
A Photochemical Overview of Molecular Solar Thermal Energy Storage

 , , and
, , and 
Abstract
:1. Introduction
2. Requisites for MOST Systems: Optical Properties
- A high quantum yield (Φ = 1) for the photogeneration of the metastable photoisomer.
- A photochemically inactive (or non-absorbing) photoisomer.
- Negligible degradation of both the photoactive molecule and its photoisomer after multiple cycles, especially moving towards higher temperatures.
2.1. Solar Match
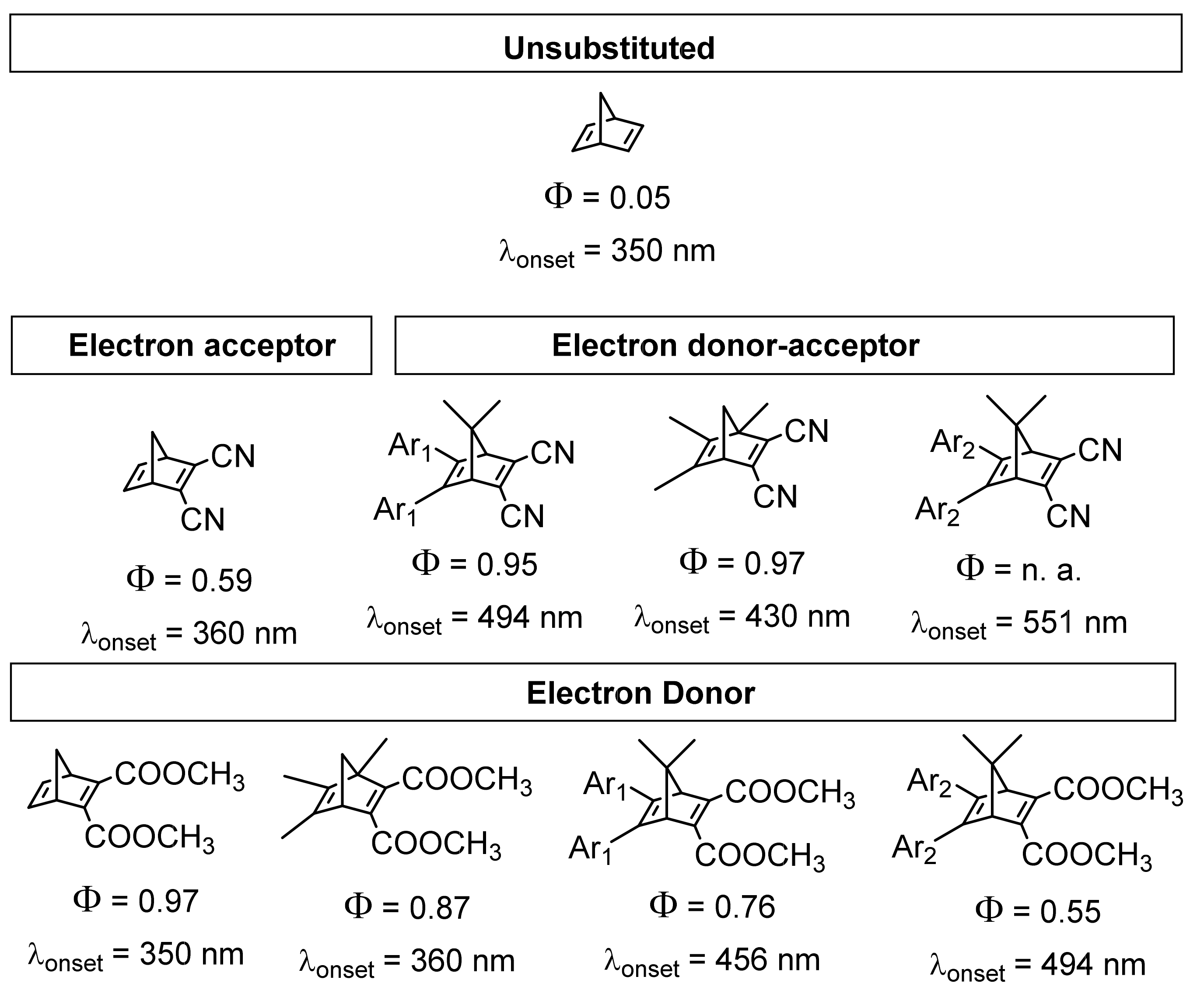
2.2. Quantum Yield
2.3. Storage Energy Density
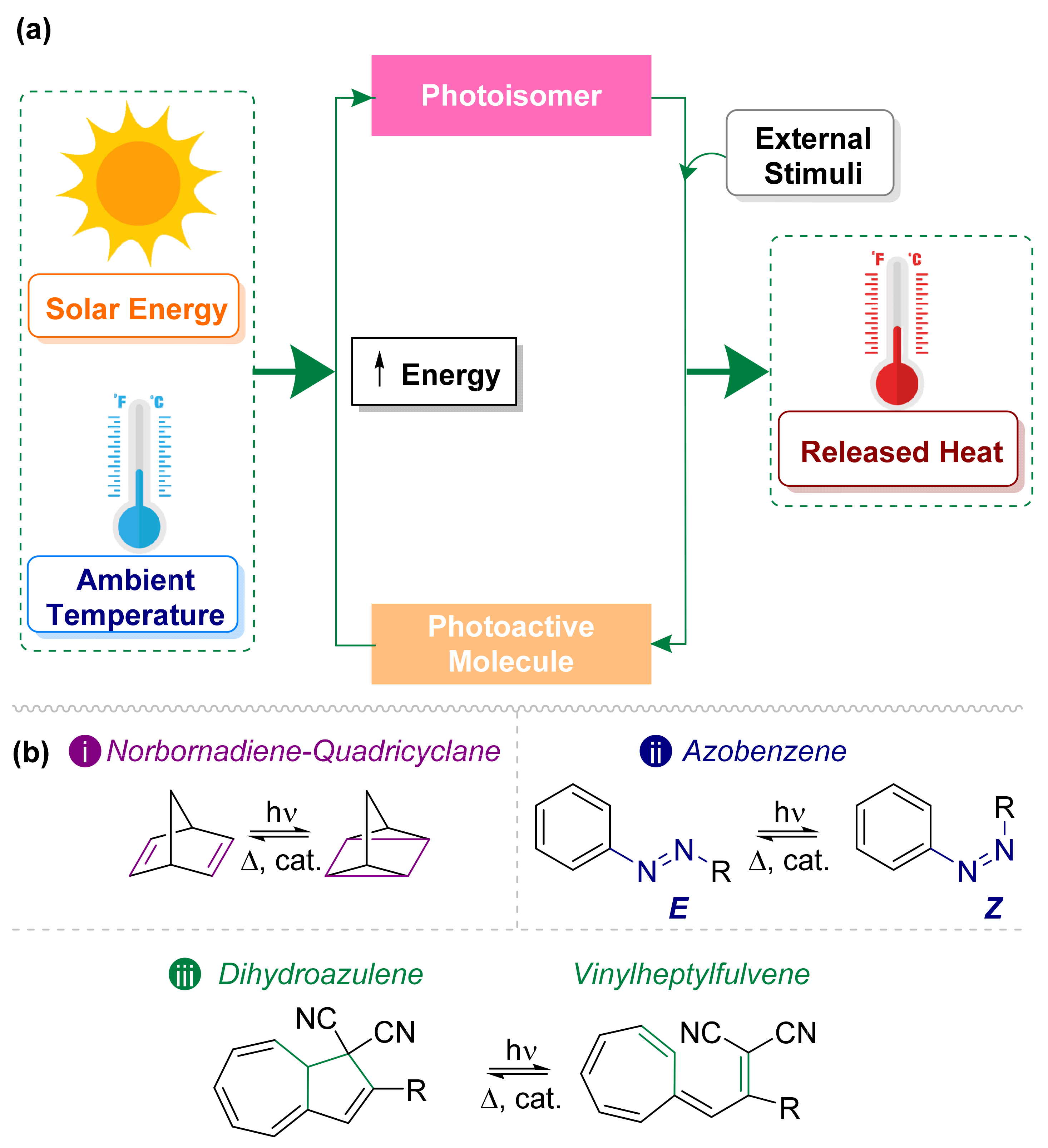
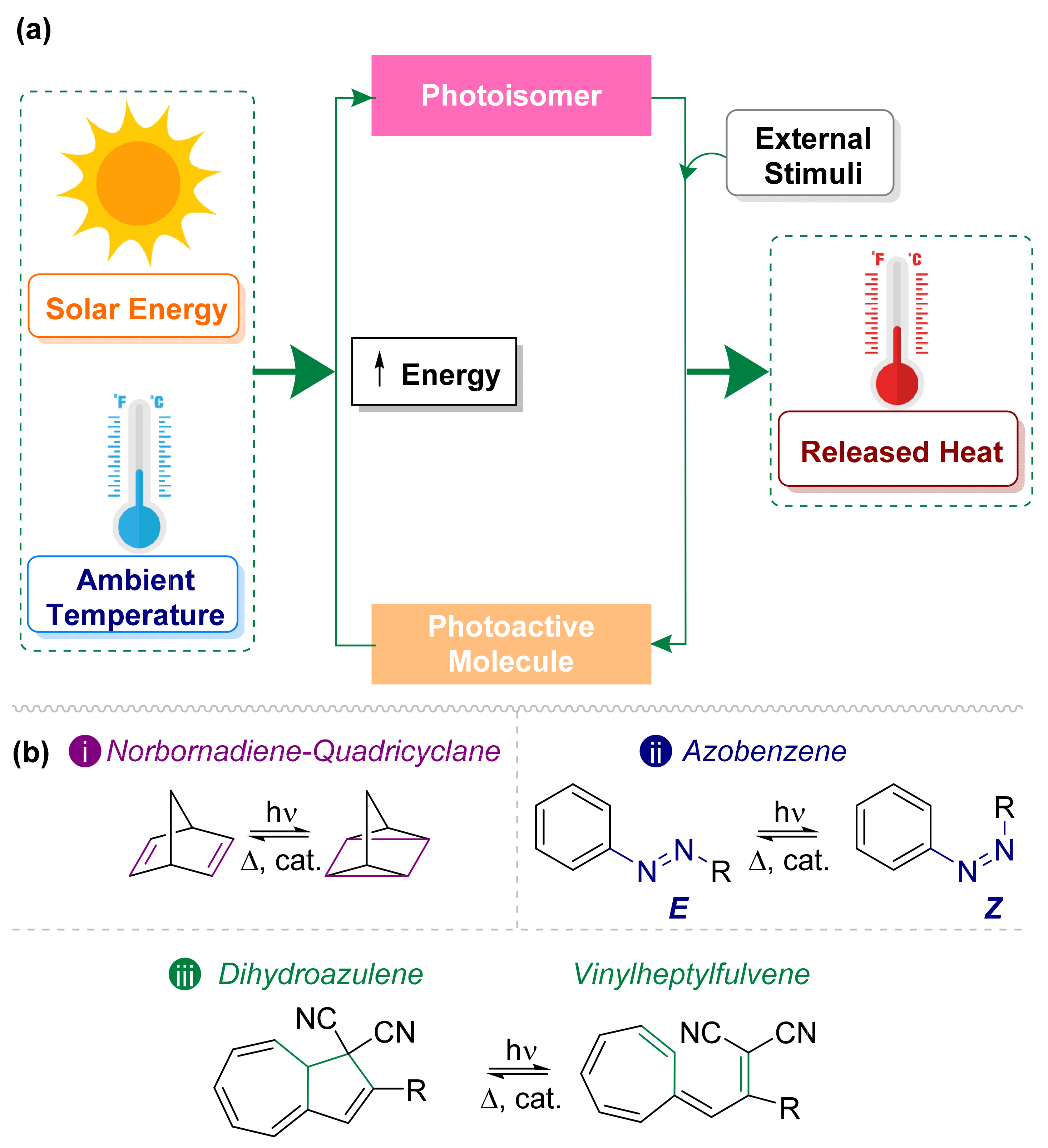
3. Photoswitches Used in MOST Technology
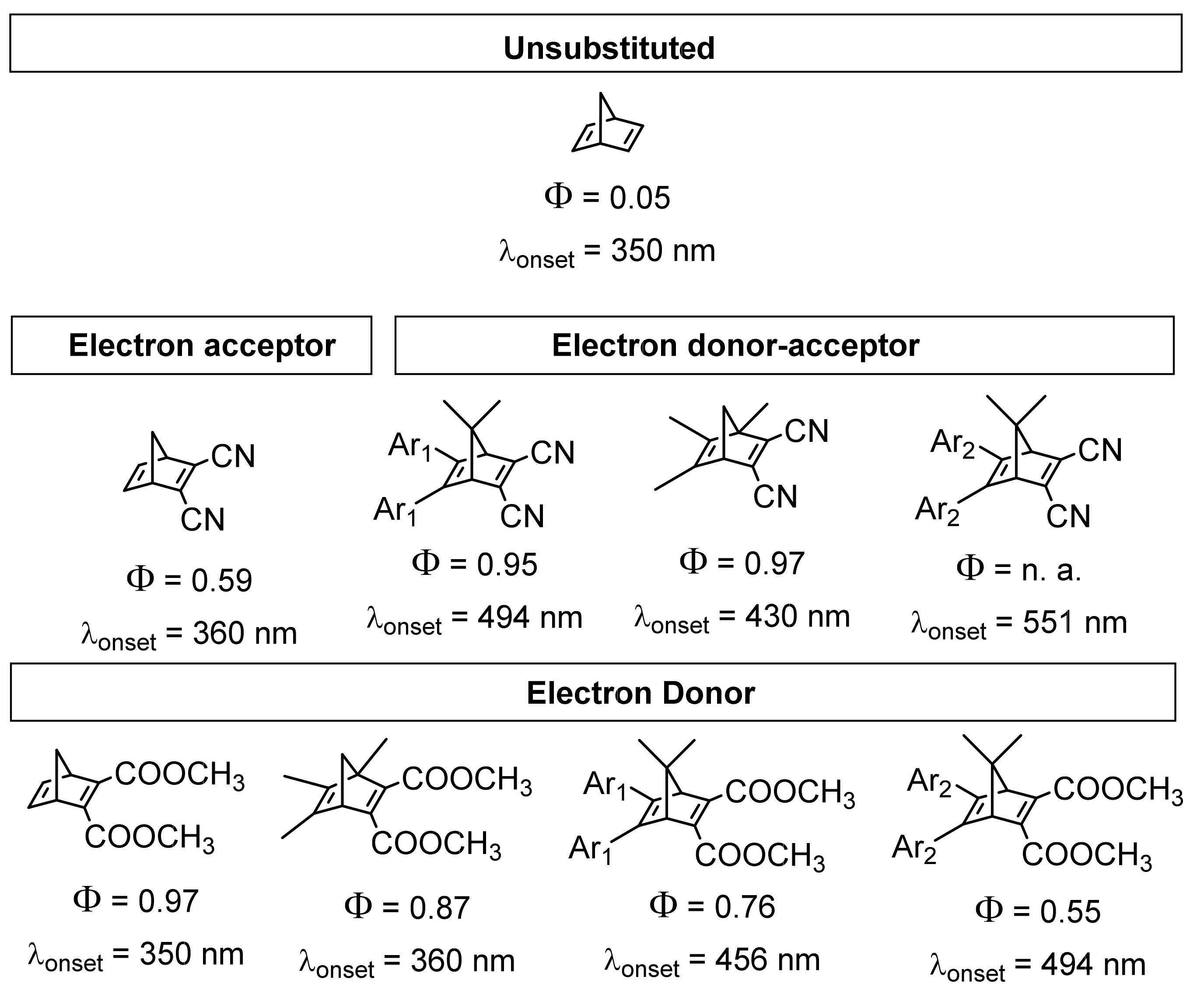
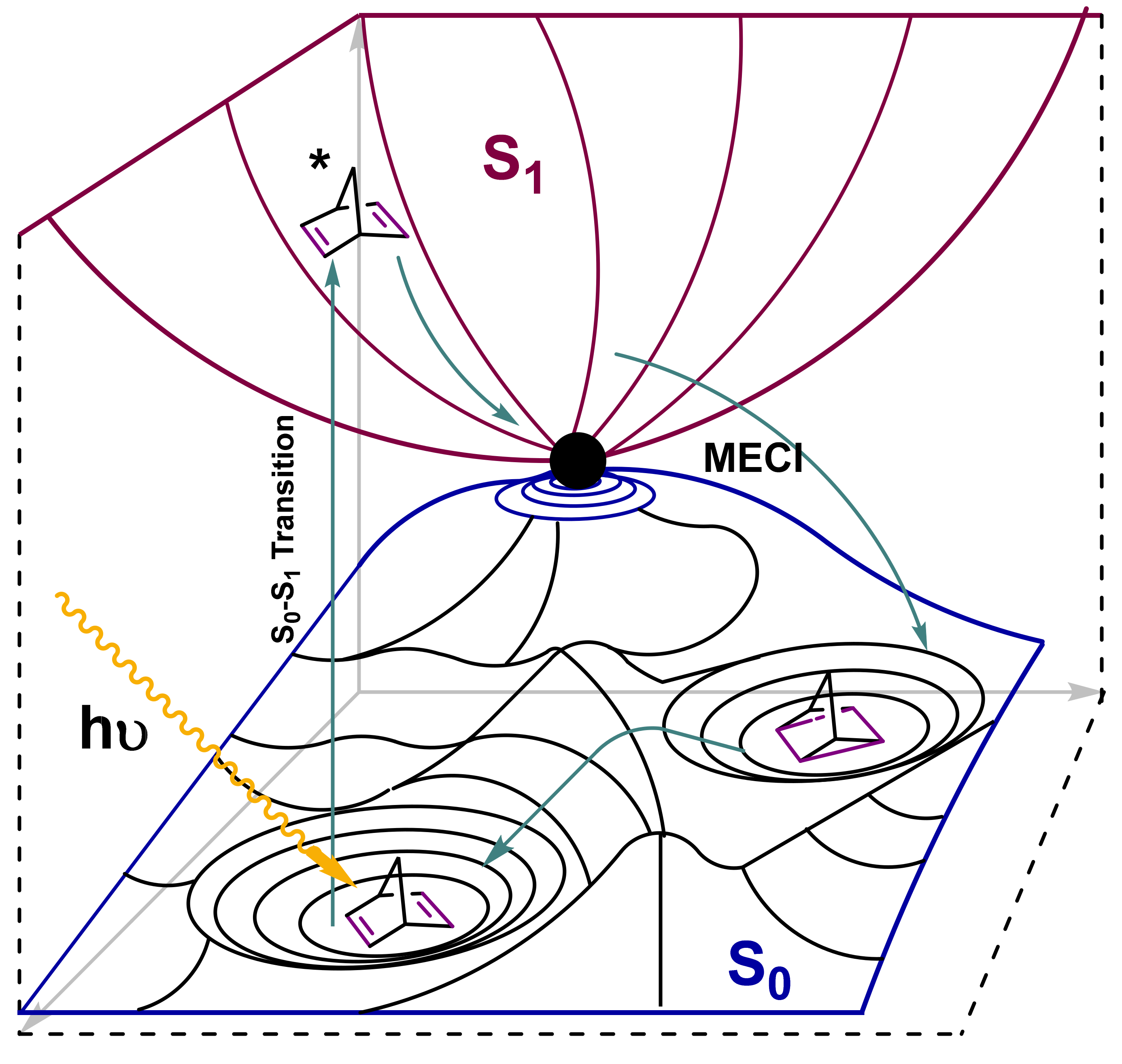
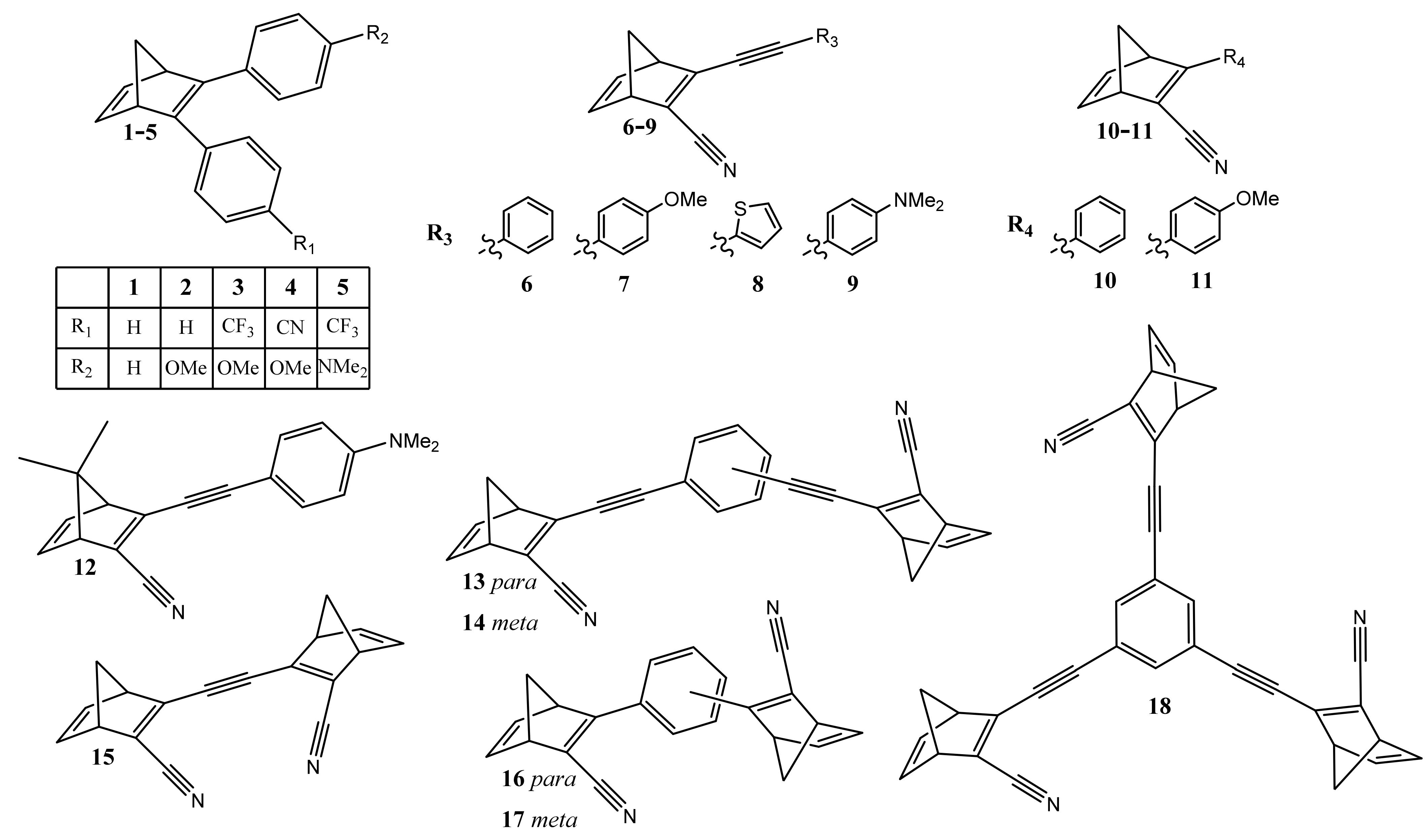
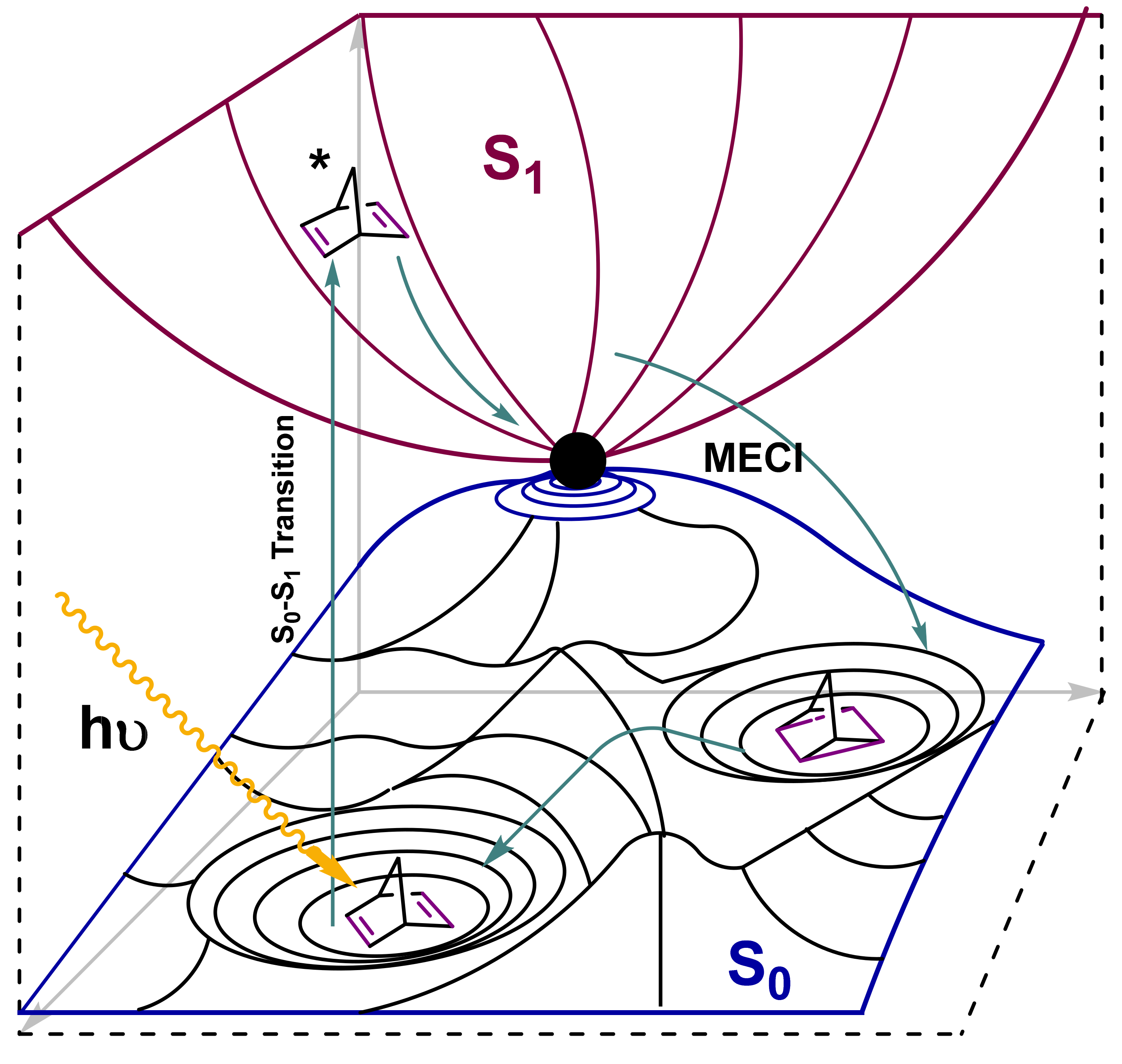
3.1. Norbornadiene/Quadricyclane Couple
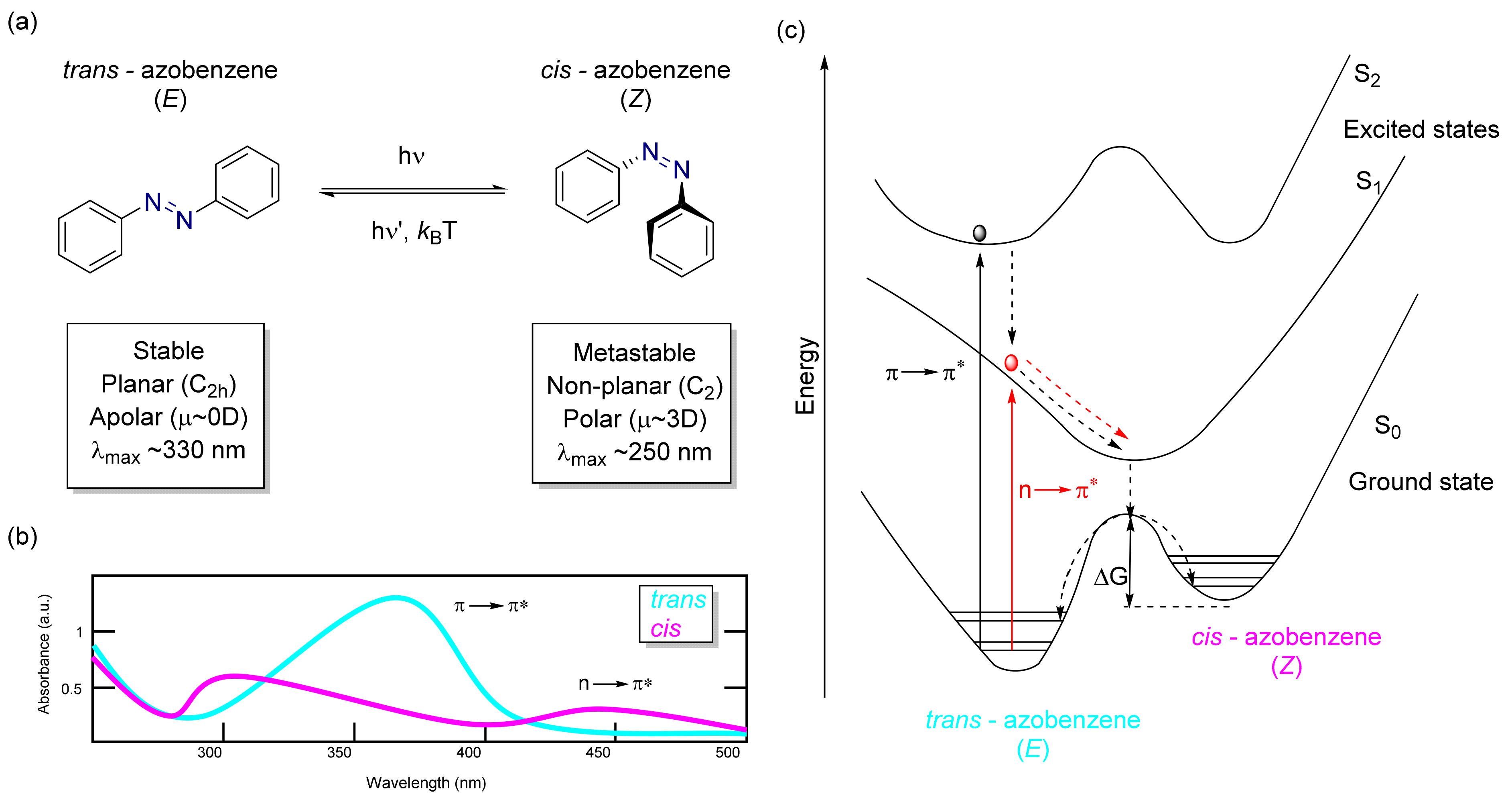
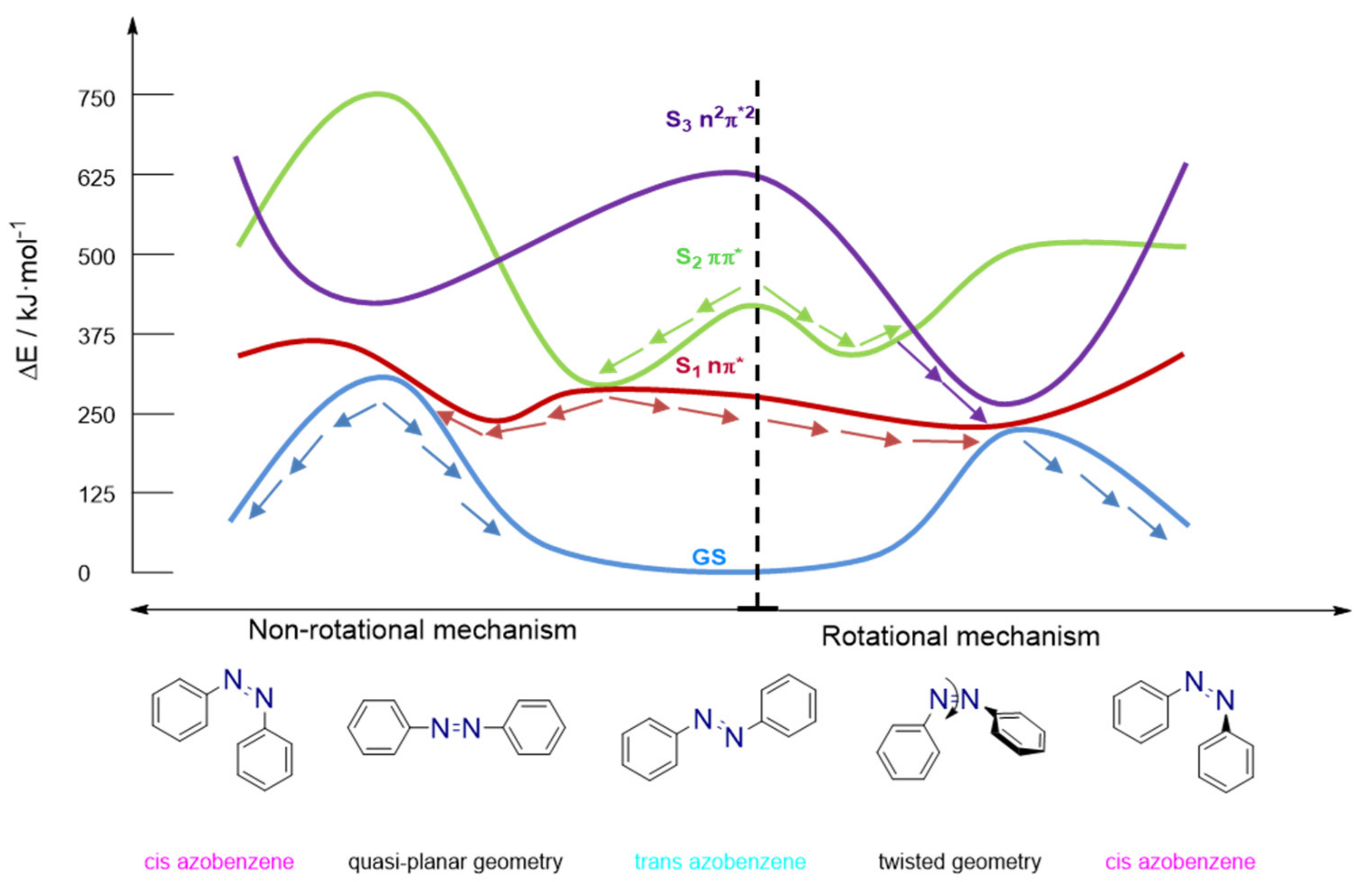
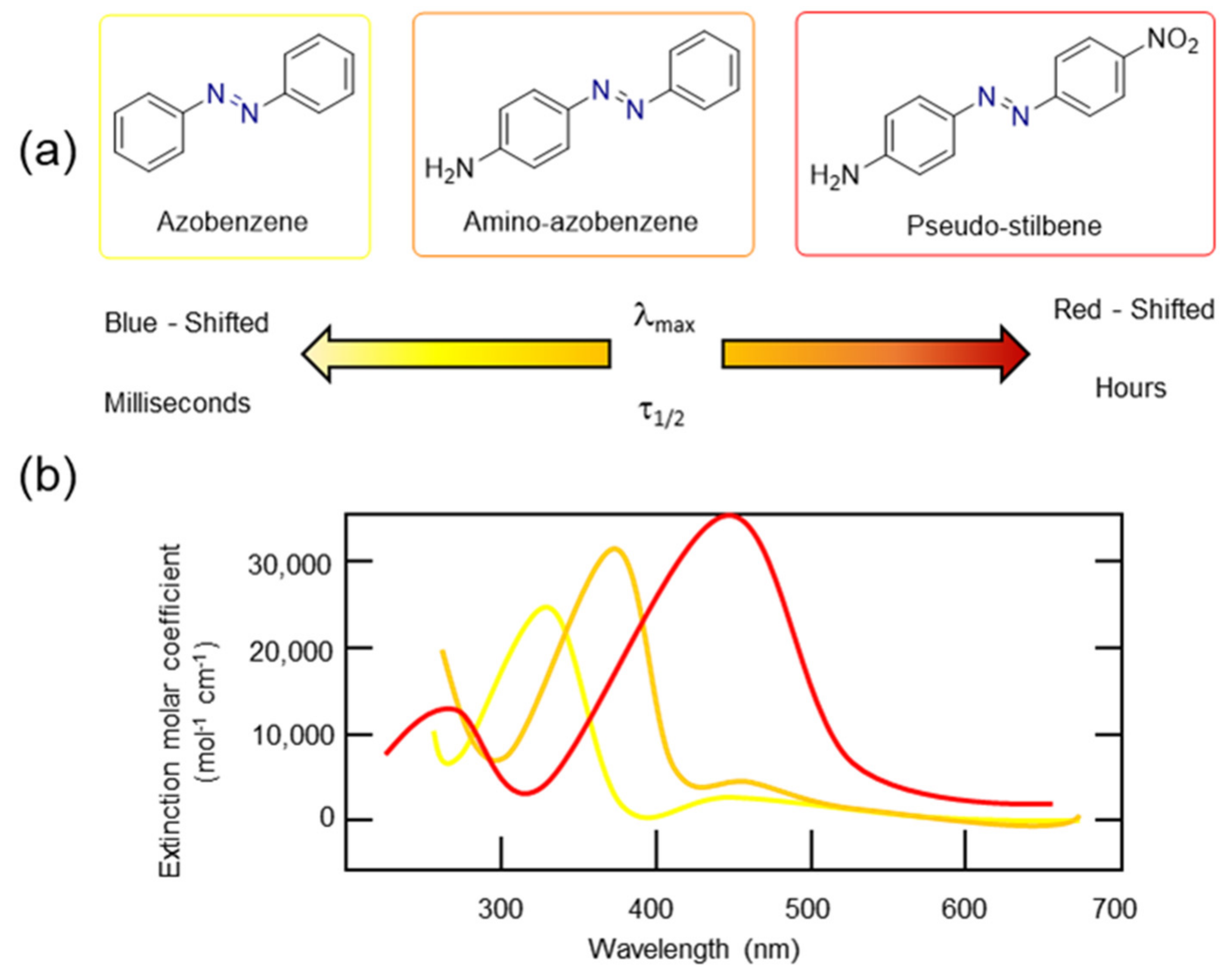
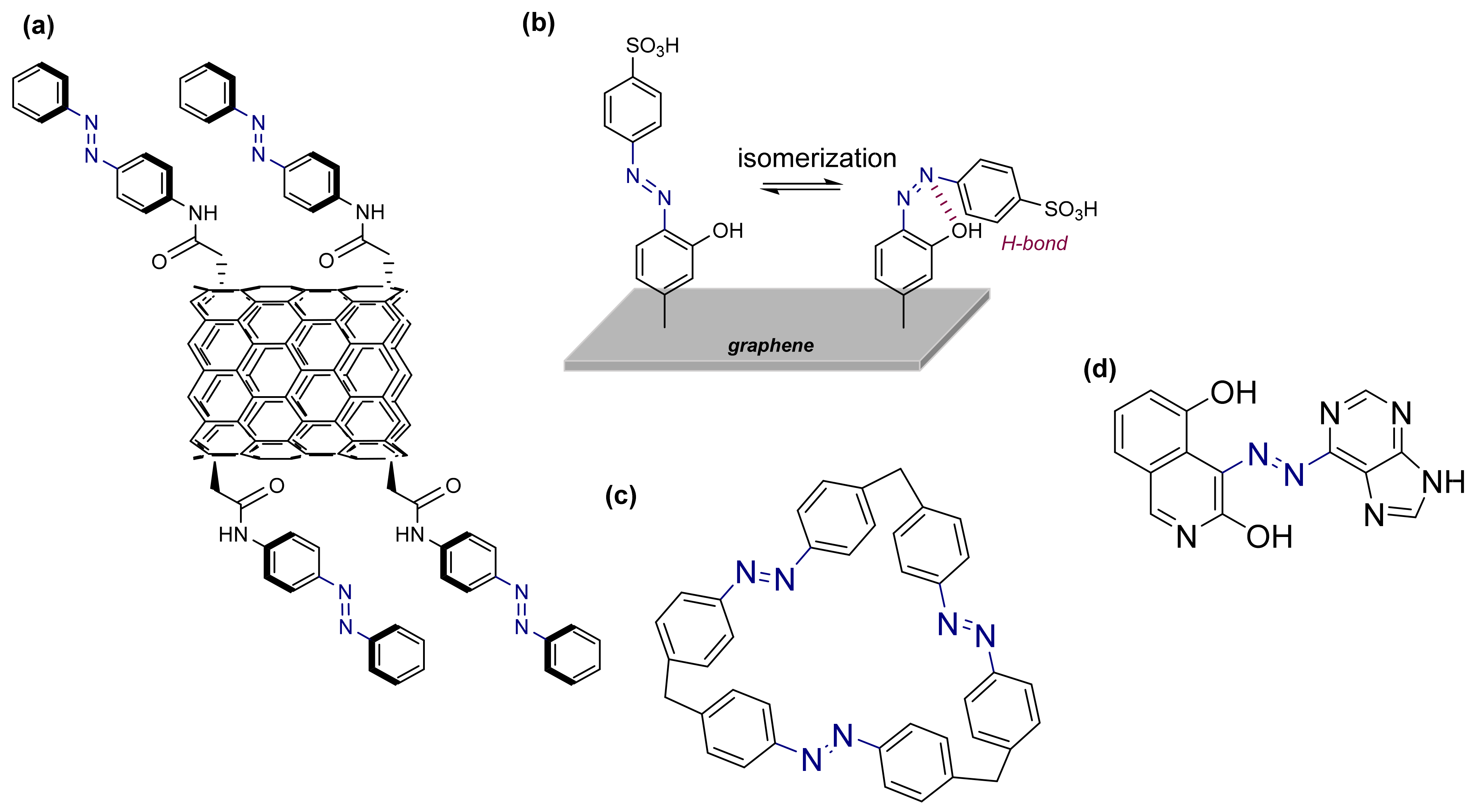
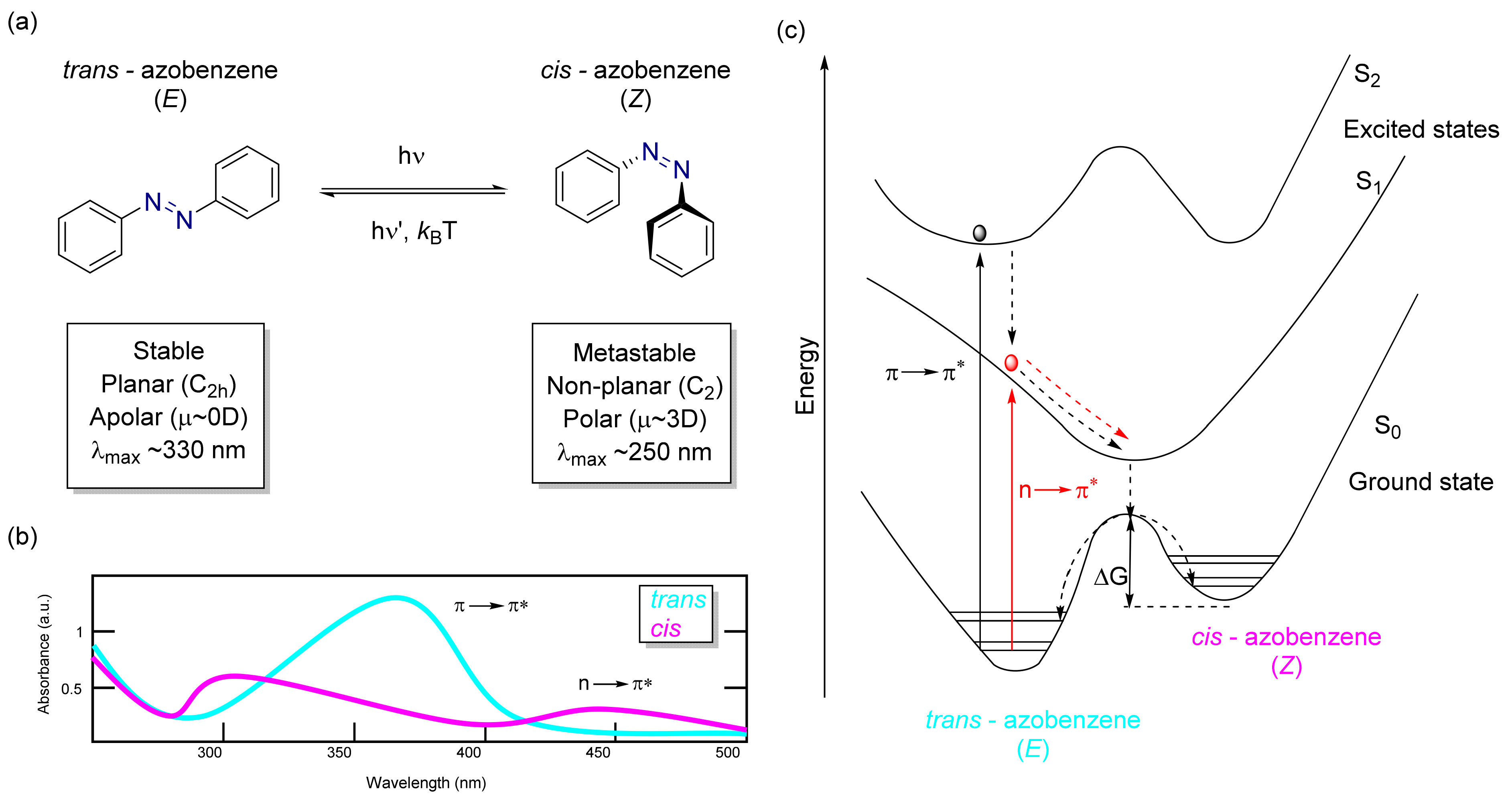
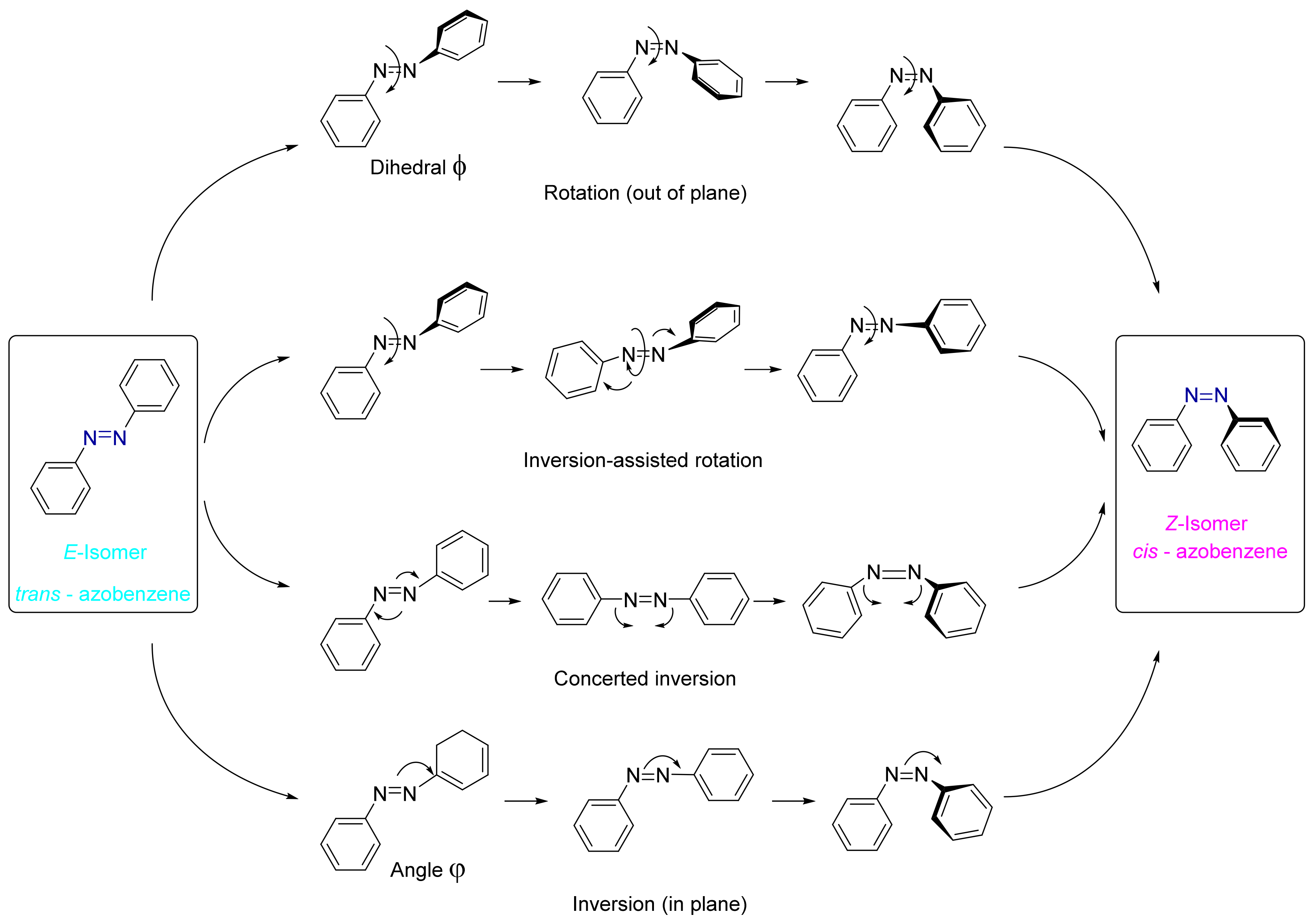
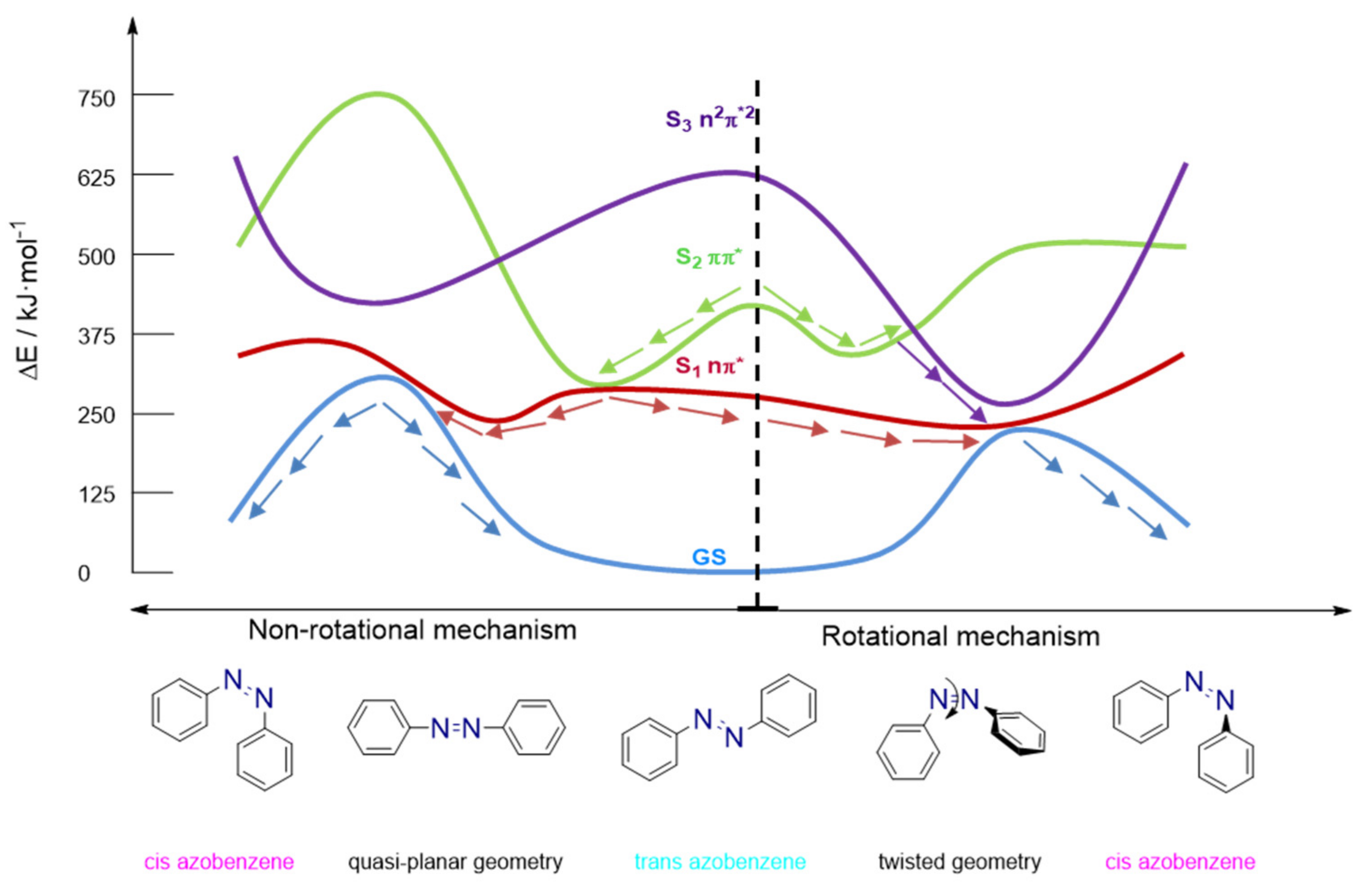
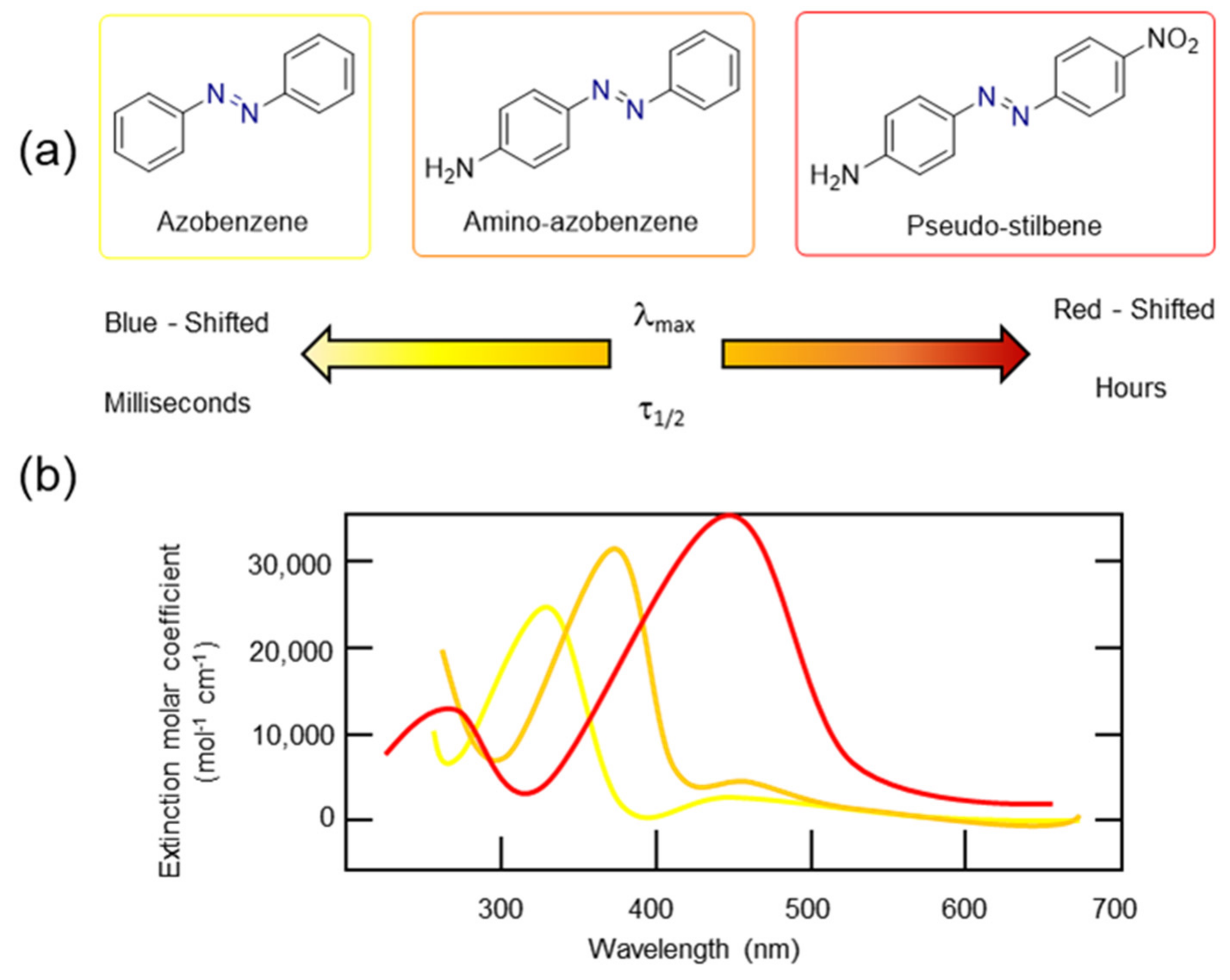
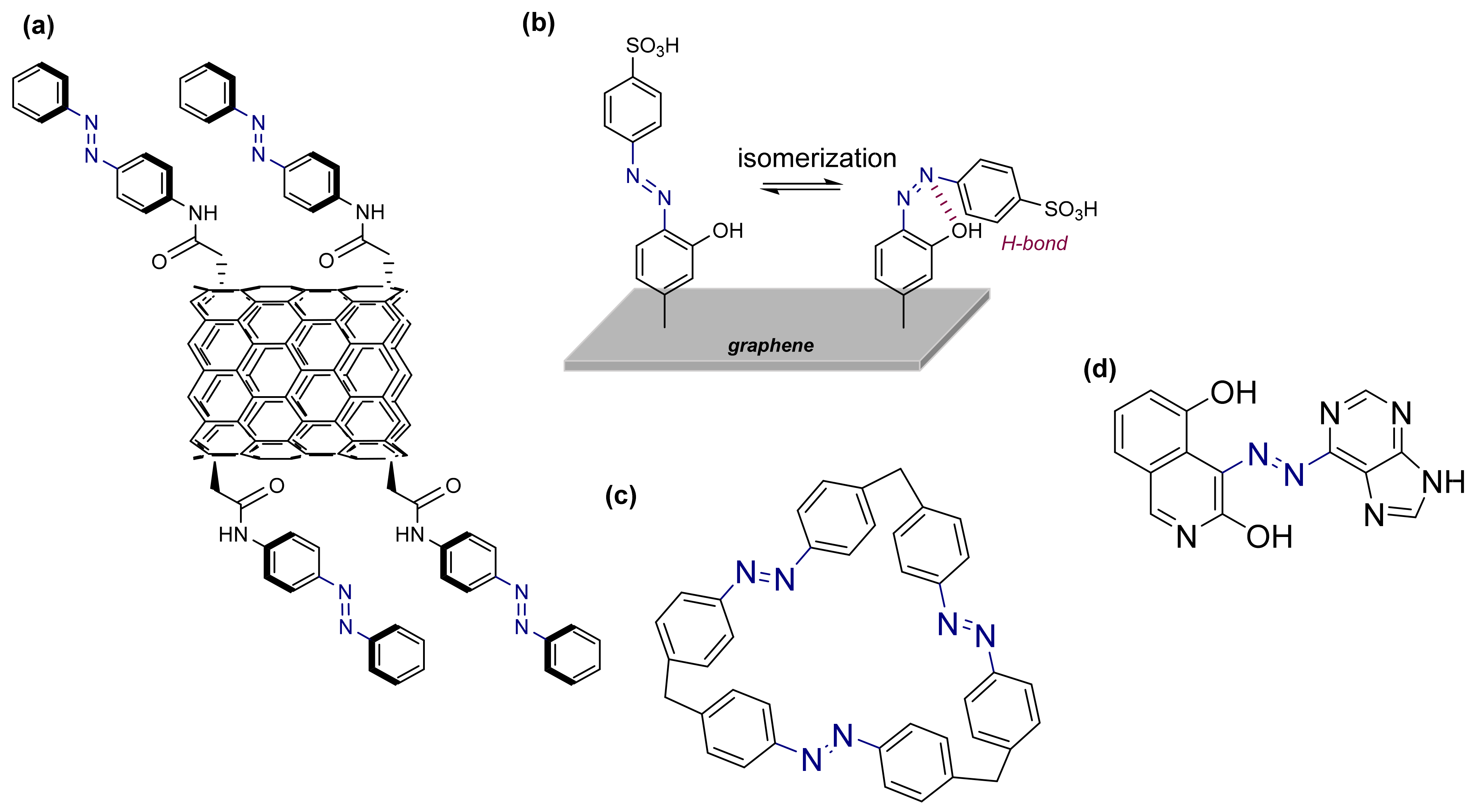
3.2. Azobenzene Photoswitches
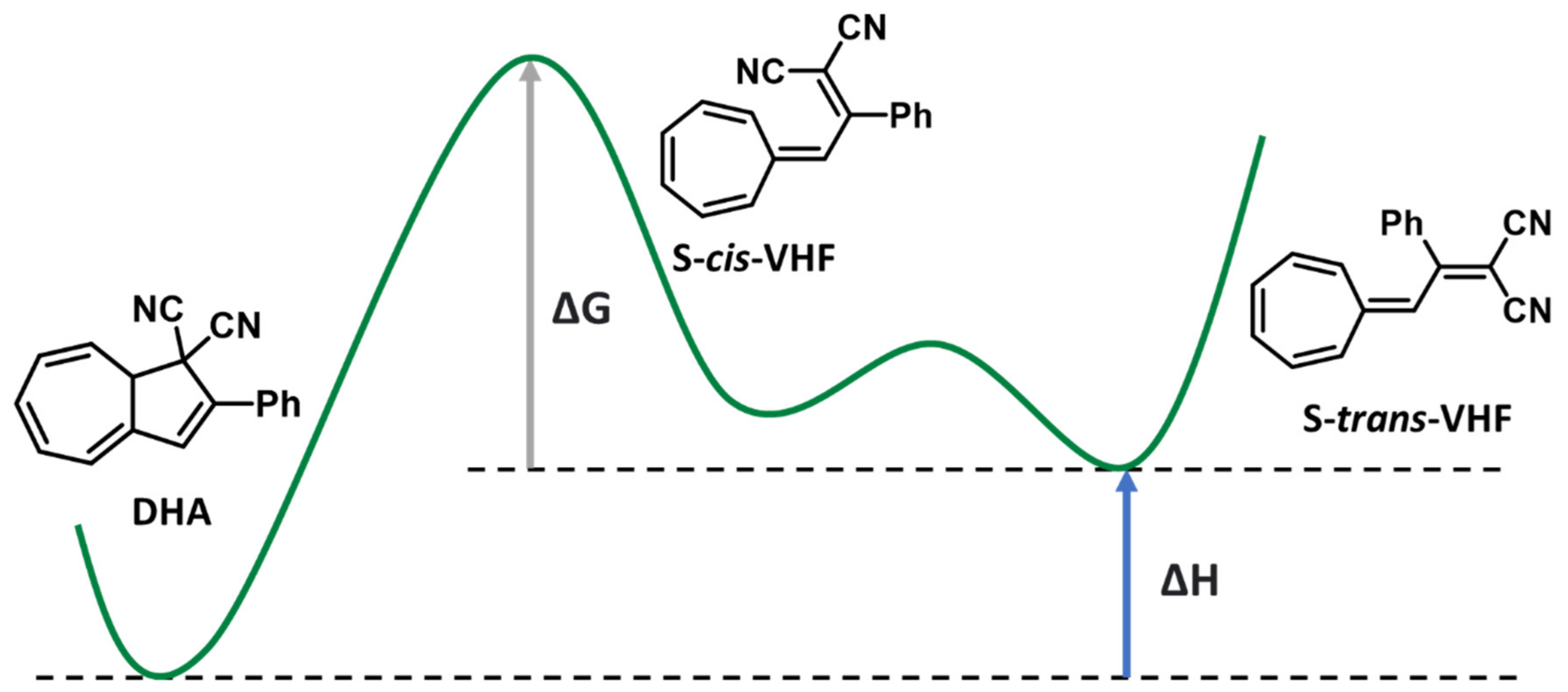
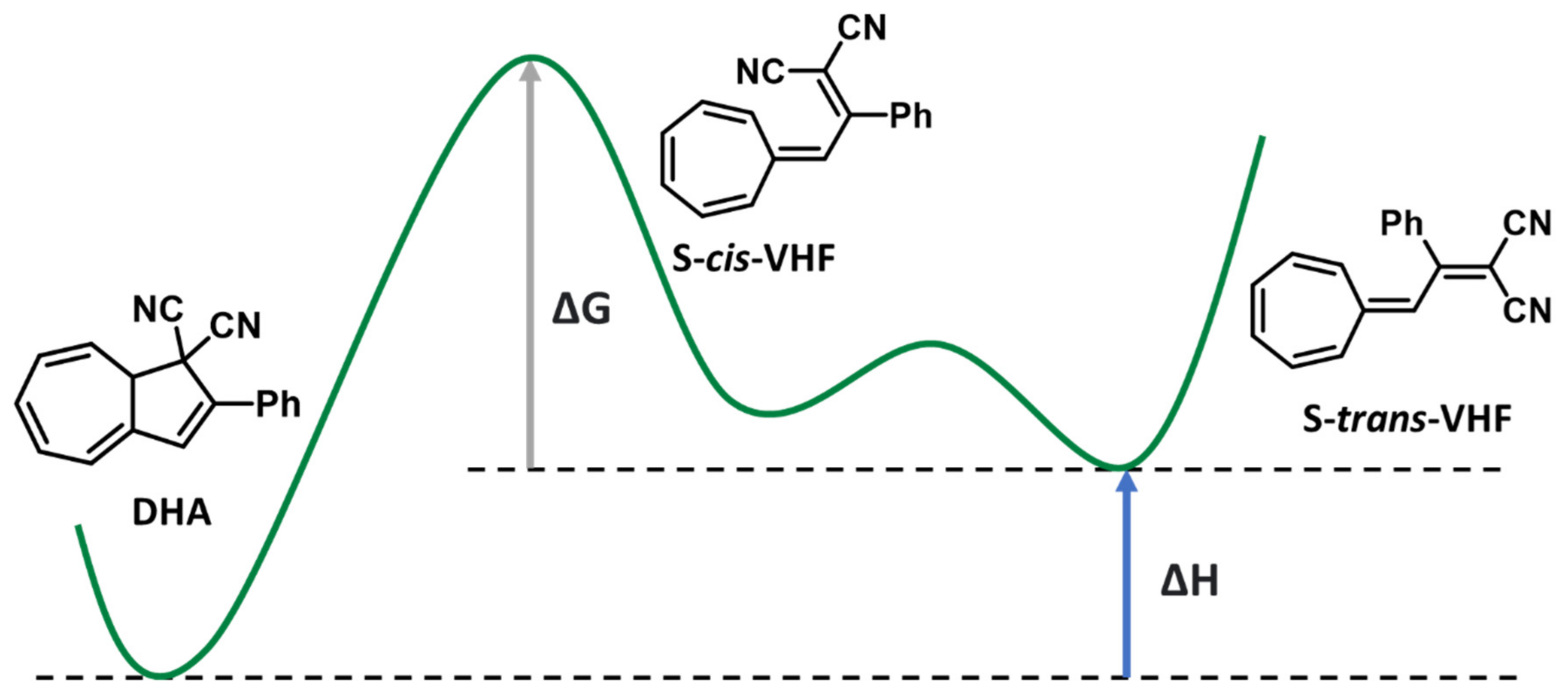
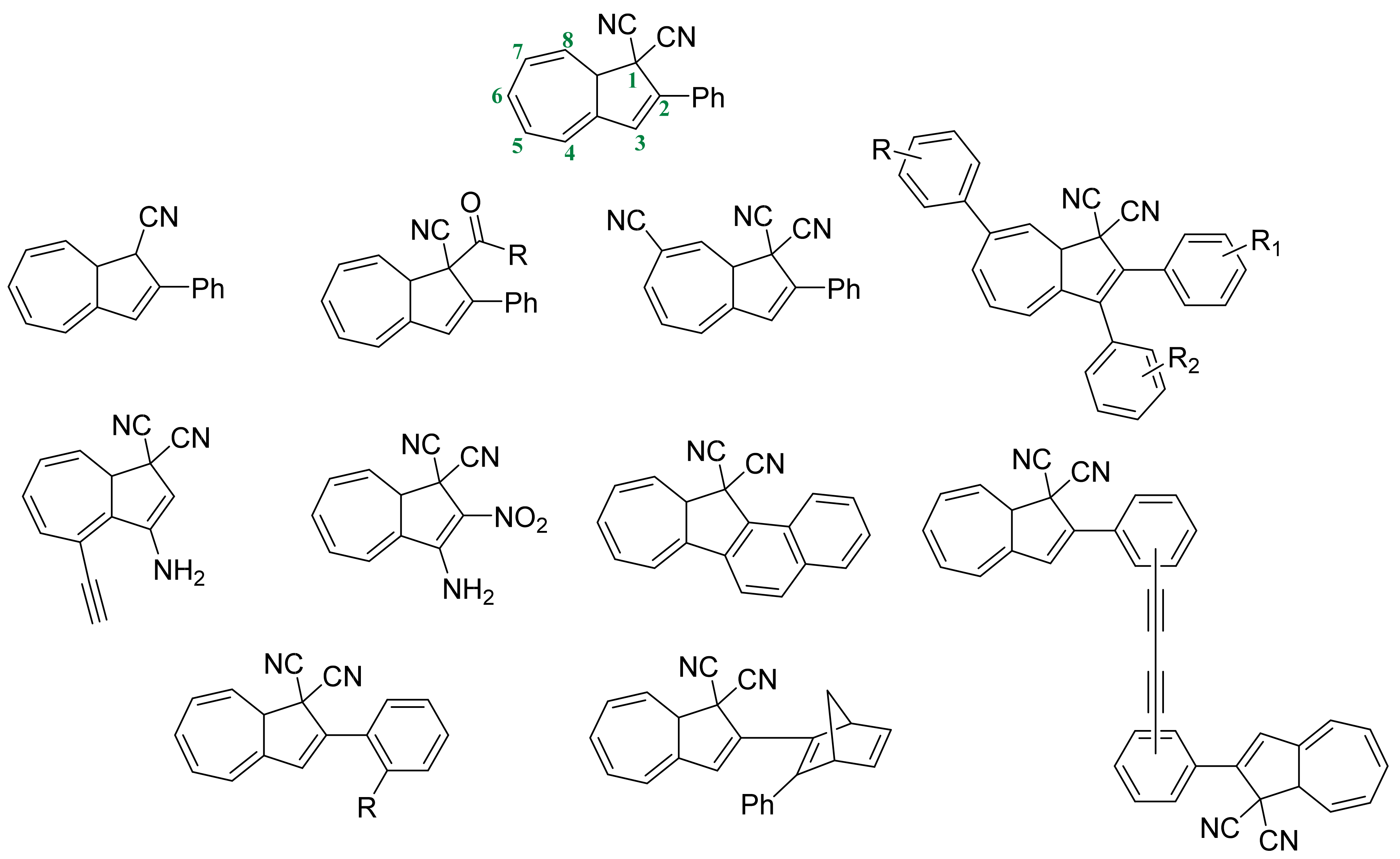
3.3. Dihdroazulene–Vinylheptafluvene (DHA–VHF)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Amorim, W.S.; Valduga, I.B.; Ribeiro, J.M.P.; Williamson, V.G.; Krauser, G.E.; Magtoto, M.K.; de Andrade Guerra, J.B.S.O. The nexus between water, energy, and food in the context of the global risks: An analysis of the interactions between food, water, and energy security. Environ. Impact Assess. Rev. 2018, 72, 1–11. [Google Scholar] [CrossRef]
- Sun, C.-L.; Wang, C.; Boulatov, R. Applications of Photoswitches in the Storage of Solar Energy. ChemPhotoChem 2019, 3, 268–283. [Google Scholar] [CrossRef]
- Wang, Z.; Erhart, P.; Li, T.; Zhang, Z.-Y.; Sampedro, D.; Hu, Z.; Wegner, H.A.; Brummel, O.; Libuda, J.; Nielsen, M.B.; et al. Storing energy with molecular photoisomers. Joule 2021, 5, 3116–3136. [Google Scholar] [CrossRef]
- Lee, D.S.; Fahey, D.W.; Skowron, A.; Allen, M.R.; Burkhardt, U.; Chen, Q.; Doherty, S.J.; Freeman, S.; Forster, P.M.; Fuglestvedt, J.; et al. The contribution of global aviation to anthropogenic climate forcing for 2000 to 2018. Atmos. Environ. 2021, 244, 117834. [Google Scholar] [CrossRef]
- IEA. Global Status Report for Buildings and Construction; International Energy Agency: Paris, France, 2019. [Google Scholar]
- Rabaia, M.K.H.; Abdelkareem, M.A.; Sayed, E.T.; Elsaid, K.; Chae, K.J.; Wilberforce, T.; Olabi, A.G. Environmental impacts of solar energy systems: A review. Sci. Total Environ. 2021, 754, 141989. [Google Scholar] [CrossRef]
- Xu, X.; Wang, G. Molecular Solar Thermal Systems towards Phase Change and Visible Light Photon Energy Storage. Small 2022, 18, 2107473. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, P.; Fan, J.; Klar, A. Underground solar energy storage via energy piles: An experimental study. Appl. Energy 2022, 306, 118042. [Google Scholar] [CrossRef]
- Ma, Q.; Wang, P. Underground solar energy storage via energy piles. Appl. Energy 2020, 261, 114361. [Google Scholar] [CrossRef]
