Effect of the Donor/Acceptor Size on the Rate of Photo-Induced Electron Transfer


Abstract
1. Introduction
2. Results and Discussion
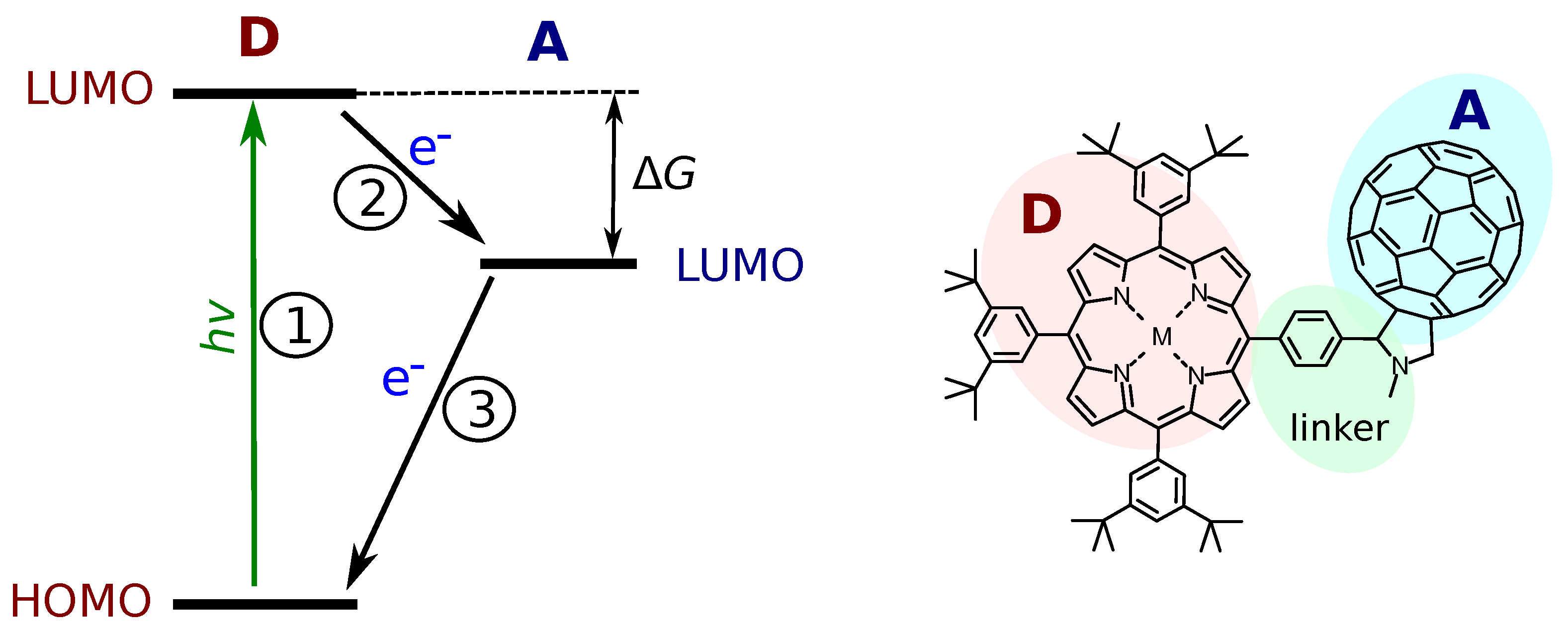
2.1. Molecular DA Systems
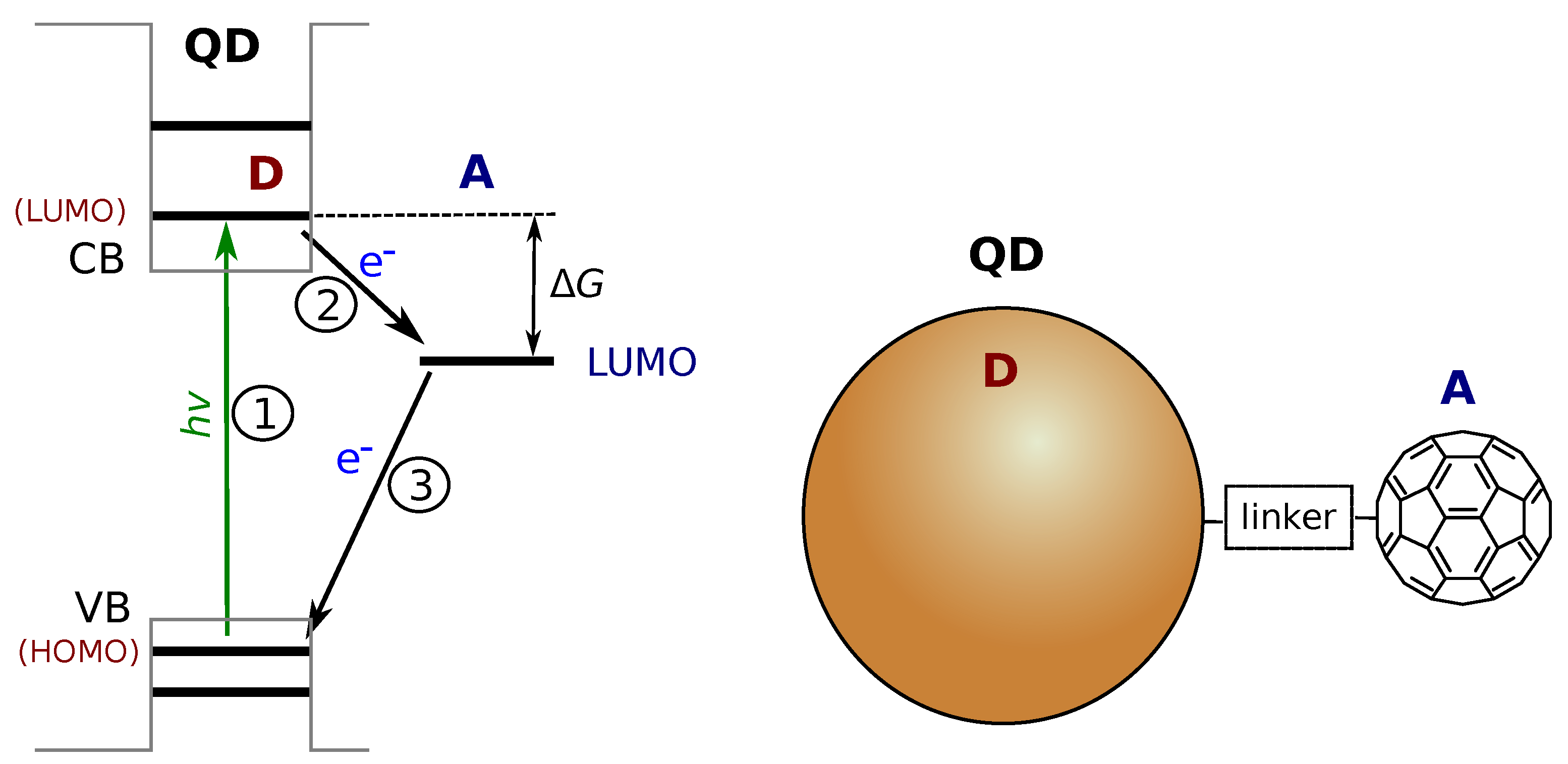
2.2. QD-Molecule Hybrids
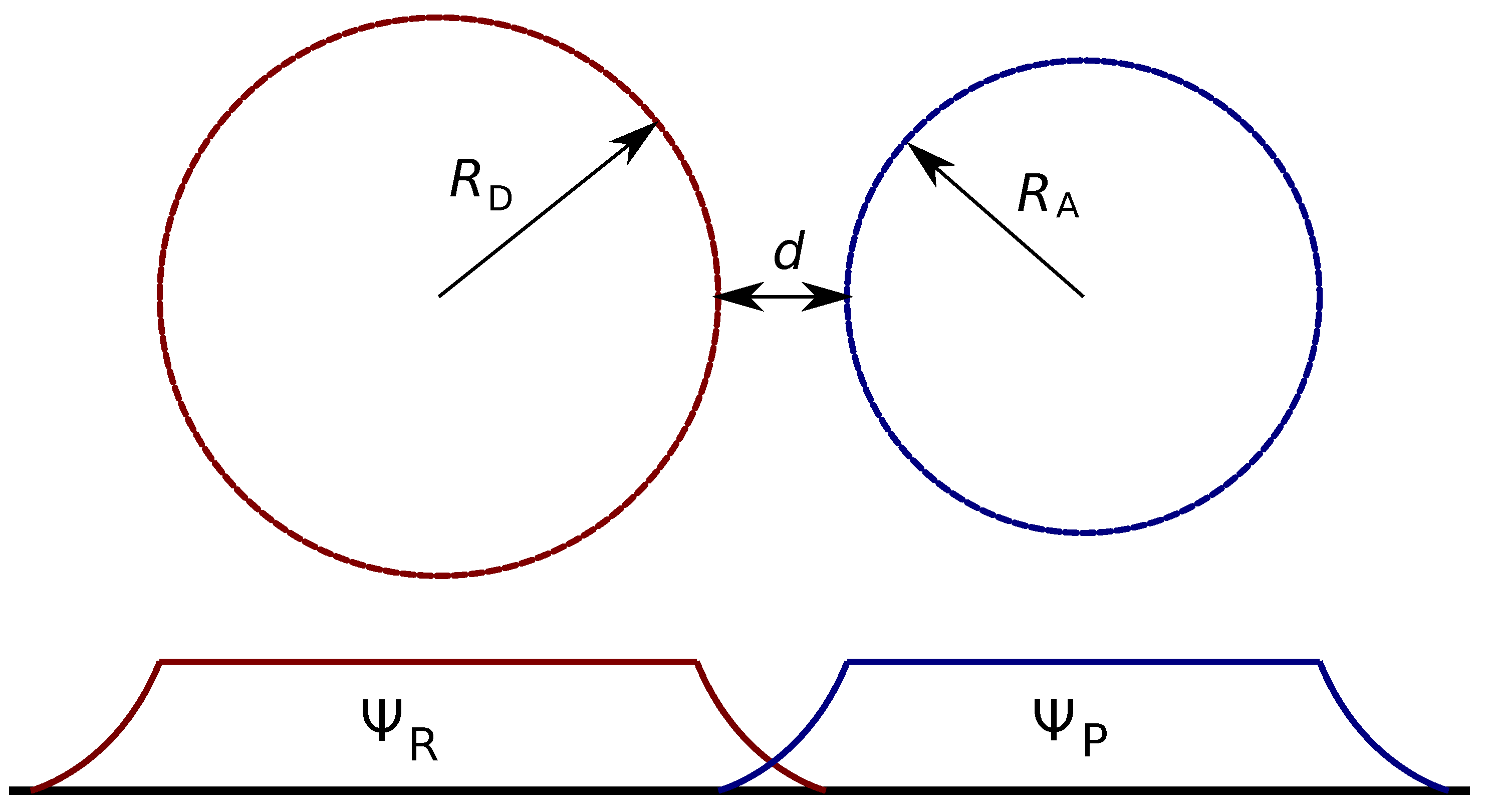
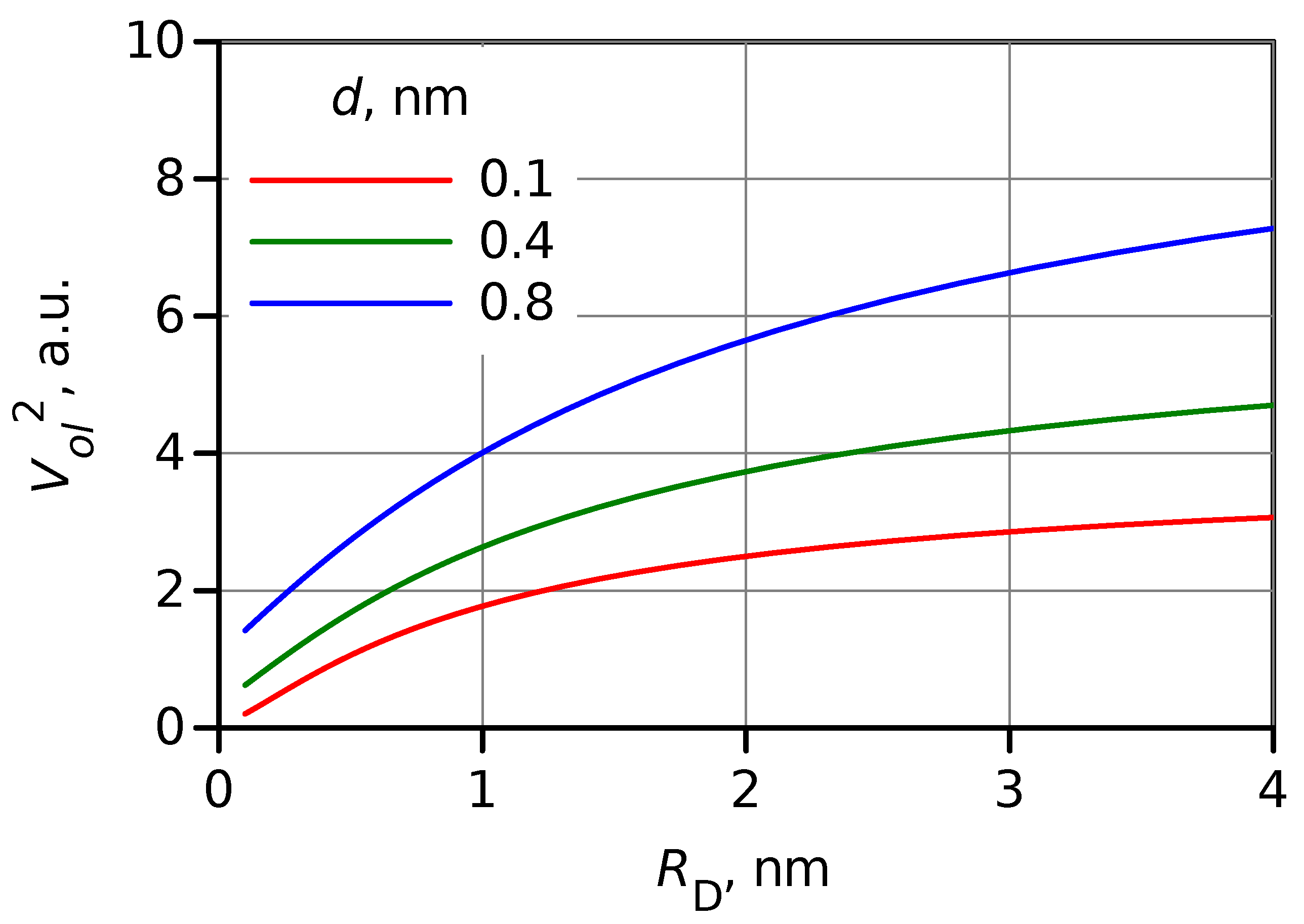
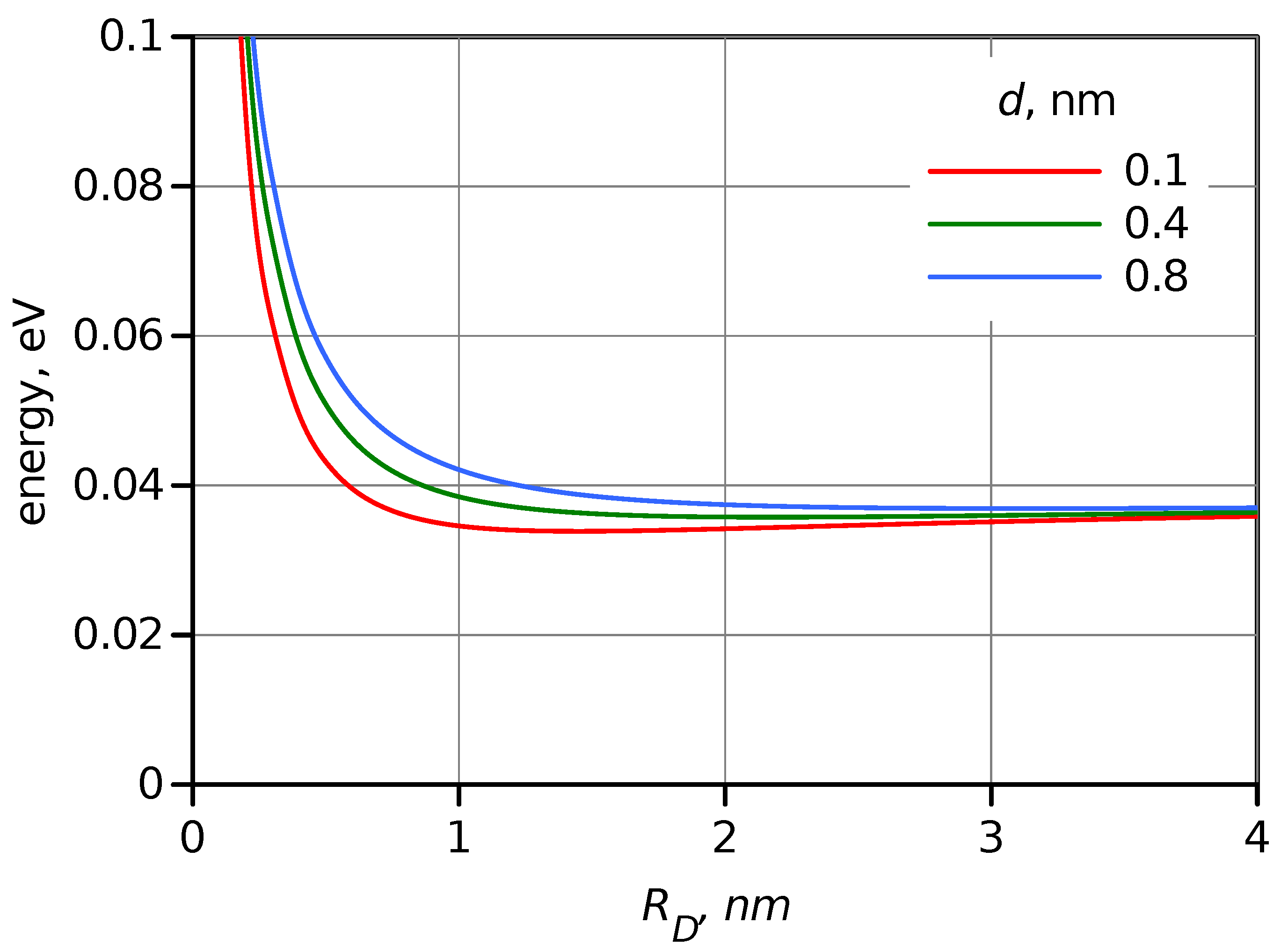
2.3. Electronic Factor in ET Rate
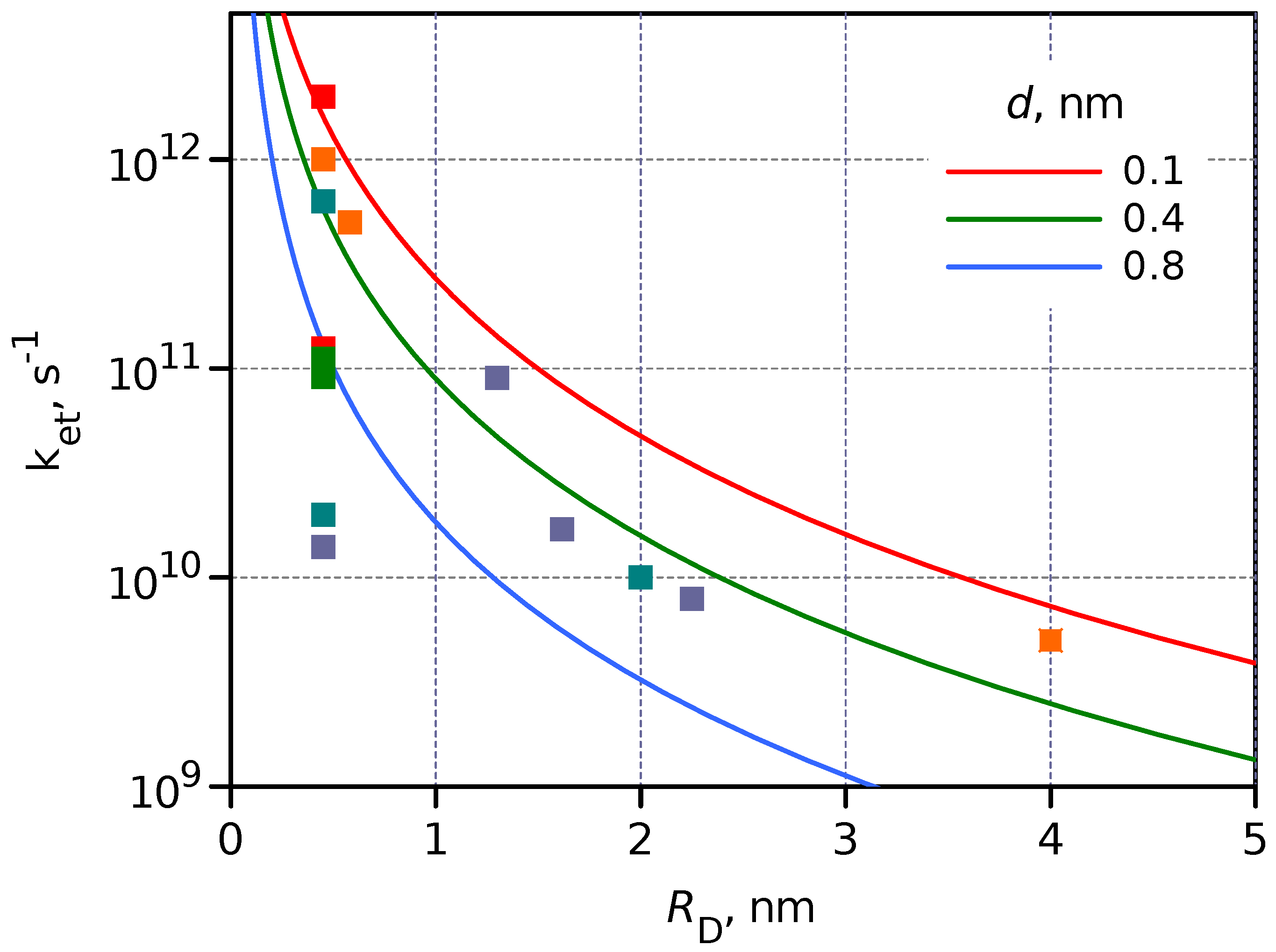
2.4. Interface between Molecular Dye and Semiconductor
3. Conclutions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| A | acceptor |
| CB | conduction band |
| CS | charge separation |
| D | donor |
| DA | donor–acceptor |
| ET | electron transfer |
| HOMO | highest occupied molecular orbital |
| LUMO | lowest unoccupied molecular orbital |
| P | porphyrin |
| Pc | phthalocyanine |
| PET | photo-induced electron transfer |
| QD | quantum dot |
| VB | valence band |
References
- Bozal-Ginesta, C.; Durrant, J.R. Artificial photosynthesis - concluding remarks. Faraday Discuss. 2019, 215, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D. Solar Fuels and Solar Chemicals Industry. Acc. Chem. Res. 2017, 50, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Bottari, G.; de la Torre, G.; Guldi, D.M.; Torres, T. Covalent and Noncovalent Phthalocyanine-Carbon Nanostructure Systems: Synthesis, Photoinduced Electron Transfer, and Application to Molecular Photovoltaics. Chem. Rev. 2010, 110, 6768–6816. [Google Scholar] [CrossRef] [PubMed]
- Barbara, P.F.; Meyer, T.J.; Ratner, M.A. Contemporary Issues in Electron Transfer Research. J. Phys. Chem. 1996, 100, 13148–13168. [Google Scholar] [CrossRef]
