaging and DNA/RNA Sensing. Chem. Eur. J. 2021, 27, 14057–14072. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Hugues, V.; Clermont, G.; Herbivo, C.; Castro, M.C.R.; Comel, A.; Raposo, M.M.M.; Blanchard-Desce, M. Fluorescence and two-photon absorption of push–pull aryl(bi)thiophenes: Structure–property relationships. Photochem. Photobiol. Sci. 2012, 11, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Amar, A.; ElKechai, A.; Halet, J.-F.; Paul, F.; Boucekkine, A. Two-photon absorption of dipolar and quadrupolar oligothiophene-cored chromophore derivatives containing terminal dimesitylboryl moieties: A theoretical (DFT) structure—Property investigation. New J. Chem. 2021, 45, 15074–15081. [Google Scholar] [CrossRef]
- Pokladek, Z.; Ripoche, N.; Betou, M.; Trolez, Y.; Mongin, O.; Olesiak-Banska, J.; Matczyszyn, K.; Samoc, M.; Humphrey, M.G.; Blanchard-Desce, M.; et al. Linear Optical and Third-Order Nonlinear Optical Properties of Some Fluorenyl- and Triarylamine-Containing Tetracyano-butadiene Derivatives. Chem. Eur. J. 2016, 22, 10155–10167. [Google Scholar] [CrossRef] [PubMed]
- Mongin, O.; Porrès, L.; Charlot, M.; Katan, C.; Blanchard-Desce, M. Synthesis, Fluorescence, and Two-Photon Absorption of a Series of Elongated Rod-like and Banana-Shaped Quadrupolar Fluorophores: A Comprehensive Study of Structure-Property Relationships. Chem. Eur. J. 2007, 13, 1481–1498. [Google Scholar] [CrossRef] [PubMed]
- Bella, A.P.; Solomon, R.V.; Vedha, S.A.; Merlin, J.P. Enhanced luminescence efficiency of structurally tailored new coumarin-based heterocyclic organic materials: A DFT/TD-DFT study. Theor. Chem. Acc. 2019, 138, 53. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6178. [Google Scholar] [CrossRef]
- Gam, S.; Messaoudi, S.; Halet, J.-F.; Boucekkine, A. How do structural factors determine the linear and non-linear optical properties of fluorene-containing quadrupolar fluorophores? A theoretical answer. New J. Chem. 2022, 46, 13431–13441. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, C.; Wu, W.; Guo, H.; Ji, S.; Song, P.; Han, K.; Zhao, J.; Zhang, X.; Wu, Y.; et al. Tuning the Emission Colour of Triphenylamine-Capped Cyclometallated Platinum(II) Complexes and Their Application in Luminescent Oxygen Sensing and Organic Light-Emitting Diodes. Eur. J. Inorg. Chem. 2010, 2010, 4683–4696. [Google Scholar] [CrossRef]
- Hwang, E.; Lusker, K.L.; Garno, J.C.; Losovyj, Y.; Nesterov, E.E. Semiconducting polymer thin films by surface-confined stepwise click polymerization. Chem. Commun. 2011, 47, 11990–11992. [Google Scholar] [CrossRef]
- The low solubility of the latter compound in common organic solvents precluded its characterization by 13C NMR.