- Wu, D.; Kong, G.; Liu, H.; Jiang, Q.; Yang, Q.; Kong, L. Performance of a full-scale energy pile for underground solar energy storage. Case Stud. Therm. Eng. 2021, 27, 101313. [Google Scholar] [CrossRef]
- Wang, H.; Qi, C. Performance study of underground thermal storage in a solar-ground coupled heat pump system for residential buildings. Energy Build. 2008, 40, 1278–1286. [Google Scholar] [CrossRef]
- Ciamician, G. The Photochemistry of the Future. Science 1912, 36, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moth-Poulsen, K. Organic Synthesis and Molecular Engineering; Nielsen, M.B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 179–196. [Google Scholar]
- Moth-Poulsen, K.; Ćoso, D.; Börjesson, K.; Vinokurov, N.; Meier, S.K.; Majumdar, A.; Vollhardt, K.P.C.; Segalman, R.A. Molecular solar thermal (MOST) energy storage and release system. Energy Environ. Sci. 2012, 5, 8534–8537. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; He, Y.; Wang, Z.; Xu, J.; Xie, M.; Tao, P.; Ji, D.; Moth-Poulsen, K.; Li, T. Photochemical Phase Transitions Enable Coharvesting of Photon Energy and Ambient Heat for Energetic Molecular Solar Thermal Batteries That Upgrade Thermal Energy. J. Am. Chem. Soc. 2020, 142, 12256–12264. [Google Scholar] [CrossRef] [PubMed]
- Lennartson, A.; Roffey, A.; Moth-Poulsen, K. Designing photoswitches for molecular solar thermal energy storage. Tetrahedron Lett. 2015, 56, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Vlasceanu, A.; Broman, S.L.; Hansen, A.S.; Skov, A.B.; Cacciarini, M.; Kadziola, A.; Kjaergaard, H.G.; Mikkelsen, K.V.; Nielsen, M.B. Solar Thermal Energy Storage in a Photochromic Macrocycle. Chem.—A Eur. J. 2016, 22, 10796–10800. [Google Scholar] [CrossRef]
- Yoshida, Z.-i. New molecular energy storage systems. J. Photochem. 1985, 29, 27–40. [Google Scholar] [CrossRef]
- Gur, I.; Sawyer, K.; Prasher, R. Searching for a Better Thermal Battery. Science 2012, 335, 1454–1455. [Google Scholar] [CrossRef]
- Bren, V.A.; Dubonosov, A.D.; Minkin, V.I.; Chernoivanov, V.A. Norbornadiene–quadricyclane—An effective molecular system for the storage of solar energy. Russ. Chem. Rev. 1991, 60, 451–469. [Google Scholar] [CrossRef]
- Philippopoulos, C.; Marangozis, J. Kinetics and efficiency of solar energy storage in the photochemical isomerization of norbornadiene to quadricyclane. Ind. Eng. Chem. Prod. Res. Dev. 1984, 23, 458–466. [Google Scholar] [CrossRef]
- Börjesson, K.; Lennartson, A.; Moth-Poulsen, K. Efficiency Limit of Molecular Solar Thermal Energy Collecting Devices. ACS Sustain. Chem. Eng. 2013, 1, 585–590. [Google Scholar] [CrossRef]
- Fei, L.; Yin, Y.; Yang, M.; Zhang, S.; Wang, C. Wearable solar energy management based on visible solar thermal energy storage for full solar spectrum utilization. Energy Storage Mater. 2021, 42, 636–644. [Google Scholar] [CrossRef]
- Mansø, M.; Petersen, A.U.; Wang, Z.; Erhart, P.; Nielsen, M.B.; Moth-Poulsen, K. Molecular solar thermal energy storage in photoswitch oligomers increases energy densities and storage times. Nat. Commun. 2018, 9, 1945. [Google Scholar] [CrossRef] [PubMed]
- Quant, M.; Lennartson, A.; Dreos, A.; Kuisma, M.; Erhart, P.; Börjesson, K.; Moth-Poulsen, K. Low Molecular Weight Norbornadiene Derivatives for Molecular Solar-Thermal Energy Storage. Chem.—A Eur. J. 2016, 22, 13265–13274. [Google Scholar] [CrossRef] [PubMed]