- Ito, A.; Meyer, T.J. The Golden Rule. Application for fun and profit in electron transfer, energy transfer, and excited-state decay. Phys. Chem. Chem. Phys. 2012, 14, 13731–13745. [Google Scholar] [CrossRef]
- Wasielewski, M. Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 1992, 92, 435–461. [Google Scholar] [CrossRef]
- Bolton, J.R.; Archer, M.D. Basic electron-transfer theory. Adv. Chem. Ser. 1991, 228, 7–23. [Google Scholar]
- Rumble, C.A.; Vauthey, E. Molecular Dynamics Simulations of Bimolecular Electron Transfer: The Distance-Dependent Electronic Coupling. J. Phys. Chem. B 2021, 125, 10527–10537. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Efimov, A.; Lemmetyinen, H. Porphyrin-Based Donor–Acceptor Dyads—Engineering the Linker and Tuning the Photoinduced Electron Transfer. In Handbook of Porphyrin Science; World Scientific publishing Co.: Singapore, 2016; Chapter 204; pp. 121–171. [Google Scholar] [CrossRef]
- Zieleniewska, A.; Lodermeyer, F.; Roth, A.; Guldi, D.M. Fullerenes—How 25 years of charge transfer chemistry have shaped our understanding of (interfacial) interactions. Chem. Soc. Rev. 2018, 47, 702–714. [Google Scholar] [CrossRef]
- KC, C.B.; D’Souza, F. Design and photochemical study of supramolecular donor–acceptor systems assembled via metal–ligand axial coordination. Coord. Chem. Rev. 2016, 322, 104–141. [Google Scholar] [CrossRef]
- Bottari, G.; de la Torre, G.; Guldi, D.M.; Torres, T. An exciting twenty-year journey exploring porphyrinoid-based photo- and electro-active systems. Coord. Chem. Rev. 2021, 428, 213605. [Google Scholar] [CrossRef]
- Kundu, S.; Patra, A. Nanoscale Strategies for Light Harvesting. Chem. Rev. 2017, 117, 712–757. [Google Scholar] [CrossRef] [PubMed]
- Ponseca, C.S.; Chábera, P.; Uhlig, J.; Persson, P.; Sundström, V. Ultrafast Electron Dynamics in Solar Energy Conversion. Chem. Rev. 2017, 117, 10940–11024. [Google Scholar] [CrossRef]