- Szczepanski, C.R.; Darmanin, T.; Godeau, G.; Guittard, F. Nanofold-decorated surfaces from the electrodeposition of di-alkyl-cyclopentadithiophenes. Polym. Adv. Technol. 2018, 29, 1170–1181. [Google Scholar] [CrossRef]
- Grelaud, G.; Cifuentes, M.P.; Schwich, T.; Argouarch, G.; Petrie, S.; Stranger, R.; Paul, F.; Humphrey, M.G. Multi-State Redox-Active Metallated-Triarylamines. Eur. J. Inorg. Chem. 2012, 2012, 65–75. [Google Scholar] [CrossRef]
- Kuzyk, M.G. Fundamental limits on two-photon absorption cross-sections. J. Chem. Phys. 2003, 119, 8327–8334. [Google Scholar] [CrossRef]
- Bazourkas, M.; Blanchard-desce, M. Molecular engineeing of push-pull dipolar and quadrupolar molecules for two-photon absorption: A multi valence-bond states approach. J. Chem. Phys. 2000, 113, 3951–3959. [Google Scholar]
- Katan, C.; Tretiak, S.; Werts, M.H.V.; Bain, A.J.; Marsh, R.J.; Leonczek, N.; Nicolaou, N.; Badaeva, E.; Mongin, O.; Blanchard-Desce, M. Two-Photon Transitions in Quadrupolar and Branched Chromophores: Experiment and Theory. J. Phys. Chem. B 2007, 111, 9468–9483. [Google Scholar] [CrossRef]
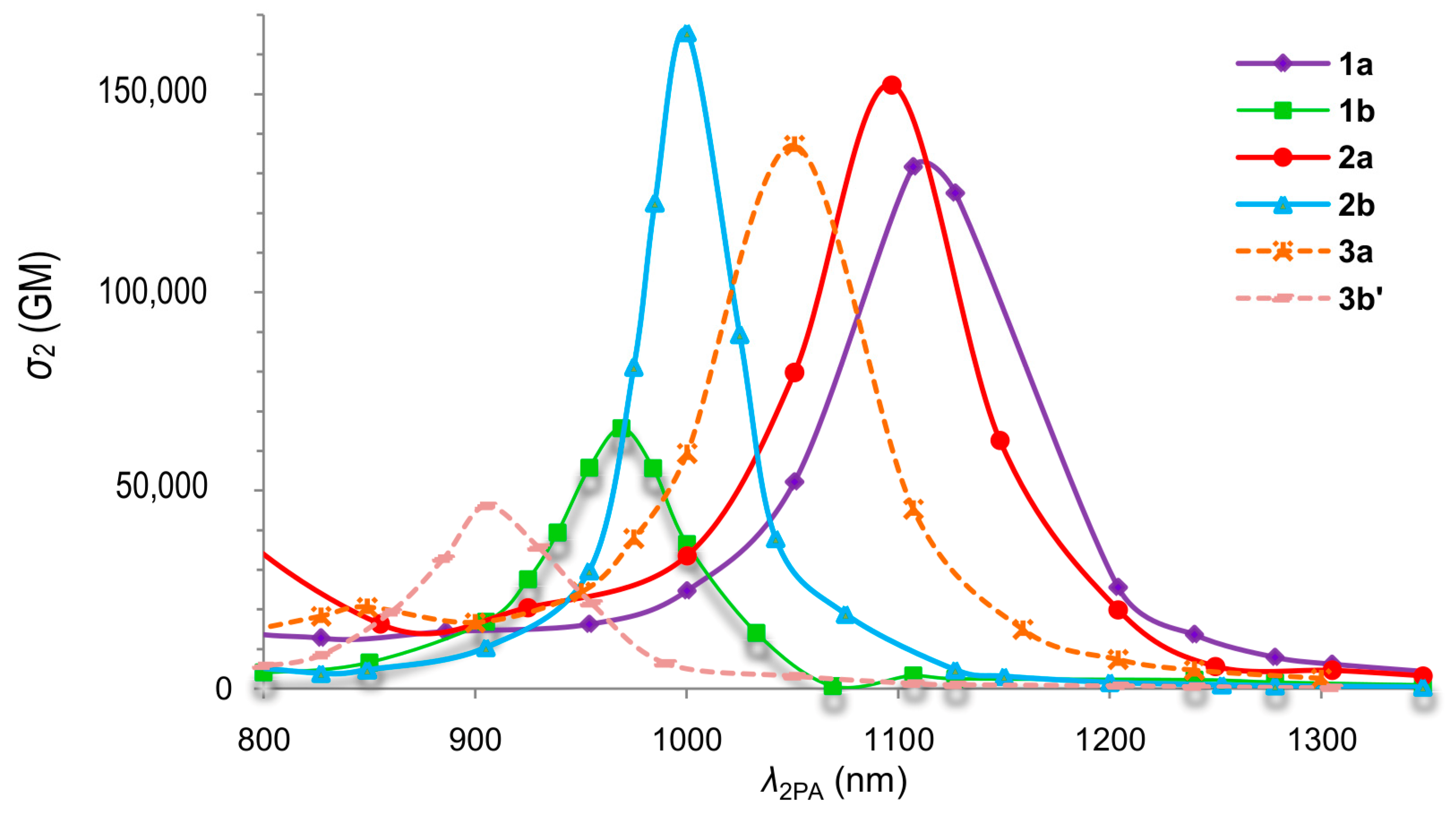
- Comparison with 3b’ should however be made with more caution, since the 2PA maximum detected for 3b’ does not correspond to the maximum of the allowed 2PA band. Actually, it is closer to that of the forbidden 2PA band and therefore is not appropriate for comparison with the allowed 2PA band of 1a,b and 2a,b, as indicated by a footnote in Table 1.