- Gray, V.; Lennartson, A.; Ratanalert, P.; Börjesson, K.; Moth-Poulsen, K. Diaryl-substituted norbornadienes with red-shifted absorption for molecular solar thermal energy storage. Chem. Commun. 2014, 50, 5330–5332. [Google Scholar] [CrossRef]
- Kuisma, M.; Lundin, A.; Moth-Poulsen, K.; Hyldgaard, P.; Erhart, P. Optimization of Norbornadiene Compounds for Solar Thermal Storage by First-Principles Calculations. ChemSusChem 2016, 9, 1786–1794. [Google Scholar] [CrossRef]
- Solmuş, İ.; Kaftanoğlu, B.; Yamalı, C.; Baker, D. Experimental investigation of a natural zeolite–water adsorption cooling unit. Appl. Energy 2011, 88, 4206–4213. [Google Scholar] [CrossRef]
- Liu, Y.; Leong, K.C. Numerical modeling of a zeolite/water adsorption cooling system with non-constant condensing pressure. Int. Commun. Heat Mass Transf. 2008, 35, 618–622. [Google Scholar] [CrossRef]
- Vollhardt, K.P.C.; Weidman, T.W. Synthesis, structure, and photochemistry of tetracarbonyl(fulvalene)diruthenium. Thermally reversible photoisomerization involving carbon-carbon bond activation at a dimetal center. J. Am. Chem. Soc. 1983, 105, 1676–1677. [Google Scholar] [CrossRef]
- Schwerzel, R.E.; Klosterman, N.E.; Kelly, J.R.; Hillenbrand, L.J. Catalytic Extraction of Stored Solar Energy from Photochemicals. U.S. Patent 4105014/1978, 1978. [Google Scholar]
- Blanco-Lomas, M.; Martínez-López, D.; Campos, P.J.; Sampedro, D. Tuning of the properties of rhodopsin-based molecular switches. Tetrahedron Lett. 2014, 55, 3361–3364. [Google Scholar] [CrossRef]
- Martinez-Lopez, D.; Yu, M.L.; Garcia-Iriepa, C.; Campos, P.J.; Frutos, L.M.; Golen, J.A.; Rasapalli, S.; Sampedro, D. Hydantoin-based molecular photoswitches. J. Org. Chem. 2015, 80, 3929–3939. [Google Scholar] [CrossRef]
- Bastianelli, C.; Caia, V.; Cum, G.; Gallo, R.; Mancini, V. Thermal isomerization of photochemically synthesized (Z)-9-styrylacridines. An unusually high enthalpy of Z→E conversion for stilbene-like compounds. J. Chem. Soc. Perkin Trans. 2 1991, 22, 679–683. [Google Scholar] [CrossRef]
- Losantos, R.; Sampedro, D. Design and Tuning of Photoswitches for Solar Energy Storage. Molecules 2021, 26, 3796. [Google Scholar] [CrossRef] [PubMed]
- Orrego-Hernández, J.; Hölzel, H.; Wang, Z.; Quant, M.; Moth-Poulsen, K. Norbornadiene/Quadricyclane (NBD/QC) and Conversion of Solar Energy. In Molecular Photoswitches; Wiley: Hoboken, NJ, USA, 2022; pp. 351–378. [Google Scholar]
- Jones, G.; Reinhardt, T.E.; Bergmark, W.R. Photon energy storage in organic materials—The case of linked anthracenes. Sol. Energy 1978, 20, 241–248. [Google Scholar] [CrossRef]
- Wang, Z.; Udmark, J.; Börjesson, K.; Rodrigues, R.; Roffey, A.; Abrahamsson, M.; Nielsen, M.B.; Moth-Poulsen, K. Evaluating Dihydroazulene/Vinylheptafulvene Photoswitches for Solar Energy Storage Applications. ChemSusChem 2017, 10, 3049–3055. [Google Scholar] [CrossRef] [PubMed]
- Broman, S.L.; Brand, S.L.; Parker, C.R.; Petersen, M.Å.; Tortzen, C.G.; Kadziola, A.; Kilså, K.; Nielsen, M.B. Optimized synthesis and detailed NMR spectroscopic characterization of the 1,8a-dihydroazulene-1,1-dicarbonitrile photoswitch. ARKIVOC 2011, 2011, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Koerstz, M.; Christensen, A.S.; Mikkelsen, K.V.; Nielsen, M.B.; Jensen, J.H. High throughput virtual screening of 230 billion molecular solar heat battery candidates. PeerJ Phys. Chem. 2021, 3, e16. [Google Scholar] [CrossRef]