- Harris, R.D.; Bettis Homan, S.; Kodaimati, M.; He, C.; Nepomnyashchii, A.B.; Swenson, N.K.; Lian, S.; Calzada, R.; Weiss, E.A. Electronic Processes within Quantum Dot-Molecule Complexes. Chem. Rev. 2016, 116, 12865–12919. [Google Scholar] [CrossRef]
- Jasieniak, J.; Califano, M.; Watkins, S.E. Size-Dependent Valence and Conduction Band-Edge Energies of Semiconductor Nanocrystals. ACS Nano 2011, 5, 5888–5902. [Google Scholar] [CrossRef]
- Bang, J.H.; Kamat, P.V. CdSe Quantum Dot–Fullerene Hybrid Nanocomposite for Solar Energy Conversion: Electron Transfer and Photoelectrochemistry. ACS Nano 2011, 5, 9421–9427. [Google Scholar] [CrossRef]
- Virkki, K.; Demir, S.; Lemmetyinen, H.; Tkachenko, N.V. Photoinduced Electron Transfer in CdSe/ZnS Quantum Dot–Fullerene Hybrids. J. Phys. Chem. C 2015, 119, 17561–17572. [Google Scholar] [CrossRef]
- Xu, Z.; Cotlet, M. Quantum Dot-Bridge-Fullerene Heterodimers with Controlled Photoinduced Electron Transfer. Angew. Chem. Intern. Ed. 2011, 50, 6079–6083. [Google Scholar] [CrossRef]
- Itskos, G.; Papagiorgis, P.; Tsokkou, D.; Othonos, A.; Hermerschmidt, F.; Economopoulos, S.P.; Yarema, M.; Heiss, W.; Choulis, S. Size-Dependent Charge Transfer in Blends of PbS Quantum Dots with a Low-Gap Silicon-Bridged Copolymer. Adv. Energy Mater. 2013, 3, 1490–1499. [Google Scholar] [CrossRef]
- Stewart, M.H.; Huston, A.L.; Scott, A.M.; Oh, E.; Algar, W.R.; Deschamps, J.R.; Susumu, K.; Jain, V.; Prasuhn, D.E.; Blanco-Canosa, J.; et al. Competition between Förster Resonance Energy Transfer and Electron Transfer in Stoichiometrically Assembled Semiconductor Quantum Dot–Fullerene Conjugates. ACS Nano 2013, 7, 9489–9505. [Google Scholar] [CrossRef] [PubMed]