- Kuzyk, M.G. Using fundamental principles to understand and optimize nonlinear-optical materials. J. Mater. Chem. 2009, 19, 7444–7465. [Google Scholar] [CrossRef]
- Kim, H.M.; Cho, B.R. Two-photon materials with large two-photon cross sections. Structure—Property relationship. Chem. Commun. 2009, 45, 153–164. [Google Scholar] [CrossRef]
- Thiophene. Available online: https://www.wikiwand.com/en/Thiophene#/citenoteCSD8 (accessed on 15 June 2022).
- Xu, L.; Zhao, J.; Liu, R.; Liu, H.; Liu, J.; Wang, H. Electrosyntheses and characterizations of novel electrochromic and fluorescentcopolymers based on 2,2-bithiophene and pyrene. Electrochim. Acta 2010, 55, 8855–8862. [Google Scholar] [CrossRef]
- Zotti, G.; Schiavon, G.; Berlin, A.; Fontana, G.; Pagani, G. Novel, Highly Conducting, and Soluble Polymers from Anodic Coupling of Alkyl-Substituted Cyclopentadithiophene Monomers. Macromolecules 1994, 27, 1938–1942. [Google Scholar] [CrossRef]
- Hapiot, P.; Lagrost, C.; Le Floch, F.; Raoult, E.; Rault-Berthelot, J. Comparative Study of the Oxidation of Fluorene and 9,9-Disubstituted Fluorenes and Their Related 2,7’-Dimers and Trimer. Chem. Mater. 2005, 17, 2003–2012. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Compare also to redox potentials reported for 15 in CH3CN (+0.108/+1.50 vs. Fc).
- Le Bahers, T.; Adamo, C.; Ciofini, I. A Qualitative Index of Spatial Extent in Charge-Transfer Excitations. J. Chem. Theory Comput. 2011, 7, 2498–2506. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1758. [Google Scholar] [CrossRef] [PubMed]
- Siedbrand, W. Radiationless Transitions in Polyatomic Molecules. II. Triplet-Ground-State Transitions in Aromatic Hydrocarbons. J. Chem. Phys. 1967, 46, 2411–2422. [Google Scholar] [CrossRef]
- Valeur, B. Invitation à la Fluorescence Moléculaire; De Boeck & Larcier: Bruxelles, Belgium, 2004. [Google Scholar]
- Amar, A.; Boucekkine, A.; Paul, F.; Mongin, O. DFT study of two-photon absorption of octupolar molecules. Theor. Chem. Acc. 2019, 138, 105. [Google Scholar] [CrossRef]
- We have presently checked that the extension of the basis set from DZP to TZP had only a small effect on the computed 2PA cross-sections.
- Ripoche, N.; Betou, M.; Philippe, C.; Trolez, Y.; Mongin, O.; Dudek, M.; Pokladek, Z.; Matczyszyn, K.; Samoc, M.; Sahnoune, H.; et al. Two-photon absorption properties of multipolar triarylamino/tosylamido 1,1,4,4-tetracyanobutadienes. Phys. Chem. Chem. Phys. 2021, 23, 22283–22297. [Google Scholar] [CrossRef]
- The Vexp values derived using this method for 1a,b and 2a,b (Table 5) are within 5% of those derived from the energy difference between the allowed and forbidden 2PA peak/shoulder at low energy
- Note however that the value reported at 700 nm for the compound 15, closely related to 1a, is measured at a single wavelength and does not necessarily correspond to the maximum.