- Börjesson, K.; Ćoso, D.; Gray, V.; Grossman, J.C.; Guan, J.; Harris, C.B.; Hertkorn, N.; Hou, Z.; Kanai, Y.; Lee , D.; et al. Exploring the Potential of Fulvalene Dimetals as Platforms for Molecular Solar Thermal Energy Storage: Computations, Syntheses, Structures, Kinetics, and Catalysis. Chem. Eur. J. 2014, 20, 15587–15604. [Google Scholar] [CrossRef]
- Kucharski, T.J.; Tian, Y.; Akbulatov, S.; Boulatov, R. Chemical solutions for the closed-cycle storage of solar energy. Energy Environ. Sci. 2011, 4, 4449–4472. [Google Scholar] [CrossRef]
- International, A. A International in Standard Tables for Reference Solar Spectral Irradiances: Direct Normal and Hemispherical on 37° Tilted Surface; ASTM International: West Conshohocken, PA, USA, 2012. [Google Scholar]
- Jorner, K.; Dreos, A.; Emanuelsson, R.; El Bakouri, O.; Galván, I.F.; Börjesson, K.; Feixas, F.; Lindh, R.; Zietz, B.; Moth-Poulsen, K.; et al. Unraveling factors leading to efficient norbornadiene–quadricyclane molecular solar-thermal energy storage systems. J. Mater. Chem. A 2017, 5, 12369–12378. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Z.; Cole, J.M. Molecular Design of UV–vis Absorption and Emission Properties in Organic Fluorophores: Toward Larger Bathochromic Shifts, Enhanced Molar Extinction Coefficients, and Greater Stokes Shifts. J. Phys. Chem. C 2013, 117, 16584–16595. [Google Scholar] [CrossRef]
- Skov, A.B.; Broman, S.L.; Gertsen, A.S.; Elm, J.; Jevric, M.; Cacciarini, M.; Kadziola, A.; Mikkelsen, K.V.; Nielsen, M.B. Aromaticity-Controlled Energy Storage Capacity of the Dihydroazulene-Vinylheptafulvene Photochromic System. Chem.—A Eur. J. 2016, 22, 14567–14575. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.U.; Hofmann, A.I.; Fillols, M.; Mansø, M.; Jevric, M.; Wang, Z.; Sumby, C.J.; Müller, C.; Moth-Poulsen, K. Solar Energy Storage by Molecular Norbornadiene–Quadricyclane Photoswitches: Polymer Film Devices. Adv. Sci. 2019, 6, 1900367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bas, E.E.; Ulukan, P.; Monari, A.; Aviyente, V.; Catak, S. Photophysical Properties of Benzophenone-Based TADF Emitters in Relation to Their Molecular Structure. J. Phys. Chem. A 2022, 4, 473–484. [Google Scholar] [CrossRef]
- Meng, F.-Y.; Chen, I.-H.; Shen, J.-Y.; Chang, K.-H.; Chou, T.-C.; Chen, Y.-A.; Chen, Y.-T.; Chen, C.-L.; Chou, P.-T. A new approach exploiting thermally activated delayed fluorescence molecules to optimize solar thermal energy storage. Nat. Commun. 2022, 13, 797. [Google Scholar] [CrossRef]
- Wang, Z.; Roffey, A.; Losantos, R.; Lennartson, A.; Jevric, M.; Petersen, A.U.; Quant, M.; Dreos, A.; Wen, X.; Sampedro, D.; et al. Macroscopic heat release in a molecular solar thermal energy storage system. Energy Environ. Sci. 2019, 12, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Dubonosov, A.D.; Bren, V.A.; Chernoivanov, V.A. Norbornadiene–quadricyclane as an abiotic system for the storage of solar energy. Russ. Chem. Rev. 2002, 71, 917–927. [Google Scholar] [CrossRef]
