Abstract
UV-induced transformations were studied for monomers of 2,3-diamino-2-butenedinitrile (DAMN) isolated in argon matrices. Photoinduced hydrogen-atom transfer was found to be the major process occurring upon UV (λ > 320 nm or λ > 295 nm) excitation of matrix-isolated DAMN monomers. As a result of the transfer of a hydrogen atom from an amino group to a nitrile fragment, a tautomer of DAMN involving a ketenimine group was generated. Identification of this photo-produced species was based on comparison of its experimental IR spectrum with the spectrum theoretically predicted for the ketenimine form. Another product photogenerated upon UV (λ > 320 nm, λ > 295 nm, or λ > 270 nm) irradiation of DAMN isolated in Ar matrices was identified as 4-amino-1H-imidazole-5-carbonitrile (AICN). The structure of this photoproduct was unambiguously assigned on the basis of an exact match of wavenumbers of the bands in the IR spectrum of this photogenerated species and the wavenumbers of IR bands of AICN trapped (in a separate experiment) from the gas phase into an Ar matrix.
Keywords:
photochemistry; IR spectra; matrix isolation; DAMN; AICN; ketenimine; hydrogen-atom transfer 1. Introduction
2,3-Diaminomaleonitrile (DAMN) and 2,3-diaminofumaronitrile (DAFN) are the cis and trans isomers of 2,3-diamino-2-butenedinitrile (Scheme 1). According to their formula, C4N4H4, these compounds can be treated as tetramers of HCN. Indeed, DAMN has been identified as one of the products of HCN polymerization [,,,,,]. Moreover, it was demonstrated that DAMN transforms to DAFN in the very well-known cis–trans photoisomerization process []. Then, this trans isomer (DAFN) can photochemically convert [,,,] into the cyclic imidazole derivative (4-amino-1H-imidazole-5-carbonitrile AICN, Scheme 1). The photoinduced DAMN-to-DAFN isomerization and formation of AICN are believed to play a key role in the formation of purine nucleobases on early Earth [,,,,].
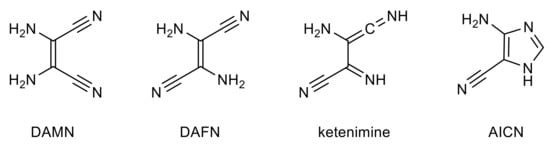
Scheme 1.
The cis (DAMN) and trans (DAFN) isomers of 2,3-diamino-2-butenedinitrile, their ketenimine tautomer, and 4-amino-1H-imidazole-5-carbonitrile (AICN).
The mechanistic description of the photoprocess transforming DAMN/DAFN into AICN was the subject of several experimental and theoretical studies [,]. Different molecular structures were proposed as potential intermediates on the route from DAMN/DAFN to AICN. One of such structures is ketenimine (Scheme 1) resulting from the photoinduced intramolecular hydrogen-atom transfer in DAFN. However, conversion of an enaminonitrile (such as DAMN/DAFN) to a structure with the imidazole ring must involve not only hydrogen-atom transfer processes, but also cleavage of one of the CC bonds and formation of a new CN bond. That is why structures with four-membered ring (azetines) or with three-membered-ring (azirines) should also be considered as potential intermediates involved in the photoinduced DAMN/DAFN → AICN process.
In the current work, the photoinduced transformations were studied for monomeric DAMN molecules isolated in low-temperature argon matrices. One of the goals of the present study was a reliable identification of a ketenimine tautomer (Scheme 1) as a pro-duct of phototransformations of the DAMN molecules. The experimental investigation was also aimed at finding convincing spectral indications of the photochemical DAMN/DAFN → AICN transformation.
2. Experimental Methods
Samples of the compounds investigated in the current study, 2,3-diamino-maleonitrile (DAMN) purity > 98%, and 4-amino-1H-imidazole-5-carbonitrile (AICN) purity > 95%, were purchased from Sigma-Aldrich. Before a matrix-isolation experiment, a solid sample of a studied compound was placed in a miniature glass oven located inside the vacuum chamber of a cryostat with a Sumitomo SRDK-408D2 closed-cycle cooler. Then, the cryostat was evacuated, and the solid compound was heated by a resistive wire wrapped around the glass oven. The vapor of the compound was deposited, together with large excess of argon, onto a CsI window cooled to 12 K. The mid-IR spectra were recorded in the 4000–500 cm−1 range with 0.5 cm−1 resolution using a Thermo Nicolet iS50R FTIR spectrometer equipped with a KBr beam splitter and a DTGS detector with a KBr window. The spectra in the lower-wavenumber 700–300 cm−1 range were recorded using the same spectrometer but with a “solid substrate” beam splitter and a DTGS detector with a polyethylene window. Matrix-isolated monomers of the studied compounds were irradiated with UV light from a high-pressure mercury lamp fitted with a water filter and an appropriate cut-off Schott filter (WG360, WG335, WG320, WG295, or UG11 transmitting light with λ > 360 nm, λ > 335 nm, λ > 320 nm, λ > 295 nm, or λ > 270 nm, respectively).
3. Computational Details
Geometries of DAMN and DAFN molecules, as well as geometries of the species that may be generated upon UV irradiation of DAMN/DAFN, were fully optimized at the DFT(B3LYP) level of theory [,,]. At the optimized geometries, harmonic vibrational frequencies and infrared intensities were computed at the same DFT (B3LYP) level. To approximately correct for the neglected anharmonicity, the harmonic DFT frequencies were scaled by factors of 0.950 (for wavenumbers higher than 3200 cm−1) and 0.980 (for wavenumbers lower than 1800 cm−1), used in our previous works [,]. For wavenumbers from the 3200–1800 cm−1 range, we used a factor of 0.957, obtained from the ratio between the frequency of the standalone band due to the C≡N stretching mode observed in this work for matrix-isolated DAMN (2225 cm−1) and the frequency of the respective mode computed for the DAMN monomer (2324 cm−1). These data were used to simulate the IR spectra using the ChemCraft software []. The peaks centered at the scaled wavenumbers were convoluted with Lorentzian functions having a full width at half-maximum (fwhm) equal to 2 cm−1 and peak heights equal to the computed infrared intensities. The above theoretical computations were carried out with the Gaussian 09 set of programs [], using the standard 6-311++G(d,p) basis set [,,].