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Argouarch, G.; Veillard, R.; Roisnel, T.; Amar, A.; Meghezzi, H.; Boucekkine, A.; Hugues, V.; Mongin, O.; Blanchard-Desce, M.; Paul, F. Triaryl-1,3,5-Triazinane-2,4,6-Triones (Isocyanurates) Peripherally Functionalized by Donor Groups: Synthesis and Study of their Linear and Nonlinear Optical Properties. Chem. Eur. J. 2012, 18, 11811–11827. [Google Scholar] [CrossRef]
- Griesbeck, S.; Michail, E.; Wang, C.; Ogasawara, H.; Lorenzen, S.; Gerstner, L.; Zang, T.; Nitsch, J.; Sato, Y.; Bertermann, R.; et al. Tuning the π-bridge of quadrupolar triarylborane chromophores for one- and two-photon excited fluorescence imaging of lysosomes in live cells. Chem. Sci. 2019, 10, 5405–5422. [Google Scholar] [CrossRef]
- Breitzer, J.G.; Dlott, D.D.; Iwaki, L.K.; Kirkpatrick, S.M.; Rauchfuss, T.B. Third-Order Nonlinear Optical Properties of Sulfur-Rich Compounds. J. Phys. Chem. A 1999, 103, 6930–6937. [Google Scholar] [CrossRef]
- In that respect, note that the 2PA cross-section reported for 17c, albeit more closely related to 2a than 17a, should be treated with caution. The latter was measured at a single wavelength, so that the maximum cross-section of this compound was possibly not reached. Furthermore, measurements were conducted in tetrachloroethane (TCE) using another method (nonlinear transmission) so the σ2 values are perhaps not strictly comparable.
- Lavis, L.D.; Raines, R.T. Bright Ideas for Chemical Biology. ACS Chem. Biol. 2008, 3, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Shriver, D.F.; Drezdzon, M.A. The Manipulation of Air-Sensitive Compound; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Gottlieb, H.E.; Kotlyer, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1977, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Demas, N.; Crosby, G.A. Measurement of photoluminescence quantum yields. J. Phys. Chem. 1971, 75, 991–1024. [Google Scholar]
- Eaton, G.R. Reference Materials for Fluorescence Measurement. Pure Appl. Chem. 1988, 60, 1107–1114. [Google Scholar] [CrossRef]
- Xu, C.; Webb, W.W. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J. Opt. Soc. Am. B 1996, 13, 481–491. [Google Scholar] [CrossRef]
- Werts, M.H.V.; Nerambourg, N.; Pélégry, D.; Le Grand, Y.; Blanchard-Desce, M. Action cross sections of two-photon excited luminescence of some Eu(III) and Tb(III) complexes. Photochem. Photobiol. Sci. 2005, 4, 531–538. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, M.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–119. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange—Correlation functional using the Coulomb—Attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Barone, V.; Bloino, J.; Biczysko, M.; Santoro, F. Fully integrated approach to compute vibrationally resolved optical spectra: From small molecules to macrosystems. J. Chem. Theory Comput. 2009, 5, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Baiardi, A.; Biczysko, M.; Bloino, J.; Cappelli, C.; Lipparini, F. Implementation and validation of a multi-purpose virtual spectrometer for large systems in complex environments. Phys. Chem. Chem. Phys. 2012, 14, 12404–12422. [Google Scholar] [CrossRef]