- Harel, Y.; Adamson, A.W.; Kutal, C.; Grutsch, P.A.; Yasufuku, K. Photocalorimetry. 6. Enthalpies of isomerization of norbornadiene and of substituted norbornadienes to corresponding quadricyclenes. J. Phys. Chem. 1987, 91, 901–904. [Google Scholar] [CrossRef]
- Dilling, W.L. Intramolecular Photochemical Cycloaddition of Nonconjugated Olefins. Chem. Rev. 1966, 66, 373–393. [Google Scholar] [CrossRef]
- Sadao, M.; Yoshinobu, A.; Zen-ichi, Y. Photochromic Solid Films Prepared by Doping with Donor–Acceptor Norbornadienes. Chem. Lett. 1987, 16, 195–198. [Google Scholar]
- Jevric, M.; Petersen, A.U.; Mansø, M.; Kumar Singh, S.; Wang, Z.; Dreos, A.; Sumby, C.; Nielsen, M.B.; Börjesson, K.; Erhart, P.; et al. Norbornadiene-Based Photoswitches with Exceptional Combination of Solar Spectrum Match and Long-Term Energy Storage. Chem.—A Eur. J. 2018, 24, 12767–12772. [Google Scholar] [CrossRef]
- Mitscherlich, E. Ueber das Stickstoffbenzid. Ann. Der Phys. 1834, 108, 225–227. [Google Scholar] [CrossRef] [Green Version]
- Noble, A. III. Zur Geschichte des Azobenzols und des Benzidins. Justus Liebigs Ann. Der Chem. 1856, 98, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Hartley, G.S. The Cis-form of Azobenzene. Nature 1937, 140, 281. [Google Scholar] [CrossRef]
- Durr, H.; Bouas-Laurent, H. Photochromism: Molecules and Systems; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Turanský, R.; Konôpka, M.; Doltsinis, N.L.; Ŝtich, I.; Marx, D. Switching of functionalized azobenzene suspended between gold tips by mechanochemical, photochemical, and opto-mechanical means. Phys. Chem. Chem. Phys. 2010, 12, 13922–13932. [Google Scholar] [CrossRef] [PubMed]
- Henzl, J.; Mehlhorn, M.; Gawronski, H.; Rieder, K.H.; Morgenstern, K. Reversible cis-trans isomerization of a single azobenzene molecule. Angew. Chem.-Int. Ed. 2006, 45, 603–606. [Google Scholar] [CrossRef]
- Tong, X.; Pelletier, M.; Lasia, A.; Zhao, Y. Fast cis-trans isomerization of an azobenzene derivative in liquids and liquid crystals under a low electric field. Angew. Chem.-Int. Ed. 2008, 47, 3596–3599. [Google Scholar] [CrossRef]
- Crecca, C.R.; Roitberg, A.E. Theoretical study of the isomerization mechanism of azobenzene and disubstituted azobenzene derivatives. J. Phys. Chem. A 2006, 110, 8188–8203. [Google Scholar] [CrossRef]
- Brown, C.J. A refinement of the crystal structure of azobenzene. Acta Crystallogr. 1966, 21, 146–152. [Google Scholar] [CrossRef]
- Mostad, A.; Romming, C. A refinement of the crystal structure of cis-azobenzene. Acta Chem. Scand. 1971, 25, 3561–3568. [Google Scholar] [CrossRef]
- Gagliardi, L.; Orlandi, G.; Bernardi, F.; Cembran, A.; Garavelli, M. A theoretical study of the lowest electronic states of azobenzene: The role of torsion coordinate in the cis–trans photoisomerization. Theor. Chem. Acc. 2004, 111, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Sudesh, G.; Road, T.B.; Sciences, P.; Green, B. Photochemistry of Azobenzene-Containing Polymers. Chem. Rev. 1989, 89, 1915–1925. [Google Scholar]
- Sension, R.J.; Repinec, S.T.; Szarka, A.Z.; Hochstrasser, R.M. Femtosecond laser studies of the cis-stilbene photoisomerization reactions. J. Chem. Phys. 1993, 98, 6291–6315. [Google Scholar] [CrossRef]
- Hamm, P.; Ohline, S.M.; Zinth, W. Vibrational cooling after ultrafast photoisomerization of azobenzene measured by femtosecond infrared spectroscopy. J. Chem. Phys. 1997, 106, 519–529. [Google Scholar] [CrossRef]