For the estimation of the isomerization barriers, the geometries of the respective transition states (TS) were fully optimized [,]. The harmonic vibrational frequencies were computed at the DFT (B3LYP)/6-311++G(d,p) level of theory to verify that the found stationary point has one imaginary frequency. Thereafter, we mapped out the intrinsic reaction coordinate (IRC) [], starting from the TS in both directions, until a local minimum was detected on each side. For the IRC following, we employed the Hessian-based predictor-corrector integrator in the Cartesian (non-mass-weighted) coordinate system as implemented in the Gaussian 16 electronic structure package []. This allowed a comparison of the shapes of different barriers, expressed in the generalized multidimensional coordinates.
Vertical excitation energies of the low-energy electronic excited singlet states were computed at the same DFT(B3LYP)/6-311++G(d,p) level, using time-dependent DFT approach [,]. For the graphical representation, each computed electronic transition was convoluted with a Lorentzian function having a half width at half-maximum (hwhm) equal to 0.248 eV (2000 cm−1).
4. Results
4.1. Molecular Structures of DAMN and DAFN
Optimization of the structure of DAMN (the cis isomer of 2,3-diamino-2-butenedinitrile), performed at the DFT(B3LYP)/6-311++G(d,p) level, led to identification of only one minimum on the potential energy surface (PES). This minimum corresponds to the structure with the C2 symmetry axis (Figure 1). In such a structure, the amino groups are symmetrically involved in a cyclic hydrogen-bond-like intramolecular interaction. This interaction may be similar to that found in the ammonia dimer [,,,].
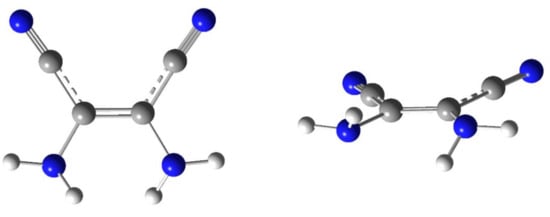
Figure 1.
Optimized geometry of the DAMN molecule: two projections of the lowest-energy form with C2 symmetry. Color codes: blue—nitrogen, gray—carbon, white—hydrogen.
Two low-energy minima were found on the PES of DAFN (the trans isomer of 2,3-diamino-2-butenedinitrile). These minima correspond to nearly planar structures of the molecule; however, in both of them, the amino groups are slightly pyramidal. In the lowest-energy structure, the lone electron pairs of the NH2 nitrogen atoms point to the same side of the “plane” formed by the heavy atoms. This structure belongs to the C2 symmetry group (Figure 2 and Appendix A, Figure A1). The other structure has a center of symmetry and belongs to the Ci symmetry group. In this form, the lone electron pairs of the NH2 nitrogen atoms point to the opposite sides of the approximate plane of the DAFN molecule. The energy of this latter Ci form is only slightly higher (by 0.46 kJ mol−1, including the zero-point vibrational energy correction) than the energy of the C2 structure.
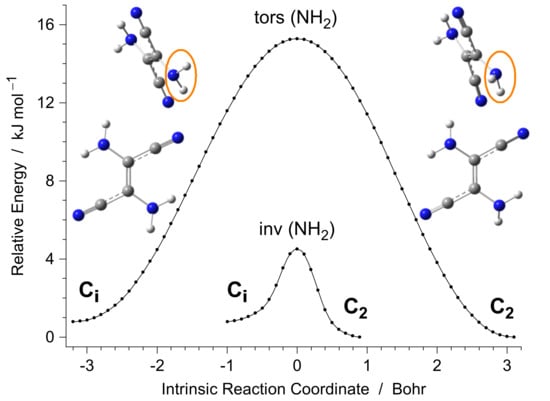
Figure 2.
Potential energy minima due to Ci and C2 structures of DAFN. The barriers separating these minima were computed at the DFT(B3LYP)/6-311++G(d,p) level by following the PES of the DAFN system along the torsional (tors) or inversional (inv) intrinsic reaction coordinate. The insets show the geometries of two DAFN minima, with emphasis (orange ovals) on the one amino group which changes the pyramidalization. Color codes: blue—nitrogen, gray—carbon, white—hydrogen.
Transition of one of these forms into the other by rotation of one of the amino groups around the C−N bond is related with a moderately high (~15 kJ mol−1) barrier. However, the transition between the C2 and Ci structures by inversion of one of the amino groups is related with such a low barrier (4.5 kJ mol−1) that its crossing would occur on the subpicosecond time scale. Hence, both the C2 and Ci structures can be treated as a single quasiplanar form of the real DAFN molecule. Existence of the two closely lying C2 and Ci minima on the PES of DAFN should result in distortions of the shape of the multidimensional potential energy well. This is expected to make some vibrations of the molecule significantly anharmonic.
4.2. Infrared Spectrum of DAMN Isolated in Low-Temperature Ar Matrices
The infrared spectrum of DAMN monomers isolated in an Ar matrix is presented in Figure 3. This spectrum is well reproduced by the results of theoretical calculation carried out at the DFT (B3LYP)/6-311++G(d,p) level for DAMN structure with C2 symmetry.
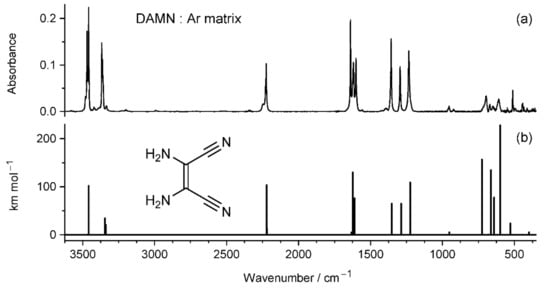
Figure 3.