- Massuyeau, F.; Faulques, E.; Latouche, C.; Barone, V. New insights into the vibrational and optical signatures of trans-stilbene via integrated experimental and quantum mechanical approaches. Phys. Chem. Chem. Phys. 2016, 18, 19378–19385. [Google Scholar] [CrossRef]
- Ayache, H.; Hammoutène, D.; Fritsch, E.; Elkechai, A.; Boucekkine, A.; Latouche, C. Comprehensive approach to simulate vibrationally resolved phosphorescence spectra of gold(III) complexes using DFT including temperature effects. Theor. Chem. Acc. 2017, 136, 108. [Google Scholar] [CrossRef]
- Massuyeau, F.; Faulques, E.; Latouche, C. New Insights To Simulate the Luminescence Properties of Pt(II) Complexes Using Quantum Calculations. J. Chem. Theory Comput. 2017, 13, 1748–1755. [Google Scholar] [CrossRef]
- Vazart, F.; Latouche, C.; Bloino, J.; Barone, V. Vibronic coupling investigation to compute phosphorescence spectra of Pt(II) complexes. Inorg. Chem. 2015, 54, 5588–5595. [Google Scholar] [CrossRef]
- Belaidi, H.; Rauch, F.; Zhang, Z.; Latouche, C.; Boucekkine, A.; Marder, T.B.; Halet, J.-F. Insights into the optical properties of triarylboranes with strongly electron-accepting bis(fluoromesityl)boryl groups: When theory meets experiment. ChemPhotoChem 2020, 4, 173–180. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Hu, Z.; Autschbach, J.; Jensen, L. Simulating Third-Order Nonlinear Optical Properties Using Damped Cubic Response Theory within Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2016, 12, 1294–1304. [Google Scholar] [CrossRef]
- ADF2019. Vrije Universiteit: Amsterdam: Amsterdam, The Netherlands, 2019. Available online: https://www.scm.com/product/adf/ (accessed on 15 June 2022).
- Te Velde, G.; Bickelhaupt, F.M.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Baerends, E.J.; Snijders, J.; Ziegler, T. Time dependant density functional theory. Theor. Chem. Acc. 2001, 22, 931–967. [Google Scholar]
- Schipper, P.R.T.; Gritsenko, O.V.; van Gisbergen, S.J.A.; Baerends, E.J. Molecular calculations of excitation energies and (hyper)polarizabilities with a statistical average of orbital model exchange-correlation potentials. J. Chem. Phys. 2000, 112, 1344–1352. [Google Scholar] [CrossRef]
- Jensen, J.F.; van Duijnen, P.T.; Snijders, J.G.J. A discrete solvent reaction field model for calculating frequency-dependent hyperpolarizabilities of molecules in solution. J. Chem. Phys. 2003, 119, 12998–13006. [Google Scholar] [CrossRef]
- Jensen, J.F.; Autschbach, J.; Schatz, J. Finite lifetime effects on the polarizability within time-dependent density-functional theory. J. Chem. Phys. 2005, 122, 224115. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Autschbach, J.; Jensen, L. Simulation of resonance hyper-Rayleigh scattering of molecules and metal clusters using a time-dependent density functional theory approach. J. Chem. Phys. 2014, 141, 124305. [Google Scholar] [CrossRef] [PubMed]