- Georgiev, A.; Bubev, E.; Dimov, D.; Yancheva, D.; Zhivkov, I.; Krajčovič, J.; Vala, M.; Weiter, M.; Machkova, M. Synthesis, structure, spectral properties and DFT quantum chemical calculations of 4-aminoazobenzene dyes. Effect of intramolecular hydrogen bonding on photoisomerization. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 175, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Schulze, F.W.; Petrick, H.J.; Cammenga, H.K.; Klinge, H.Z. Thermodynamic properties of the structural analogues benzo[c]-cinnoline, trans-azobenzene and cis-azobenzene. Z. Phys. Chem. Neue Fol. 1997, 1, 107. [Google Scholar] [CrossRef]
- Brown, E.V.; Granneman, G.R. Cis-Trans Isomerism in the Pyridyl Analogs of Azobenzene. A Kinetic and Molecular Orbital Analysis. J. Am. Chem. Soc. 1975, 97, 621–627. [Google Scholar] [CrossRef]
- Tamai, N.; Miyasaka, H. Ultrafast Dynamics of Photochromic Systems. Chem. Rev. 2000, 100, 1875–1890. [Google Scholar] [CrossRef]
- Casellas, J.; Bearpark, M.J.; Reguero, M. Excited-State Decay in the Photoisomerisation of Azobenzene: A New Balance between Mechanisms. ChemPhysChem 2016, 17, 3068–3079. [Google Scholar] [CrossRef] [Green Version]
- Rau, H. Further evidence for rotation in the π,π* and inversion in the n,π* photoisomerization of azobenzenes. J. Photochem. 1984, 26, 221–225. [Google Scholar] [CrossRef]
- Fujino, T.; Arzhantsev, S.Y.; Tahara, T. Femtosecond Time-Resolved Fluorescence Study of Photoisomerization of trans-Azobenzene. J. Phys. Chem. A 2001, 105, 8123–8129. [Google Scholar] [CrossRef]
- Rau, H. Photochromism: Molecules and Systems, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1990; pp. 165–192. [Google Scholar]
- Kunz, A.; Heindl, A.H.; Dreos, A.; Wang, Z.; Moth-Poulsen, K.; Becker, J.; Wegner, H.A. Intermolecular London Dispersion Interactions of Azobenzene Switches for Tuning Molecular Solar Thermal Energy Storage Systems. ChemPlusChem 2019, 84, 1145–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Losantos, R.; Sampedro, D.; Morikawa, M.-a.; Börjesson, K.; Kimizuka, N.; Moth-Poulsen, K. Demonstration of an azobenzene derivative based solar thermal energy storage system. J. Mater. Chem. A 2019, 7, 15042–15047. [Google Scholar] [CrossRef] [Green Version]
- Shigeru, Y.; Hiroshi, O.; Osamu, T. The cis-trans Photoisomerization of Azobenzene. Bull. Chem. Soc. Jpn. 1962, 35, 1849–1853. [Google Scholar]
- Zimmerman, G.; Chow, L.-Y.; Paik, U.-J. The Photochemical Isomerization of Azobenzene1. J. Am. Chem. Soc. 1958, 80, 3528–3531. [Google Scholar] [CrossRef]
- Ladányi, V.; Dvořák, P.; Al Anshori, J.; Vetráková, Ľ.; Wirz, J.; Heger, D. Azobenzene photoisomerization quantum yields in methanol redetermined. Photochem. Photobiol. Sci. 2017, 16, 1757–1761. [Google Scholar] [CrossRef]
- Mahimwalla, Z.; Yager, K.G.; Mamiya, J.-i.; Shishido, A.; Priimagi, A.; Barrett, C.J. Azobenzene photomechanics: Prospects and potential applications. Polym. Bull. 2012, 69, 967–1006. [Google Scholar] [CrossRef]
- Kolpak, A.M.; Grossman, J.C. Azobenzene-Functionalized Carbon Nanotubes As High-Energy Density Solar Thermal Fuels. Nano Lett. 2011, 11, 3156–3162. [Google Scholar] [CrossRef]
