
Phosphorescence and Photophysical Parameters of Porphycene in Cryogenic Matrices


Abstract
:1. Introduction
2. Materials and Methods
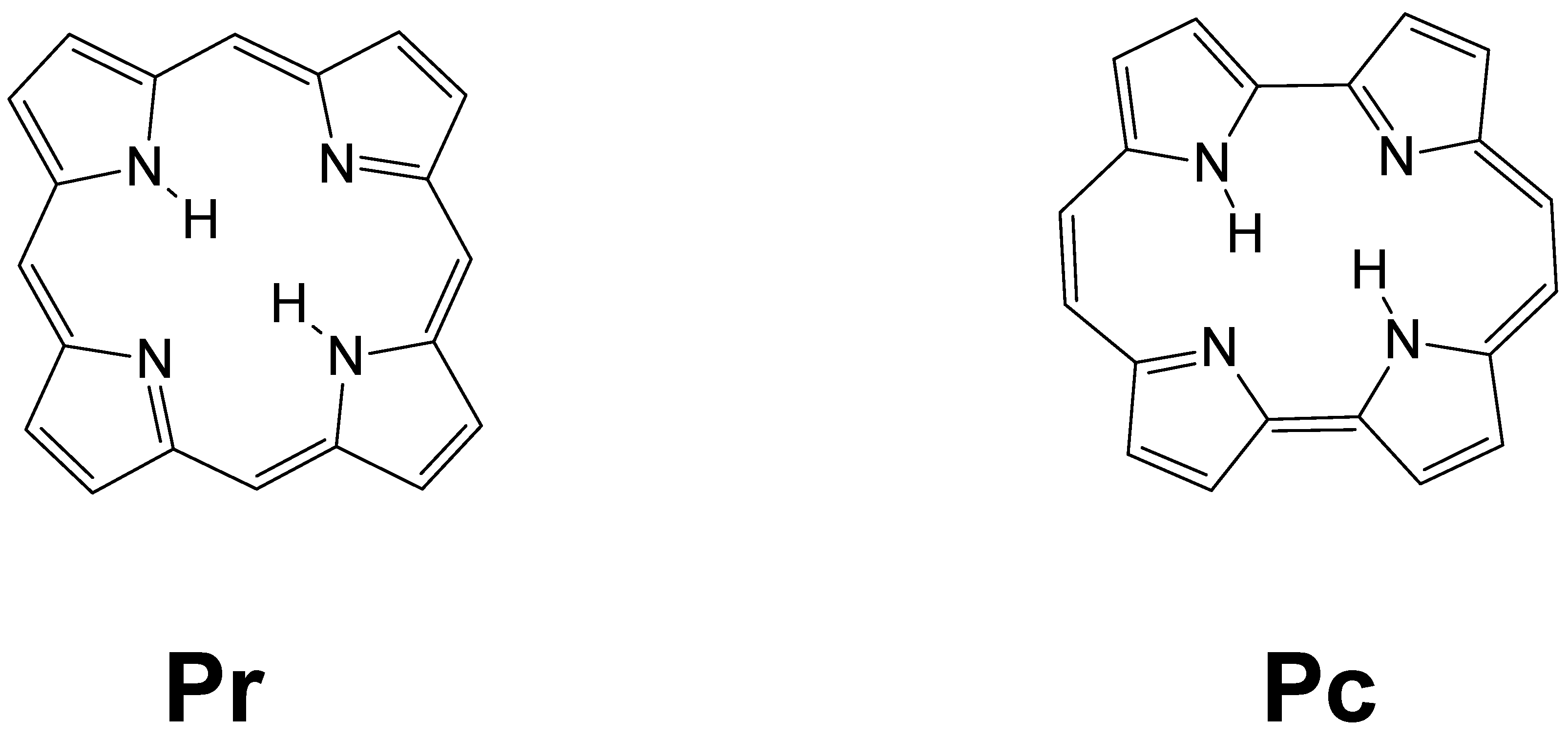