(a) Infrared spectrum of 2,3-diaminomaleonitrile (DAMN) monomers isolated in an Ar matrix at 12 K, compared with (b) theoretical IR spectrum of this compound computed at the DFT (B3LYP)/6-311++G(d,p) level.
In the high-wavenumber region of the experimental spectrum, the bands due to antisymmetric stretching vibrations of the NH2 groups appear at 3470 and 3459 cm−1, whereas the bands due to symmetric stretching vibrations of the NH2 groups appear at 3367 and 3362 cm−1. In the above descriptions, “antisymmetric” and “symmetric” terms refer to local symmetry of each of the NH2 groups. The characteristic band due to the stretching vibration of the triple C≡N bond appears at 2225 cm−1 in the spectral region free of any other IR bands. Typically, the stretching modes of the triple C≡N bonds in nitriles give rise to weak bands in IR spectra []. However, a relatively high IR intensity (103.7 km mol−1) of the C≡N stretching mode in DAMN results from the antisymmetric coupling of stretching modes due to two nitrile groups. The other (symmetric) coupling gives rise to the mode with much lower IR-intensity (12.8 km mol−1). Strong IR bands assigned to the NH2 scissoring vibrations are found in the experimental spectrum at 1640, 1620, and 1601 cm−1. A fingerprint pattern of the bands at 1357, 1295, and 1234 cm−1 is formed by three IR absorptions due to the NH2 rocking vibrations coupled with the stretching vibrations of the single C−N bonds. This pattern of three bands is very well reproduced by the theoretical calculations. In the spectral range below 750 cm−1, there are significant discrepancies between the bands observed in the experimental spectrum and the pattern of bands predicted by theoretical calculation carried out within the harmonic approximation. The reason for that is the contribution of the NH2 vibrations in the inversional direction (together with the C−C≡N bending vibrations) to the normal modes with frequencies lower than 750 cm−1. The vibrational modes involving NH2 inversional movement are usually quite poorly reproduced by the calculations performed within the harmonic approximation. Moreover, the commonly used perturbational approach to correct for anharmonicity fails to help in such cases.
4.3. Photoproducts Generated upon UV Excitation of Matrix-Isolated DAMN
Monomers of DAMN isolated in an Ar matrix at 12 K were exposed to UV light of different wavelengths, limited from the short-wavelength side by an appropriate cut-off filter. Irradiation of matrix-isolated DAMN with UV light (λ > 360 nm or λ > 335 nm) did not result in any photochemical transformations of the compound. Exposure of the matrix to UV (λ > 320 nm) radiation induced a very slow decrease of the IR bands due to the DAMN reactant and appearance of a set of new IR bands. The observed photochemical changes were more vigorous when the matrix was exposed to UV (λ > 295 nm) light.
In the spectrum of the photoproduced species, the most appealing is the intense IR absorption at 2026 cm−1 (Figure 4). The wavenumber of this band is characteristic of the “antisymmetric” stretching vibration of the C=C=N fragment in the −C=C=N−H ketenimine group [,,,,,,]. The frequency of this vibration is well predicted by the theoretical calculations carried out for the ketenimine tautomer of DAMN/DAFN. In addition, a high IR intensity (605–535 km mol−1) was theoretically predicted in agreement with the experimental observation. This suggests that ketenimine photoproduct was generated upon UV excitation by hydrogen-atom transfer from an amino group to the nitrogen atom of one of the C≡N groups. Another intense IR band appears in the spectrum of the photoproduced species at 906 cm−1. This band can be assigned to the bending of the C=N−H fragment of the −C=C=N−H ketenimine group. A medium-intensity band found at 1572 cm−1 is due to the stretching vibration of the C=N bond in the –C=NH imine group. The overall agreement between the IR spectrum theoretically predicted for the ketenimine form of DAMN/DAFN and the spectrum of the new IR bands appearing after UV irradiation of matrix isolated DAMN confirms that the ketenimine tautomer is the main product generated in this process.
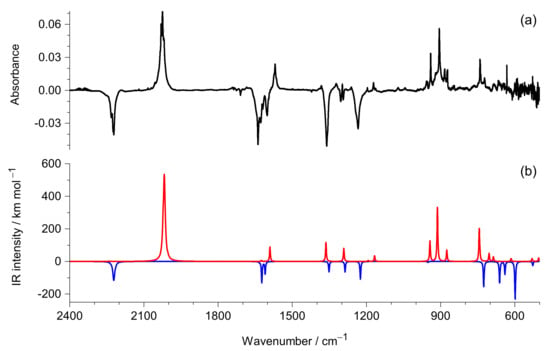
Figure 4.
(a) Infrared spectrum recorded after UV (λ > 295 nm) irradiation of DAMN monomers isolated in an Ar matrix minus infrared spectrum recorded immediately after deposition of the matrix; (b) Theoretical spectra of DAMN (blue) and ketenimine (red) Zt isomer (see Scheme 2) computed at the DFT (B3LYP)/6-311++G(d,p) level. The intensities of the theoretical bands predicted for the DAMN molecule were multiplied by −1.
In all possible structures of the ketenimine form of DAMN/DAFN, the hydrogen atom of the −C=C=N−H group always adopts a perpendicular orientation (designated as a wedge NH bond in Scheme 2) with respect to the approximate plane of the heavy atoms [,]. Apart from this, in the molecules adopting ketenimine tautomeric form, there are three further degrees of conformational freedom: (i) flip of the N−H bond in the −C=N−H imine group, which can assume Z or E orientation with respect to the central C−C bond, (ii) torsion around the central C−C bond, which can assume trans or cis orientation with respect to the position of the −NH2 and =NH groups, (iii) torsional (or inversional) movements of hydrogen atoms in the −NH2 group (see Scheme 2).