- Kucharski, T.J.; Ferralis, N.; Kolpak, A.M.; Zheng, J.O.; Nocera, D.G.; Grossman, J.C. Templated assembly of photoswitches significantly increases the energy-storage capacity of solar thermal fuels. Nat. Chem. 2014, 6, 441–447. [Google Scholar] [CrossRef]
- Wu, S.; Butt, H.-J. Solar-Thermal Energy Conversion and Storage Using Photoresponsive Azobenzene-Containing Polymers. Macromol. Rapid Commun. 2020, 41, 1900413. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Luo, W.; Feng, Y. Photo-responsive carbon nanomaterials functionalized by azobenzene moieties: Structures, properties and application. Nanoscale 2012, 4, 6118–6134. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, H.; Luo, W.; Liu, E.; Zhao, N.; Yoshino, K.; Feng, W. Covalent functionalization of graphene by azobenzene with molecular hydrogen bonds for long-term solar thermal storage. Sci. Rep. 2013, 3, 3260. [Google Scholar] [CrossRef] [PubMed]
- Norikane, Y.; Kitamoto, K.; Tamaoki, N. Novel crystal structure, cis-trans isomerization, and host property of meta-substituted macrocyclic azobenzenes with the shortest linkers. J. Org. Chem. 2003, 68, 8291–8304. [Google Scholar] [CrossRef]
- Durgun, E.; Grossman, J.C. Photoswitchable Molecular Rings for Solar-Thermal Energy Storage. J. Phys. Chem. Lett. 2013, 4, 854–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, D.; Cho, E.; Grossman, J.C. Solid-State Solar Thermal Fuels for Heat Release Applications. Adv. Energy Mater. 2016, 6, 1502006. [Google Scholar] [CrossRef]
- Lv, J.-A.; Liu, Y.; Wei, J.; Chen, E.; Qin, L.; Yu, Y. Photocontrol of fluid slugs in liquid crystal polymer microactuators. Nature 2016, 537, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.P.; Renna, L.A.; Boyle, C.J.; Kwak, H.S.; Harder, E.; Damm, W.; Venkataraman, D. High Energy Density in Azobenzene-based Materials for Photo-Thermal Batteries via Controlled Polymer Architecture and Polymer-Solvent Interactions. Sci. Rep. 2017, 7, 17773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, D.; Grossman, J.C. Conformal Electroplating of Azobenzene-Based Solar Thermal Fuels onto Large-Area and Fiber Geometries. ACS Appl. Mater. Interfaces 2016, 8, 26319–26325. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.N.; Zhitomirsky, D.; Han, G.G.D.; Liu, Y.; Grossman, J.C. Molecularly Engineered Azobenzene Derivatives for High Energy Density Solid-State Solar Thermal Fuels. ACS Appl. Mater. Interfaces 2017, 9, 8679–8687. [Google Scholar] [CrossRef]
- Calbo, J.; Weston, C.E.; White, A.J.P.; Rzepa, H.S.; Contreras-García, J.; Fuchter, M.J. Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design. J. Am. Chem. Soc. 2017, 139, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Saritas, K.; Grossman, J.C. Accurate Isomerization Enthalpy and Investigation of the Errors in Density Functional Theory for Dihydroazulene/Vinylheptafulvene Photochromism Using Diffusion Monte Carlo. J. Phys. Chem. C 2017, 121, 26677–26685. [Google Scholar] [CrossRef]
- Goerner, H.; Fischer, C.; Gierisch, S.; Daub, J. Dihydroazulene/vinylheptafulvene photochromism: Effects of substituents, solvent, and temperature in the photorearrangement of dihydroazulenes to vinylheptafulvenes. J. Phys. Chem. 1993, 97, 4110–4117. [Google Scholar] [CrossRef]