- ADF-GUI 2012; SCM: Amsterdam, The Netherlands. Available online: https://www.scm.com/product/gui/ (accessed on 15 June 2022).
- Beerepoot, M.T.P.; Friese, D.H.; List, N.H.; Kongsted, J.; Ruud, K. Benchmarking two-photon absorption cross sections: Performance of CC2 and CAM-B3LYP. Phys. Chem. Chem. Phys. 2015, 17, 19306–19314. [Google Scholar] [CrossRef]
- Silverstein, D.W.; Jensen, L. Vibronic coupling simulations for linear and nonlinear optical processes: Theory. J. Chem. Phys. 2012, 136, 064111. [Google Scholar] [CrossRef]
- Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 1931, 9, 273–294. [Google Scholar] [CrossRef]
- Pérez-Moreno, J.; Kuzyk, M.G. Fundamental limits of the dispersion of the two-photon absorption cross section. J. Chem. Phys. 2005, 123, 194101. [Google Scholar] [CrossRef]
- Pérez-Moreno, J.; Clays, K.; Kuzyk, M.G. A new dipole-free sum-over-states expression for the second hyperpolarizability. J. Chem. Phys. 2008, 128, 084109. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | λabs (nm) | εmax (M−1 cm−1) | λem (nm) | ∆ω a (cm−1) | ФFb | Ф∆c | 1–ФF–Ф∆ | εmaxФF d (M−1 cm−1) | σ2max e (GM) | σ2max ФF f (GM) | σ2max /Mg (GM.mol/g) | σ2max/(Neff)2 (GM) | σ2Ф∆ (GM) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 414 305 | 65,000 33,900 | 475, 507 | 3102 | 0.35 | 0.54 | 0.11 | 22,750 | 900 | 315 | 1.831 | 1.236 | 486 |
| 1b | 413 | 50,200 | 467, 496 | 2800 | 0.28 | 0.65 | 0.07 | 14,056 | 610 | 171 | 1.537 | 0.770 | 397 |
| 2a | 438 303 | 69,800 30,500 | 485, 517 | 2212 | 0.34 | 0.49 | 0.17 | 23,732 | 1300 | 442 | 1.910 | 1.786 | 637 |
| 2b | 446 | 59,200 | 496, 520 | 2260 | 0.39 | 0.39 | 0.22 | 23,088 | 1510 | 589 | 2.649 | 1.911 | 589 |
| 3ah | 390 301 | 126,800 55,000 | 428 | 2277 | 0.85 | nd i | <0.15 | 107,780 | 980 | 833 | 1.574 | 1.047 | / |
| 3b′j | 372 | 69,200 | 404 | 2129 | 0.98 | nd i | <0.02 | 67,816 | >68 k | >67 | >0.132 | >0.066 k | / |
| Cmpd | τ (ns) | kR (s−1) | kNR (s−1) | τRDFT a (ns) |
|---|---|---|---|---|
| 1a | 0.29 | 1.2 × 109 | 2.2 × 109 | 1.54 |
| 1b | 0.27 | 1.0 × 109 | 2.7 ×109 | 1.63 |
| 2a | 0.56 | 0.6 × 109 | 1.2 × 109 | 1.64 |
| 2b | 0.80 | 0.5 ×109 | 0.8 × 109 | 1.81 |
| 3a b | 1.10 | 0.8 × 109 | 0.1 × 109 | 2.08 |
| 3b′ c | 0.79 | 1.2 × 109 | 0.03 × 109 | 1.83 |
| Cmpd | S0n | E0n | λcalc | f0n | Main MO Transition Percentage |
|---|---|---|---|---|---|
| 1a | 1 | 2.88 | 433 | 2.89 | HOMO → LUMO (79%) |
| 2 * | 3.50 | 354 | 0.008 | HOMO-1 → LUMO (48%) | |
| 3 | 3.97 | 313 | 0.37 | HOMO-2 → LUMO (48%) | |
| 8 | 4.33 | 286 | 0.54 | HOMO-1 → LUMO + 6 (26%) | |
| HOMO-1 → LUMO + 5 (17%) | |||||
| 27 | 5.26 | 236 | 0.23 | HOMO-11 → LUMO (42%) | |
| 1b | 1 | 2.74 | 426 | 2.44 | HOMO → LUMO (89%) |
| 2 * | 3.74 | 331 | 0.003 | HOMO → LUMO + 1 (65%) | |
| 4 | 4.48 | 277 | 0.38 | HOMO → LUMO + 2 (58%) | |
| HOMO-1 → LUMO + 1 (21%) | |||||
| 10 | 4.97 | 250 | 0.23 | HOMO-3 → LUMO (33%) | |
| 18 | 5.55 | 223 | 0.26 | HOMO → LUMO + 3 (53%) | |
| 2a | 1 | 2.73 | 454 | 2.80 | HOMO → LUMO (85%) |
| 2 * | 3.51 | 353 | 0.13 | HOMO-1 → LUMO (46%) | |