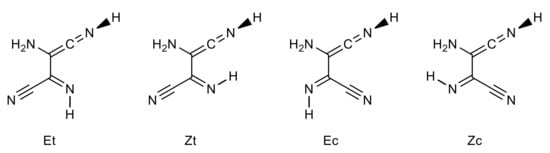
Scheme 2.
Structures of main isomers of ketenimine; (i) E/Z stands for the anti/syn orientations of the imine group; (ii) t/c stands for the trans/cis configuration around the central CC bond (see text for details). See Appendix B, Figure A2 for the optimized geometries, including the pairs of structures with different orientations of the amino group.
Different orientation with respect to the (i) and (ii) degrees of freedom can give rise to four different isomers/conformers of the molecule adopting the ketenimine tautomeric form (see Appendix B, Figure A2). In each of these four structures, an easy tunneling of light hydrogen atoms with respect to the (iii) degree of freedom should readily lead to collapse of the higher-energy formal conformer to the lower-energy structure differing by torsion (or inversion) of the hydrogen atoms in the −NH2 group. Therefore, under the experimental conditions, the molecule in the ketenimine tautomeric form can adopt four different isomers. The infrared bands appearing at 2055, 2030, and 2019 cm−1 as satellites of the most intense band at 2026 cm−1 can be the spectral indications of different isomers/conformers of the molecule adopting the ketenimine tautomeric form. Alternatively, the multiplet structure of the ketenimine IR band appearing in the 2055–2019 cm−1 range may be attributed to the site-splitting. Similar split bands of conformationally rigid ketenimines were reported earlier [,,].
Upon irradiation of an Ar matrix containing the DAMN monomers at wavelengths from the 270 < λ < 320 nm range, a set of new IR bands emerged at 3528, 3488, 3430, 2219, 1622, 1574, 1458, 1439, 1375, 1247, 1184, and 1109 cm−1. The spectral positions of these bands were compared with the positions of the bands observed in the spectrum of 4-amino-1H-imidazole-5-carbonitrile (AICN) monomers trapped (in a separate experiment) from the gas phase into an Ar matrix. Very good match (Figure 5) of wavenumbers of the IR bands observed in both experiments demonstrates that AICN was generated upon UV excitation of DAMN.
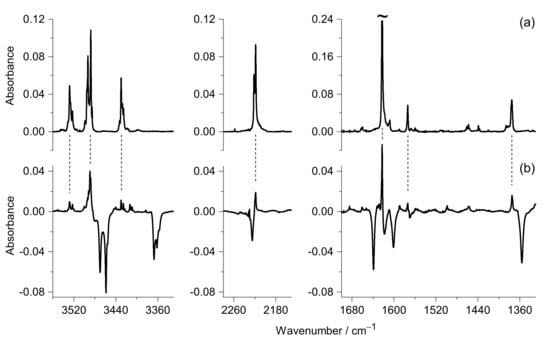
Figure 5.
Experimental identification of AICN. (a) IR spectrum of AICN monomers trapped (in a separate experiment) from the gas phase into an Ar matrix; (b) Difference spectrum: the spectrum recorded after prolonged UV (λ > 295 nm) irradiation of matrix-isolated DAMN monomers minus the spectrum recorded before that irradiation. In this difference spectrum, the positive bands are due to photogenerated products and the negative bands are due to the consumed DAMN reactant. Dashed lines between the spectra in the top and bottom frames connect the strongest bands of AICN and their counterparts observed in the spectrum of products photogenerated from DAMN. Tilde (top-right frame) indicates that the strongest IR band of AICN is truncated.
AICN may adopt two tautomeric forms differing in the position of the hydrogen atom attached to one of the imidazole ring nitrogen atoms. In the literature, there are discrepancies regarding the nature of the AICN tautomer resulting from photo-transformations of DAMN. In early and influential works of Ferris and co-authors [,,,,,,,], the AICN tautomer is graphically presented as having the labile H-atom attached to the ring N-atom vicinal to the amino group (see Figure 6c). In other works [,,], the AICN tautomer is graphically presented as having the labile H-atom attached to the ring N-atom vicinal to the nitrile group (see Figure 6b). Therefore, it seemed to us instructive to verify the tautomeric structure of AICN in the context of the present study, both experimentally and computationally.
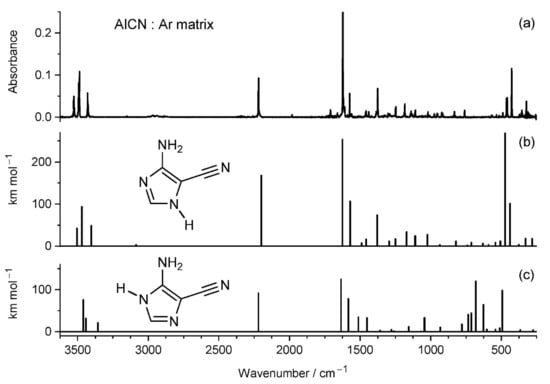
Figure 6.
Comparison of (a) the experimental infrared spectrum of AICN monomers isolated in an Ar matrix at 12 K with theoretical infrared spectra of two tautomers of the compound computed at the DFT (B3LYP)/6-311++G(d,p) level: (b) 4-amino-1H-imidazole-5-carbonitrile, and (c) 5-amino-1H-imidazole-4-carbonitrile.
Comparison of the IR spectra theoretically computed for the two tautomeric forms of AICN with the experimental spectrum of AICN monomers isolated in an Ar matrix (Figure 6a) shows that the compound trapped from the gas phase adopts the lower-energy structure with the labile hydrogen atom attached to the nitrogen atom vicinal to the nitrile group. Thus, this tautomeric form was generated upon excitation of matrix-isolated DAMN with UV (270 < λ < 320 nm) light.
5. Discussion
The photochemical generation of ketenimine and AICN isomers was observed within the current work to occur upon UV excitation of DAMN. It looks quite probable that these photochemical processes should rely upon the generation of DAFN as an intermediate (see Scheme 3).