- Ragoussi, M.E.; de la Torre, G.; Torres, T. Tuning the Electronic Properties of Porphyrin Dyes: Effects of meso Substitution on Their Optical and Electrochemical Behaviour. Eur. J. Org. Chem. 2013, 2013, 2832–2840. [Google Scholar] [CrossRef]
- Li, R.; Zhang, X.; Zhu, P.; Ng, D.K.P.; Kobayashi, N.; Jiang, J. Electron-Donating or -Withdrawing Nature of Substituents Revealed by the Electrochemistry of Metal-Free Phthalocyanines. Inorg. Chem. 2006, 45, 2327–2334. [Google Scholar] [CrossRef]
- Imahori, H.; Tkachenko, N.V.; Vehmanen, V.; Tamaki, K.; Lemmetyinen, H.; Sakata, Y.; Fukuzumi, S. An Extremely Small Reorganization Energy of Electron Transfer in Porphyrin-Fullerene Dyad. JPCA 2001, 105, 1750–1756. [Google Scholar] [CrossRef]
- Guldi, D.M.; Prato, M. Excited-State Properties of C60 Fullerene Derivatives. Acc. Chem. Res. 2000, 33, 695–703. [Google Scholar] [CrossRef]
- Waskasi, M.M.; Kodis, G.; Moore, A.L.; Moore, T.A.; Gust, D.; Matyushov, D.V. Marcus Bell-Shaped Electron Transfer Kinetics Observed in an Arrhenius Plot. J. Am. Chem. Soc. 2016, 138, 9251–9257. [Google Scholar] [CrossRef] [PubMed]
- Kuciauskas, D.; Liddell, P.A.; Lin, S.; Stone, S.G.; Moore, A.L.; Moore, T.A.; Gust, D. Photoinduced Electron Transfer in Carotenoporphyrin-Fullerene Triads: Temperature and Solvent Effects. J. Phys. Chem. B 2000, 104, 4307–4321. [Google Scholar] [CrossRef]
- Chukharev, V.; Tkachenko, N.V.; Efimov, A.; Guldi, D.M.; Hirsch, A.; Scheloske, M.; Lemmetyinen, H. Tuning the Ground-State and Excited-State Interchromophore Interactions in Porphyrin-Fullerene π-Stacks. J. Phys. Chem. B 2004, 108, 16377–16385. [Google Scholar] [CrossRef]