| 3 | 3.95 | 314 | 0.43 | HOMO-2 → LUMO (40%) | |
| HOMO-1 → LUMO + 1 (28%) | |||||
| 8 | 4.31 | 288 | 0.55 | HOMO-1 → LUMO + 6 (42%) | |
| 20 | 5.02 | 248 | 0.24 | HOMO-5→ LUMO (58%) | |
| 2b | 1 | 2.86 | 455 | 2.40 | HOMO → LUMO (90%) |
| 2 * | 3.64 | 340 | 0.11 | HOMO → LUMO + 1 (72%) | |
| 3 | 4.42 | 281 | 0.37 | HOMO → LUMO + 2 (73%) | |
| 6 | 4.77 | 260 | 0.22 | HOMO-2 → LUMO (77%) | |
| 18 | 5.42 | 229 | 0.22 | HOMO → LUMO + 4 (47%) | |
| HOMO-6 → LUMO + 1 (28%) |
| Cmpd | Exp | Theo a | |||||
|---|---|---|---|---|---|---|---|
| σ2max b | ωOPA | ωTPA | σ2max b | ||||
| 1a | 729 | 900 | 2.22 | 1.12 | 559 (0.35 × 10−3) | 1107 | 13,168 |
| 1b | 729 | 610 | 2.55 | 1.28 | 486 (0.17 × 10−3) | 969 | 6560 |
| 2a | 738 | 1300 | 2.25 | 1.13 | 551 (0.41 × 10−2) | 1097 | 15,241 |
| 2b | 738 | 1510 | 2.49 | 1.24 | 498 (0.12 × 10−1) | 1000 | 16,579 |
| 3a c,d | 720 | 980 | 2.35 | 1.18 | 528 (0.42 × 10−2) | 1051 | 13,710 |
| 3b’ e/3b d | 730 | / | 2.73 | 1.37 | 454 (0.26 × 10−2) | 905 | 7615 |
| Cmpd | λ1PA | λ2PAmax | λ2PAmin a | σ2min a | Vexp | VCAM-B3LYP | Ref. | |
|---|---|---|---|---|---|---|---|---|
| (nm) | (nm) | (nm) | (GM) | (cm−1) | (cm−1) | (eV) | ||
| 1a | 414 | 740 | 830 | 120 | 1436 | 2500 | 0.31 | This work |
| 1b | 413 | 730 | 820 | 140 | 1592 | 4033 | 0.50 | This work |
| 2a | 438 | 740 | 880 | 60 | 2098 | 4920 | 0.61 | This work |
| 2b | 446 | 750 | 890 | 150 | 2123 | 3670 | 0.46 | This work |
| 3a | 391 | 720 | 790 | 260 | 1101 | 1653 | 0.22 | [24] |
| 3b’/3b | 372 | NA b | 730 c | 70 c | NA b | 2540 | 0.32 | [25] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richy, N.; Gam, S.; Messaoudi, S.; Triadon, A.; Mongin, O.; Blanchard-Desce, M.; Latouche, C.; Humphrey, M.G.; Boucekkine, A.; Halet, J.-F.; et al. Linear and Nonlinear Optical Properties of Quadrupolar Bithiophenes and Cyclopentadithiophenes as Fluorescent Oxygen Photosensitizers. Photochem 2023, 3, 127-154. https://doi.org/10.3390/photochem3010009
Richy N, Gam S, Messaoudi S, Triadon A, Mongin O, Blanchard-Desce M, Latouche C, Humphrey MG, Boucekkine A, Halet J-F, et al. Linear and Nonlinear Optical Properties of Quadrupolar Bithiophenes and Cyclopentadithiophenes as Fluorescent Oxygen Photosensitizers. Photochem. 2023; 3(1):127-154. https://doi.org/10.3390/photochem3010009
Chicago/Turabian StyleRichy, Nicolas, Safa Gam, Sabri Messaoudi, Amédée Triadon, Olivier Mongin, Mireille Blanchard-Desce, Camille Latouche, Mark G. Humphrey, Abdou Boucekkine, Jean-François Halet, and et al. 2023. "Linear and Nonlinear Optical Properties of Quadrupolar Bithiophenes and Cyclopentadithiophenes as Fluorescent Oxygen Photosensitizers" Photochem 3, no. 1: 127-154. https://doi.org/10.3390/photochem3010009
APA StyleRichy, N., Gam, S., Messaoudi, S., Triadon, A., Mongin, O., Blanchard-Desce, M., Latouche, C., Humphrey, M. G., Boucekkine, A., Halet, J.-F., & Paul, F. (2023). Linear and Nonlinear Optical Properties of Quadrupolar Bithiophenes and Cyclopentadithiophenes as Fluorescent Oxygen Photosensitizers. Photochem, 3(1), 127-154. https://doi.org/10.3390/photochem3010009