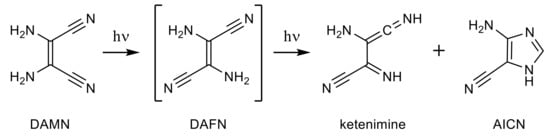
Scheme 3.
Photoinduced transformations of matrix-isolated DAMN molecules.
The photoinduced isomerizations about double bonds have been of particular interest because they represent the simplest way of converting UV light into mechanical motion [,]. The cis–trans photoisomerizations were experimentally observed [,,,,], and theoretically studied [,,,] for ethylene, stilbene, and azobenzene, where the isomers differ by orientation of the substituents about double bonds. The role of conical intersection of the ground and first excited states in the cis–trans photoisomerization is discussed in the works of Minezawa and Gordon [,]. In the particular case of the DAMN-to-DAFN transformation, the role of these conical intersections was theoretically studied by Szabla et al. []. Valiev et al. pointed out the importance of anharmonic effects for internal conversion between electronic states with the same spin multiplicity when the molecule contains accepting X–H stretching modes with a vibrational energy larger than 2000 cm−1 []. The alternative mechanism involving intersystem crossing to the triplet states is discussed for the cis-trans photoisomerizations in the works of Knuts et al. [], Plachkevytch et al. [], and Szabla et al. [].
Koch and Rodehorst [] carried out a quantitative investigation of the photochemical conversion of DAMN into DAFN in methanol solution at 30 °C. Irradiation of DAMN with RPR-3000 Å lamps through Pyrex glass led to formation of a photostationary ratio of 80% of DAFN and 20% of DAMN []. However, it should be noted that under the matrix-isolation conditions of the present study, in the course of irradiations of DAMN, the formation of DAFN has not been verified. We previously studied UV-induced photochemical reactions of six-membered heterocycles, such as α-pyrone [] or its sulfur analogues []. Upon UV irradiations of α-pyrone, we observed the ring-opening reactions leading to generation of the conjugated aldehyde-ketene, O=CH−CH=CH−CH=C=O. Subsequently, for the matrix-isolated conjugated aldehyde-ketene [] as well as for its sulfur analogue [], we observed the Z-E isomerizations around the central C=C bond. As it was demonstrated by using UV light filtered with different cut-off filters, the directions of the observed Z-E isomerization photoprocesses are dependent on the wavelength of the incident UV light. Here we should note that the molecules of aldehyde-ketene have a size similar to DAMN, DAFN, or AICN. Therefore, the lack of observation of DAFN in the present work should not be attributed to the spatial limitations induced by the cryogenic matrix, but rather to the different UV-absorptions of the involved species. With this idea in mind, we computed the lowest electronic singlet transitions of the relevant species and simulated their UV spectra, presented in Figure 7.
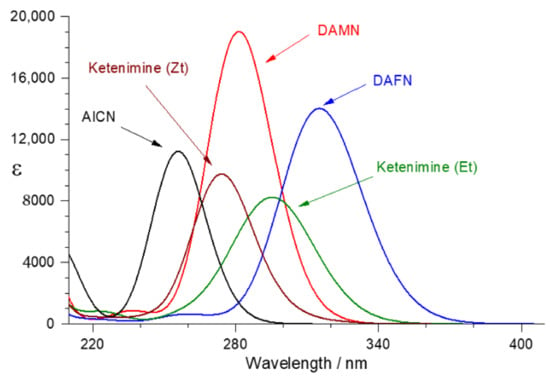
Figure 7.
Simulated UV spectra of DAMN (red), DAFN (blue), ketenimine Zt and Et forms (brown and green, respectively), as well as 4-amino-1H-imidazole-5-carbonitrile (AICN, black), obtained from vertical transition energies determined by TD-DFT calculations at the B3LYP/6-311++G(d,p) level. Each computed transition was convoluted with a Lorentzian function having an hwhm of 0.248 eV (2000 cm−1).
From the analysis of Figure 7, it immediately becomes clear why DAFN was not observed in the present work. This species has the longest wavelength absorption compared to any other isomer. Therefore, DAFN should be very photoreactive and will be consumed by the UV-light (λ > 320 nm or λ > 295 nm) which is used to promote DAMN or ketenimine to their electronically excited states. Analysis of Figure 7 also suggests that the ketenimine Zt isomer has higher probability of being accumulated in the cryomatrix (rather than the Et form, absorbing at longer wavelengths). In fact, it is the Zt isomer of ketenimine whose computed IR spectrum (Figure 4) best reproduces the observed experimental IR spectrum of the photoproduced ketenimine. The other isomer (Et) was not observed. Finally, the computed UV-spectra explain why the five-membered ring isomer (AICN) accumulated in the sample upon UV-irradiations at shorter wavelengths: it should be less affected by the filtered UV-irradiation (λ > 295 nm or λ > 270 nm) that consumes DAMN and also consumes the ketenimine at the more advanced stages of the experiment.
6. Conclusions
Experimental investigations carried out in the present work for matrix-isolated monomers of DAMN (2,3-diamino-2-butenedinitrile) led to identification of two products generated upon UV irradiation of the compound.
One of these photoproducts (the ketenimine tautomer of the compound, Scheme 3) was very reliably identified not only based on the characteristic ketenimine band appearing at 2026 cm−1 in the IR spectrum recorded after UV irradiation, but also on the basis of a good agreement between the whole IR spectrum of the photogenerated product and the theoretical spectrum predicted for the ketenimine tautomeric form. Moreover, the identification of the other photoproduct (4-amino-1H-imidazole-5-carbonitrile, AICN, Scheme 3) is unquestionable. All the strong and medium-strong bands present in the IR spectrum of AICN deposited from the gas phase into an Ar matrix have their counterparts in the spectrum recorded after UV irradiation of matrix-isolated sample containing DAMN and its isomeric ketenimine. The exact match of wavenumbers of the bands in the spectrum of the authentic AICN matrix sample and the bands emerging upon UV irradiation of DAMN/ketenimine leaves no doubt about the correctness of the identification of the AICN photoproduct.