- Vail, S.A.; Schuster, D.I.; Guldi, D.M.; Isosomppi, M.; Tkachenko, N.; Lemmetyinen, H.; Palkar, A.; Echegoyen, L.; Chen, X.; Zhang, J.Z.H. Energy and Electron Transfer in β-Alkynyl-Linked Porphyrin-[60]Fullerene Dyads. J. Phys. Chem. B 2006, 110, 14155–14166. [Google Scholar] [CrossRef]
- Higashino, T.; Yamada, T.; Yamamoto, M.; Furube, A.; Tkachenko, N.V.; Miura, T.; Kobori, Y.; Jono, R.; Yamashita, K.; Imahori, H. Remarkable Dependence of the Final Charge Separation Efficiency on the Donor–Acceptor Interaction in Photoinduced Electron Transfer. Angew. Chem. Int. Ed. 2016, 55, 629–633. [Google Scholar] [CrossRef]
- Strelnikov, A.A.; Konev, A.S.; Levin, O.V.; Khlebnikov, A.F.; Iwasaki, A.; Yamanouchi, K.; Tkachenko, N.V. Switching Competition between Electron and Energy Transfers in Porphyrin–Fullerene Dyads. J. Phys. Chem. B 2020, 124, 10899–10912. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Tkachenko, N.V.; Efimov, A.; Lehtivuori, H.; Ohkubo, K.; Fukuzumi, S.; Lemmetyinen, H. Exciplex Mediated Photoinduced Electron Transfer Reactions of Phthalocyanine-Fullerene Dyads. J. Phys. Chem. A 2008, 112, 6884–6892. [Google Scholar] [CrossRef] [PubMed]
- Gouloumis, A.; de la Escosura, A.; Vázquez, P.; Torres, T.; Kahnt, A.; Guldi, D.M.; Neugebauer, H.; Winder, C.; Drees, M.; Sariciftci, N.S. Photoinduced Electron Transfer in a New Bis(C60)-Phthalocyanine Triad. Org. Lett. 2006, 8, 5187–5190. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Zhu, H.; Jin, S.; Zhan, W.; Lian, T. Poisson-Distributed Electron-Transfer Dynamics from Single Quantum Dots to C60 Molecules. ACS Nano 2011, 5, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; George, L.; Tkachenko, N.V. Charge transfer dynamics in CsPbBr3 perovskite quantum dots–anthraquinone/fullerene (C60) hybrids. Nanoscale 2019, 11, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Fazio, M.; Durandin, A.; Tkachenko, N.; Niemi, M.; Lemmetyinen, H.; Schuster, D. Synthesis, Conformational Interconversion, and Photophysics of Tethered Porphyrin–Fullerene Dyads with Parachute Topology. Chem. Europ. J. 2009, 15, 7698–7705. [Google Scholar] [CrossRef]