- Christensen, O.; Nielsen, L.E.; Johansen, J.; Holk, K.; Udmark, J.; Nielsen, M.B.; Cacciarini, M.; Mikkelsen, K.V. Density Functional Theory Study of Carbamoyl-Substituted Dihydroazulene/Vinylheptafulvene Derivatives and Solvent Effects. J. Phys. Chem. C 2022, 126, 4815–4825. [Google Scholar] [CrossRef]
- Broman, S.L.; Jevric, M.; Nielsen, M.B. Linear Free-Energy Correlations for the Vinylheptafulvene Ring Closure: A Probe for Hammett σ Values. Chem.—A Eur. J. 2013, 19, 9542–9548. [Google Scholar] [CrossRef] [PubMed]
- Kilde, M.D.; Hansen, M.H.; Broman, S.L.; Mikkelsen, K.V.; Nielsen, M.B. Expanding the Hammett Correlations for the Vinylheptafulvene Ring-Closure Reaction. Eur. J. Org. Chem. 2017, 2017, 1052–1062. [Google Scholar] [CrossRef]
- Ranzenigo, A.; Cordero, F.M.; Cacciarini, M.; Nielsen, M.B. ortho-Substituted 2-Phenyldihydroazulene Photoswitches: Enhancing the Lifetime of the Photoisomer by ortho-Aryl Interactions. Molecules 2021, 26, 6462. [Google Scholar] [CrossRef]
- Schøttler, C.; Vegge, S.K.; Cacciarini, M.; Nielsen, M.B. Long-Term Energy Storage Systems Based on the Dihydroazulene/Vinylheptafulvene Photo-/Thermoswitch. ChemPhotoChem 2022, 6, e202200037. [Google Scholar] [CrossRef]
- Abedi, M.; Pápai, M.; Henriksen, N.E.; Møller, K.B.; Nielsen, M.B.; Mikkelsen, K.V. Theoretical Investigation on the Control of Macrocyclic Dihydroazulene/Azobenzene Photoswitches. J. Phys. Chem. C 2019, 123, 25579–25584. [Google Scholar] [CrossRef]
- Wang, Z.; Hölzel, H.; Moth-Poulsen, K. Status and challenges for molecular solar thermal energy storage system based devices. Chem. Soc. Rev. 2022. Advance Article. [Google Scholar] [CrossRef]


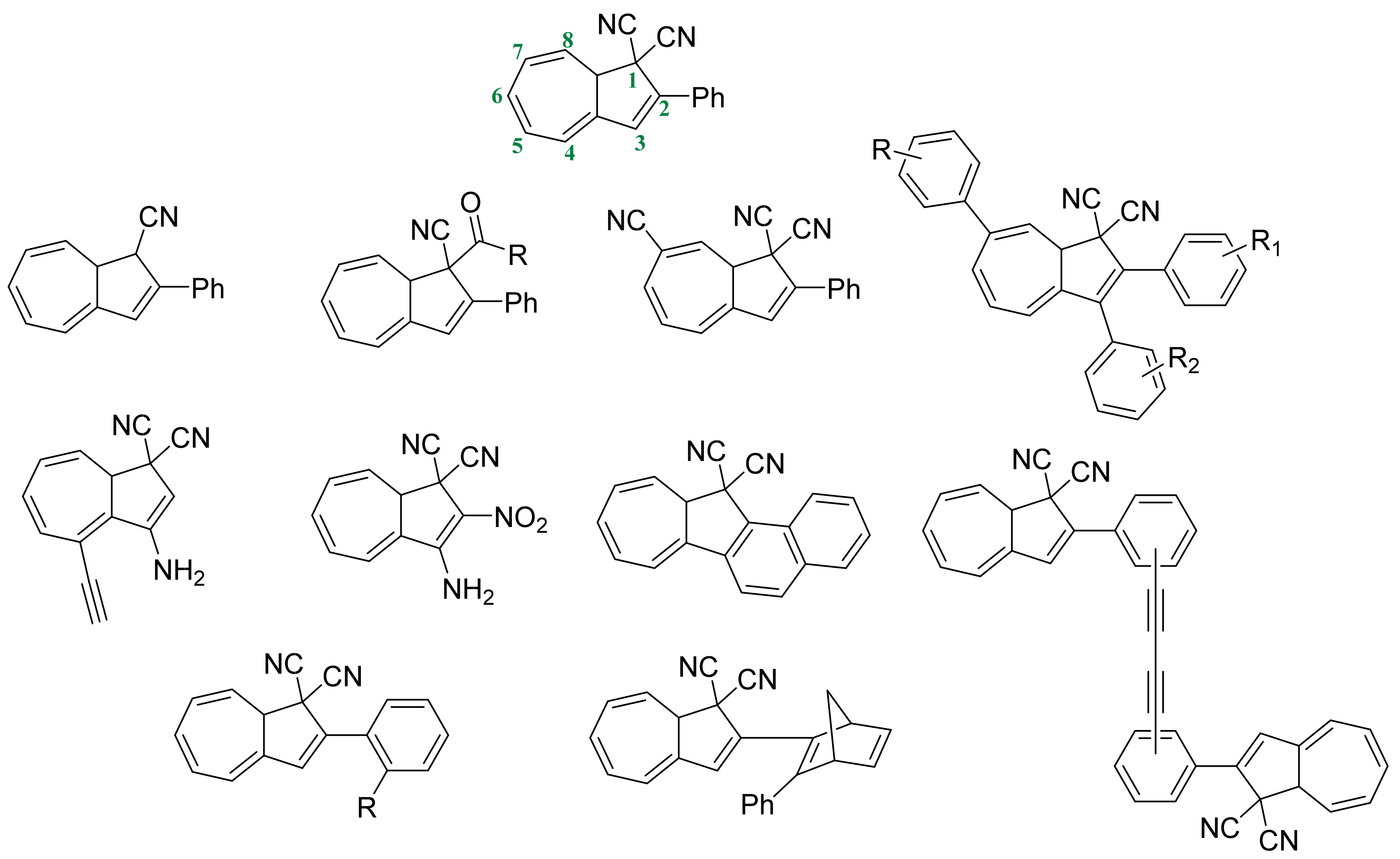

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NBD | Molar Mass (g/mol) | λmax (nm) | ∆Hisomerization (kJ/mol) | Energy Density (kJ/kg) |
|---|---|---|---|---|
| Unsubstituted | 92 | 236 | 113 | 1228 |
| 1 | 244 | 308 | 96 | 393 |
| 2 | 274 | 309 | 97 | 354 |
| 3 | 342 | 318 | 98 | 287 |
| 4 | 299 | 350 | 97 | 324 |
| 5 | 355 | 365 | 102 | 287 |
| 6 | 217 | 331 | - | - |
| 7 | 247 | 355 | - | - |
| 8 | 223 | 340 | - | - |
| 9 | 260 | 398 | 103 | 396 |
| 10 | 193 | 309 | 122 | 632 |
| 11 | 223 | 326 | 89 | 397 |
| 12 | 288 | 380 | 91 | 315 |
| 13 | 356 | 359 | 183 | 514 |
| 14 | 356 | 334 | 99 | 278 |
| 15 | 256 | 362 | - | - |
| 16 | 308 | 350 | - | - |
| 17 | 308 | 308 | 173 | 562 |
| 18 | 495 | 336 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimenez-Gomez, A.; Magson, L.; Peñin, B.; Sanosa, N.; Soilán, J.; Losantos, R.; Sampedro, D. A Photochemical Overview of Molecular Solar Thermal Energy Storage. Photochem 2022, 2, 694-716. https://doi.org/10.3390/photochem2030045
Gimenez-Gomez A, Magson L, Peñin B, Sanosa N, Soilán J, Losantos R, Sampedro D. A Photochemical Overview of Molecular Solar Thermal Energy Storage. Photochem. 2022; 2(3):694-716. https://doi.org/10.3390/photochem2030045
Chicago/Turabian StyleGimenez-Gomez, Alberto, Lucien Magson, Beatriz Peñin, Nil Sanosa, Jacobo Soilán, Raúl Losantos, and Diego Sampedro. 2022. "A Photochemical Overview of Molecular Solar Thermal Energy Storage" Photochem 2, no. 3: 694-716. https://doi.org/10.3390/photochem2030045
APA StyleGimenez-Gomez, A., Magson, L., Peñin, B., Sanosa, N., Soilán, J., Losantos, R., & Sampedro, D. (2022). A Photochemical Overview of Molecular Solar Thermal Energy Storage. Photochem, 2(3), 694-716. https://doi.org/10.3390/photochem2030045