Author Contributions
Conceptualization, I.R.; investigation, I.R., H.R. and L.L.; writing—original draft preparation, L.L.; visualization, H.R. and I.R.; writing—review and editing, I.R., H.R. and L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Funds via the Portuguese Foundation for Science and Technology (FCT). The Chemical Process Engineering and Forest Products Research Centre (CIEPQPF) is supported by FCT through projects UIDB/EQU/00102/2020 and UIDP/EQU/00102/2020.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Structures of DAMN and DAFN
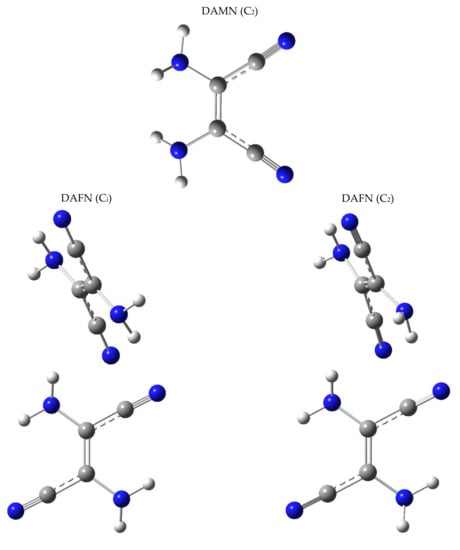
Figure A1.
Optimized geometries of DAMN/DAFN. Top: DAMN (C2 symmetry). Bottom left: DAFN (Ci symmetry). Bottom right: DAFN (C2 symmetry). Note: DAFN (Ci) differs from DAFN (C2) only by a change in pyramidalization of one of the amino groups (see also Figure 2). Color codes: blue—nitrogen, gray—carbon, white—hydrogen.
Appendix B. Structures of the Ketenimine Tautomer of DAMN/DAFN
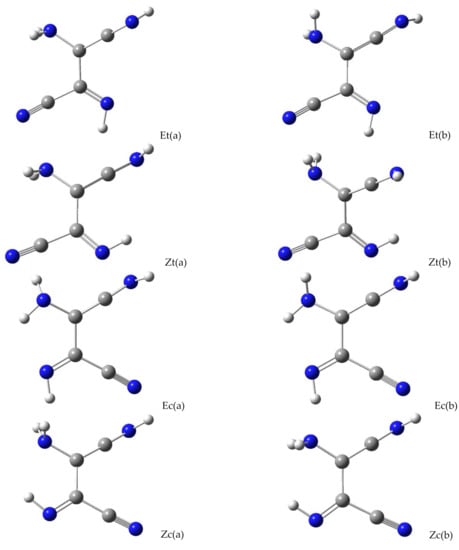
Figure A2.
Optimized geometries of the ketenimine isomers. See Scheme 2 for the adopted E/Z and c/t naming system. Symbols (a,b) stand for different orientations of the NH2 group, with all other conformational parameters equal. In the (a,b) pairs, the (a) orientation is more stable than (b). Color codes: blue—nitrogen, gray—carbon, white—hydrogen.
References
- Sanchez, R.; Ferris, J.; Orgel, L.E. Conditions for Purine Synthesis: Did Prebiotic Synthesis Occur at Low Temperatures? Science 1966, 153, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Studies in Prebiotic Synthesis. II. Synthesis of Purine Precursors and Amino Acids from Aqueous Hydrogen Cyanide. J. Mol. Biol. 1967, 30, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E.; Lohrmann, R. Prebiotic Chemistry and Nucleic Acid Replication. Acc. Chem. Res. 1974, 7, 368–377. [Google Scholar] [CrossRef]
- Erian, A.W. The Chemistry of β-Enaminonitriles as Versatile Reagents in Heterocyclic Synthesis. Chem. Rev. 1993, 93, 1991–2005. [Google Scholar] [CrossRef]
- Mamajanov, I.; Herzfeld, J. HCN polymers characterized by SSNMR: Solid state reaction of crystalline tetramer (diaminomaleo-nitrile). J. Chem. Phys. 2009, 130, 134504. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Bermejo, M.; de la Fuente, J.L.; Pérez-Fernández, C.; Mateo-Martí, E. A Comprehensive Review of HCN-Derived Polymers. Processes 2021, 9, 597. [Google Scholar] [CrossRef]
- Yamada, Y.; Nagashima, N.; Iwashita, Y.; Nakamura, A.; Kumashiro, I. Synthesis and Molecular Structure of Diaminofumaronitrile. Tetrahedron Lett. 1968, 9, 4529–4532. [Google Scholar] [CrossRef]
- Ferris, J.P.; Orgel, L.E. An Unusual Photochemical Rearrangement in the Synthesis of Adenine from Hydrogen Cyanide. J. Am. Chem. Soc. 1966, 88, 1074. [Google Scholar] [CrossRef]
- Ferris, J.P.; Kuder, J.E.; Catalano, A.W. Photochemical Reactions and Chemical Evolution of Purines and Nicotinamide Derivatives. Science 1969, 166, 765–766. [Google Scholar] [CrossRef]
- Ferris, J.P.; Kuder, J.E. Chemical Evolution. III. The Photochemical Conversion of Enaminonitriles to Imidazoles. J. Am. Chem. Soc. 1970, 92, 2527–2533. [Google Scholar] [CrossRef]
- Ferris, J.P.; Trimmer, R.W. Chemical Evolution. XXVIII. Photochemical Conversion of Enaminonitriles to Imidazoles. Scope and Mechanism. J. Org. Chem. 1976, 41, 19–24. [Google Scholar] [CrossRef]
- Ferris, J.P.; Hagan, W.J., Jr. HCN and Chemical Evolution: The Possible Role of Cyano Compounds in Prebiotic Synthesis. Tetrahedron 1984, 40, 1093–1120. [Google Scholar] [CrossRef]
- Boulanger, E.; Anoop, A.; Nachtigallova, D.; Thiel, W.; Barbatti, M. Photochemical Steps in the Prebiotic Synthesis of Purine Precursors from HCN. Angew. Chem. Int. Edit. 2013, 52, 8000–8003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Lapinski, L.; Reva, I.; Rostkowska, H.; Halasa, A.; Fausto, R.; Nowak, M.J. Conformational transformation in squaric acid induced by near-IR laser light. J. Phys. Chem. A 2013, 117, 5251–5259. [Google Scholar] [CrossRef]
- Halasa, A.; Lapinski, L.; Reva, I.; Rostkowska, H.; Fausto, R.; Nowak, M.J. Three Conformers of 2-Furoic Acid: Structure Changes Induced with Near-IR Laser Light. J. Phys. Chem. A 2015, 119, 1037–1047. [Google Scholar] [CrossRef]