- Imahori, H.; Tamaki, K.; Guldi, D.M.; Luo, C.; Fujitsuka, M.; Ito, O.; Sakata, Y.; Fukuzumi, S. Modulating Charge Separation and Charge Recombination Dynamics in Porphyrin-Fullerene Linked Dyads and Triads: Marcus-Normal versus Inverted Region. J. Am. Chem. Soc. 2001, 123, 2607–2617. [Google Scholar] [CrossRef]
- Ito, A.; Fang, Z.; Brennaman, M.K.; Meyer, T.J. Long-range photoinduced electron transfer dynamics in rigid media. Phys. Chem. Chem. Phys. 2014, 16, 4880–4891. [Google Scholar] [CrossRef]
- Chaban, V.V.; Prezhdo, V.V.; Prezhdo, O.V. Covalent Linking Greatly Enhances Photoinduced Electron Transfer in Fullerene-Quantum Dot Nanocomposites: Time-Domain Ab Initio Study. J. Phys. Chem. Lett. 2013, 4, 1–6. [Google Scholar] [CrossRef]
- Lian, S.; Kodaimati, M.S.; Dolzhnikov, D.S.; Calzada, R.; Weiss, E.A. Powering a CO2 Reduction Catalyst with Visible Light through Multiple Sub-picosecond Electron Transfers from a Quantum Dot. J. Am. Chem. Soc. 2017, 139, 8931–8938. [Google Scholar] [CrossRef]
- Ricks, A.B.; Brown, K.E.; Wenninger, M.; Karlen, S.D.; Berlin, Y.A.; Co, D.T.; Wasielewski, M.R. Exponential Distance Dependence of Photoinitiated Stepwise Electron Transfer in Donor–Bridge–Acceptor Molecules: Implications for Wirelike Behavior. J. Am. Chem. Soc. 2012, 134, 4581–4588. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.B.; O’Keeffe, M.; Ruoff, R.S. Van Der Waals Surface Areas and Volumes of Fullerenes. J. Phys. Chem. 1994, 98, 9465–9469. [Google Scholar] [CrossRef]