- Zhurko, G.A. Chemcraft—Graphical Program for Visualization of Quantum Chemistry Computations, Version 1.8; 2016. Available online: http://www.chemcraftprog.com (accessed on 15 June 2022).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. 3. The 3-21+G Basis Set for 1st Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods. 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Peng, C.Y.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Peng, C.Y.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Nelson, D.D., Jr.; Fraser, G.T.; Klemperer, W. Ammonia dimer: A surprising structure. J. Chem. Phys. 1985, 83, 6201–6208. [Google Scholar] [CrossRef]
- Nelson Jr., D. D.; Fraser, G.T.; Klemperer, W. Does Ammonia Hydrogen Bond? Science 1987, 238, 1670–1674. [Google Scholar] [CrossRef]
- Tao, F.M.; Klemperer, W. Ab Initio Search for the Equilibrium Structure of the Ammonia Dimer. J. Chem. Phys. 1993, 99, 5976–5982. [Google Scholar] [CrossRef]
- Jing, A.; Szalewicz, K.; van der Avoird, A. Ammonia dimer: Extremely fluxional but still hydrogen bonded. Nat. Commun. 2022, 13, 1470. [Google Scholar] [CrossRef]
- Reva, I.D.; Stepanian, S.G.; Adamowicz, L.; Fausto, R. Conformational Behavior of Cyanoacetic Acid: A Combined Matrix Isolation Fourier Transform Infrared Spectroscopy and Theoretical Study. J. Phys. Chem. A 2003, 107, 6351–6359. [Google Scholar] [CrossRef] [Green Version]
- Jacox, M.E.; Milligan, D.E. Infrared Study of the Reactions of CH2 and NH with C2H2 and C2H4 in Solid Argon. J. Am. Chem. Soc. 1963, 85, 278–282. [Google Scholar] [CrossRef]
- Jacox, M.E. Matrix Isolation Study of the Interaction of Excited Argon Atoms with Methyl Cyanide. Vibrational and Electronic Spectra of Ketenimine. Chem. Phys. 1979, 43, 157–172. [Google Scholar] [CrossRef]
- Finnerty, J.; Mitschke, U.; Wentrup, C. Linear ketenimines. Variable structures of C,C-dicyanoketenimines and C,C-bis-sulfonylketenimines. J. Org. Chem. 2002, 67, 1084–1092. [Google Scholar] [CrossRef]
- Inui, H.; Murata, S. Photochemistry of 2-(1-naphthyl)-2H-azirines in matrixes and in solutions: Wavelength-dependent C-C and C-N bond cleavage of the azirine ring. J. Am. Chem. Soc. 2005, 127, 2628–2636. [Google Scholar] [CrossRef]
- Nunes, C.M.; Reva, I.; Pinho e Melo, T.M.V.D.; Fausto, R.; Šolomek, T.; Bally, T. The Pyrolysis of Isoxazole Revisited: A New Primary Product and the Pivotal Role of the Vinylnitrene. A Low-Temperature Matrix Isolation and Computational Study. J. Am. Chem. Soc. 2011, 133, 18911–18923. [Google Scholar] [CrossRef]
- Nunes, C.M.; Reva, I.; Pinho e Melo, T.M.V.D.; Fausto, R. UV-Laser Photochemistry of Isoxazole Isolated in a Low-Temperature Matrix. J. Org. Chem. 2012, 77, 8723–8732. [Google Scholar] [CrossRef]
- Nunes, C.M.; Pinto, S.M.V.; Reva, I.; Fausto, R. On the Photochemistry of 1,2-Benzisoxazole: Capture of Elusive Spiro-2H-azirine and Ketenimine Intermediates. Eur. J. Org. Chem. 2016, 2016, 4152–4158. [Google Scholar] [CrossRef]
- Ito, F.; Nakanaga, T. Observation of the high-resolution spectrum of the N-H bending vibration of ketenimine CH2CNH. J. Mol. Spectrosc. 2010, 264, 100–104. [Google Scholar] [CrossRef]
- Tschöpe, M.; Schröder, B.; Erfort, S.; Rauhut, G. High-Level Rovibrational Calculations on Ketenimine. Front. Chem. 2021, 8, 623641. [Google Scholar] [CrossRef]
- Reva, I.; Lopes Jesus, A.J.; Nunes, C.M.; Roque, J.P.L.; Fausto, R. UV-Induced Photochemistry of 1,3-Benzoxazole, 2-Isocyano-phenol, and 2-Cyanophenol Isolated in Low-Temperature Ar Matrixes. J. Org. Chem. 2021, 86, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Narang, R.S.; Newton, T.A.; Rao, V.R. Mechanistic Studies on the Photochemical Conversion of Enaminonitriles to Imidazoles. J. Org. Chem. 1979, 44, 1273–1278. [Google Scholar] [CrossRef]
- Becker, R.S.; Kolc, J.; Rothman, W. Spectroscopic and Photochemical Study of Diaminomaleonitrile and Diaminofumaronitrile. J. Am. Chem. Soc. 1973, 95, 1269–1273. [Google Scholar] [CrossRef]
- Plese, M.; Jasien, P.G. The photochemical isomerization of DAMN to ACI: Stability of potential intermediates. J. Mol. Struct. (Theochem) 1996, 364, 121–130. [Google Scholar] [CrossRef]
- Jeilani, Y.A.; Williams, P.N.; Walton, S.; Nguyen, M.T. Unified reaction pathways for the prebiotic formation of RNA and DNA nucleobases. Phys. Chem. Chem. Phys. 2016, 18, 20177–20188. [Google Scholar] [CrossRef] [Green Version]