- Ye, S.; Kathiravan, A.; Hayashi, H.; Tong, Y.; Infahsaeng, Y.; Chabera, P.; Pascher, T.; Yartsev, A.P.; Isoda, S.; Imahori, H.; et al. Role of Adsorption Structures of Zn-Porphyrin on TiO2 in Dye-Sensitized Solar Cells Studied by Sum Frequency Generation Vibrational Spectroscopy and Ultrafast Spectroscopy. J. Phys. Chem. C 2013, 117, 6066–6080. [Google Scholar] [CrossRef]
- Stranius, K.; George, L.; Efimov, A.; Ruoko, T.P.; Pohjola, J.; Tkachenko, N.V. Photophysical Study of a Self-Assembled Donor-Acceptor Two-Layer Film on TiO2. Langmuir 2015, 31, 944–952. [Google Scholar] [CrossRef]
- Martín, C.; Ziółek, M.; Douhal, A. Ultrafast and fast charge separation processes in real dye-sensitized solar cells. J. Photochem. Photobiol. C 2016, 26, 1–30. [Google Scholar] [CrossRef]
- Virkki, K.; Tervola, E.; Ince, M.; Torres, T.; Tkachenko, N.V. Comparison of electron injection and recombination on TiO2 nanoparticles and ZnO nanorods photosensitized by phthalocyanine. R. Soc. Open Sci. 2018, 5, 180323. [Google Scholar] [CrossRef]
- Němec, H.; Rochford, J.; Taratula, O.; Galoppini, E.; Kužel, P.; Polívka, T.; Yartsev, A.; Sundström, V. Influence of the Electron-Cation Interaction on Electron Mobility in Dye-Sensitized ZnO and TiO2 Nanocrystals: A Study Using Ultrafast Terahertz Spectroscopy. Phys. Rev. Lett. 2010, 104, 197401. [Google Scholar] [CrossRef]
- Jiang, J.; Alsam, A.; Wang, S.; Aly, S.M.; Pan, Z.; Mohammed, O.F.; Schanze, K.S. Effect of Conjugation Length on Photoinduced Charge Transfer in π-Conjugated Oligomer-Acceptor Dyads. J. Phys. Chem. A 2017, 121, 4891–4901. [Google Scholar] [CrossRef]
- Umeyama, T.; Imahori, H. Electron transfer and exciplex chemistry of functionalized nanocarbons: Effects of electronic coupling and donor dimerization. Nanoscale Horiz. 2018, 3, 352–366. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | , nm | d, nm | , eV | Reference, Comments | |