- Koumura, N.; Zijlstra, R.W.J.; van Delden, R.A.; Harada, N.; Feringa, B.L. Light-driven monodirectional molecular rotor. Nature 1999, 401, 152–155. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.Y.; Morokuma, K. Computational Study on the Working Mechanism of a Stilbene Light-Driven Molecular Rotary Motor: Sloped Minimal Energy Path and Unidirectional Nonadiabatic Photoisomerization. J. Am. Chem. Soc. 2012, 134, 4864–4876. [Google Scholar] [CrossRef]
- Arai, T.; Tokumaru, K. Photochemical One-Way Adiabatic Isomerization of Aromatic Olefins. Chem. Rev. 1993, 93, 23–39. [Google Scholar] [CrossRef]
- Duarte, L.; Fausto, R.; Reva, I. Structural and spectroscopic characterization of E- and Z-isomers of azobenzene. Phys. Chem. Chem. Phys. 2014, 16, 16919–16930. [Google Scholar] [CrossRef]
- Duarte, L.; Khriachtchev, L.; Fausto, R.; Reva, I. Photoisomerization of azobenzenes isolated in cryogenic matrices. Phys. Chem. Chem. Phys. 2016, 18, 16802–16811. [Google Scholar] [CrossRef] [Green Version]
- Mahimwalla, Z.; Yager, K.G.; Mamiya, J.-I.; Shishido, A.; Priimagi, A.; Barrett, C.J. Azobenzene photomechanics: Prospects and potential applications. Polym. Bull. 2012, 69, 967–1006. [Google Scholar] [CrossRef]
- Quick, M.; Dobryakov, A.L.; Gerecke, M.; Richter, C.; Berndt, F.; Ioffe, I.N.; Granovsky, A.A.; Mahrwald, R.; Ernsting, N.P.; Kovalenko, S.A. Photoisomerization Dynamics and Pathways of trans- and cis- Azobenzene in Solution from Broadband Femtosecond Spectroscopies and Calculations. J. Phys. Chem. B 2014, 118, 8756–8771. [Google Scholar] [CrossRef] [PubMed]
- Han, W.G.; Lovell, T.; Liu, T.Q.; Noodleman, L. Density functional studies of the ground- and excited-state potential-energy curves of stilbene cis-trans isomerization. ChemPhysChem 2002, 3, 167–178. [Google Scholar] [CrossRef]
- Quenneville, J.; Martínez, T.J. Ab initio study of cis-trans photoisomerization in stilbene and ethylene. J. Phys. Chem. A 2003, 107, 829–837. [Google Scholar] [CrossRef]
- Levine, B.G.; Martínez, T.J. Isomerization through conical intersections. Annu. Rev. Phys. Chem. 2007, 58, 613–634. [Google Scholar] [CrossRef]
- Minezawa, N.; Gordon, M.S. Optimizing Conical Intersections by Spin-Flip Density Functional Theory: Application to Ethylene. J. Phys. Chem. A 2009, 113, 12749–12753. [Google Scholar] [CrossRef] [Green Version]
- Minezawa, N.; Gordon, M.S. Photoisomerization of Stilbene: A Spin-Flip Density Functional Theory Approach. J. Phys. Chem. A 2011, 115, 7901–7911. [Google Scholar] [CrossRef] [Green Version]
- Szabla, R.; Góra, R.W.; Šponer, J.; Šponer, J.E. Molecular Mechanism of Diaminomaleonitrile to Diaminofumaronitrile Photoisomerization: An Intermediate Step in the Prebiotic Formation of Purine Nucleobases. Chem. Eur. J. 2014, 20, 2515–2521. [Google Scholar] [CrossRef]
- Valiev, R.R.; Nasibullin, R.T.; Cherepanov, V.N.; Baryshnikov, G.V.; Sundholm, D.; Ågren, H.; Minaev, B.F.; Kurtén, T. First-principles calculations of anharmonic and deuteration effects on the photophysical properties of polyacenes and porphyrinoids. Phys. Chem. Chem. Phys. 2020, 22, 22314–22323. [Google Scholar] [CrossRef]
- Knuts, S.; Minaev, B.F.; Vahtras, O.; Ågren, H. Spin-Orbit Coupling in the Intersystem Crossing of the Ring-Opened Oxirane Biradical. Int. J. Quantum Chem. 1995, 55, 23–34. [Google Scholar] [CrossRef]
- Plachkevytch, O.; Minaev, B.; Ågren, H. Paramagnetic exchange spin-catalysis of the cis-trans isomerization of substituted ethylenes. J. Phys. Chem. 1996, 100, 8308–8315. [Google Scholar] [CrossRef]
- Koch, T.H.; Rodehorst, R.M. A Quantitative Investigation of the Photochemical Conversion of Diaminomaleonitrile to Diaminofumaronitrile and 4-Amino-5-Cyanoimidazole. J. Am. Chem. Soc. 1974, 96, 6707–6710. [Google Scholar] [CrossRef]
- Breda, S.; Reva, I.; Lapinski, L.; Fausto, R. Matrix isolation FTIR and theoretical study of α-pyrone photochemistry. Phys. Chem. Chem. Phys. 2004, 6, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Breda, S.; Reva, I.; Lapinski, L.; Cristiano, M.L.S.; Frija, L.; Fausto, R. Photochemical Ring-Opening and Intramolecular Hydrogen Shift Reactions in Sulfur Analogues of α-Pyrone. J. Phys. Chem. A 2006, 110, 6415–6425. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).