|---|---|---|---|---|---|
| Molecular | |||||
| H2P | 0.45 | 0.3 | 27 a | 0.67 | [27] |
| H2P | 0.45 | 0.7 | 91 | [28] | |
| H2P | 0.45 | 0 | 200 b | [29] ignoring exciplex | |
| ZnP | 0.45 | 0.5 | 110 b | [30] | |
| ZnP | 0.45 | 0 | 500 | [29] ignoring exciplex | |
| ZnP | 0.45 | 1.1 | 14 b | [31] | |
| H2P | 0.45 | 0.6 | 150 | 0.47 | [32] calculated |
| H2Pc | 0.58 | 0.1 | 280 | 0.55 | [33] |
| ZnPc | 0.58 | 0.1 | 190 | 0.65 | [33] |
| ZnPc | 0.58 | 0.2 | 62 | [34] | |
| QD | |||||
| CdSe | 2.3 | 0.6 | 1 | 0.4 | [20] |
| CdSe | 2.3 | 1.2 | 0.33 | 0.4 | [20] |
| CdSe | 1.3 | 1 | 1.8 | 0.29 | [18] recalculated to 1:1 |
| CdSe | 1.6 | 1 | 0.2 | 0.46 | [18] recalculated to 1:1 |
| CdSe | 2.3 | 1 | 0.05 | 0.62 | [18] recalculated to 1:1 |
| CdSe | 2.2 | 0.2 | 10 | [35] | |
| CdSe | 1.2–2.5 | 0.1–0.3 | 1.5–10 | 0.7–1.0 | [19] estimated |
| CsPbBr3 | ≈4 | 0.2 | 5 | 0.64 | [36] inaccurate d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkachenko, N.V. Effect of the Donor/Acceptor Size on the Rate of Photo-Induced Electron Transfer. Photochem 2022, 2, 918-931. https://doi.org/10.3390/photochem2040059
Tkachenko NV. Effect of the Donor/Acceptor Size on the Rate of Photo-Induced Electron Transfer. Photochem. 2022; 2(4):918-931. https://doi.org/10.3390/photochem2040059
Chicago/Turabian StyleTkachenko, Nikolai V. 2022. "Effect of the Donor/Acceptor Size on the Rate of Photo-Induced Electron Transfer" Photochem 2, no. 4: 918-931. https://doi.org/10.3390/photochem2040059
APA StyleTkachenko, N. V. (2022). Effect of the Donor/Acceptor Size on the Rate of Photo-Induced Electron Transfer. Photochem, 2(4), 918-931. https://doi.org/10.3390/photochem2040059