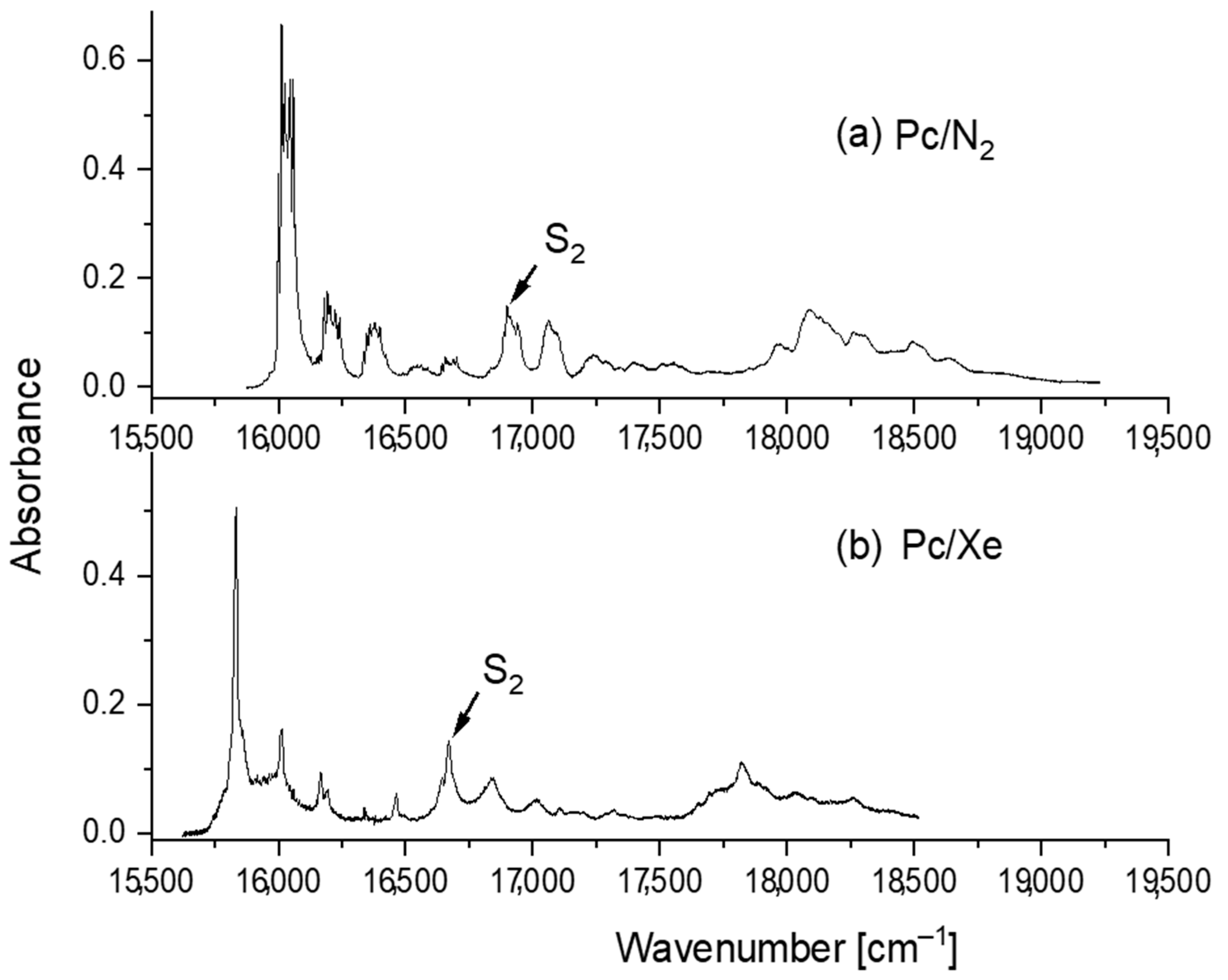
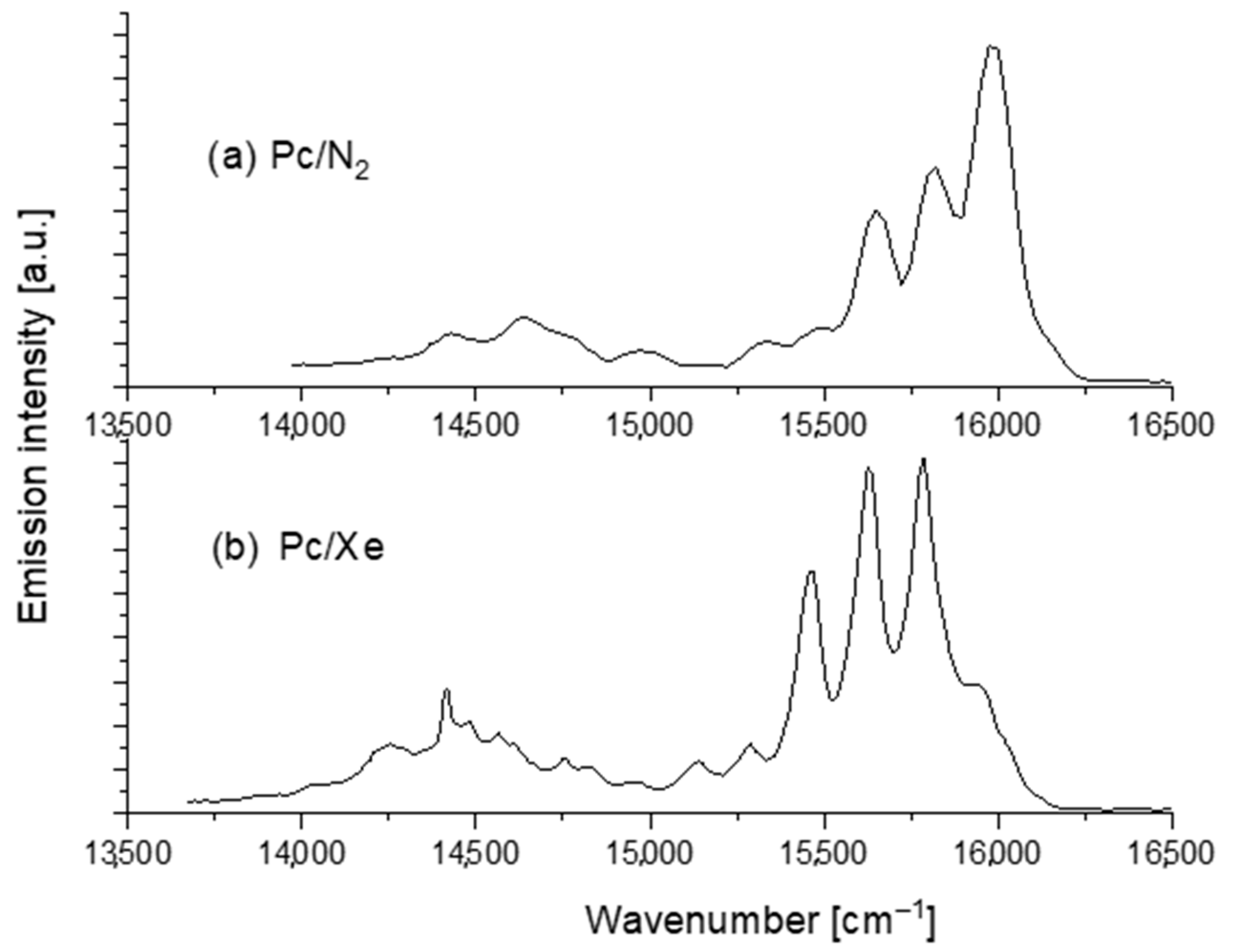
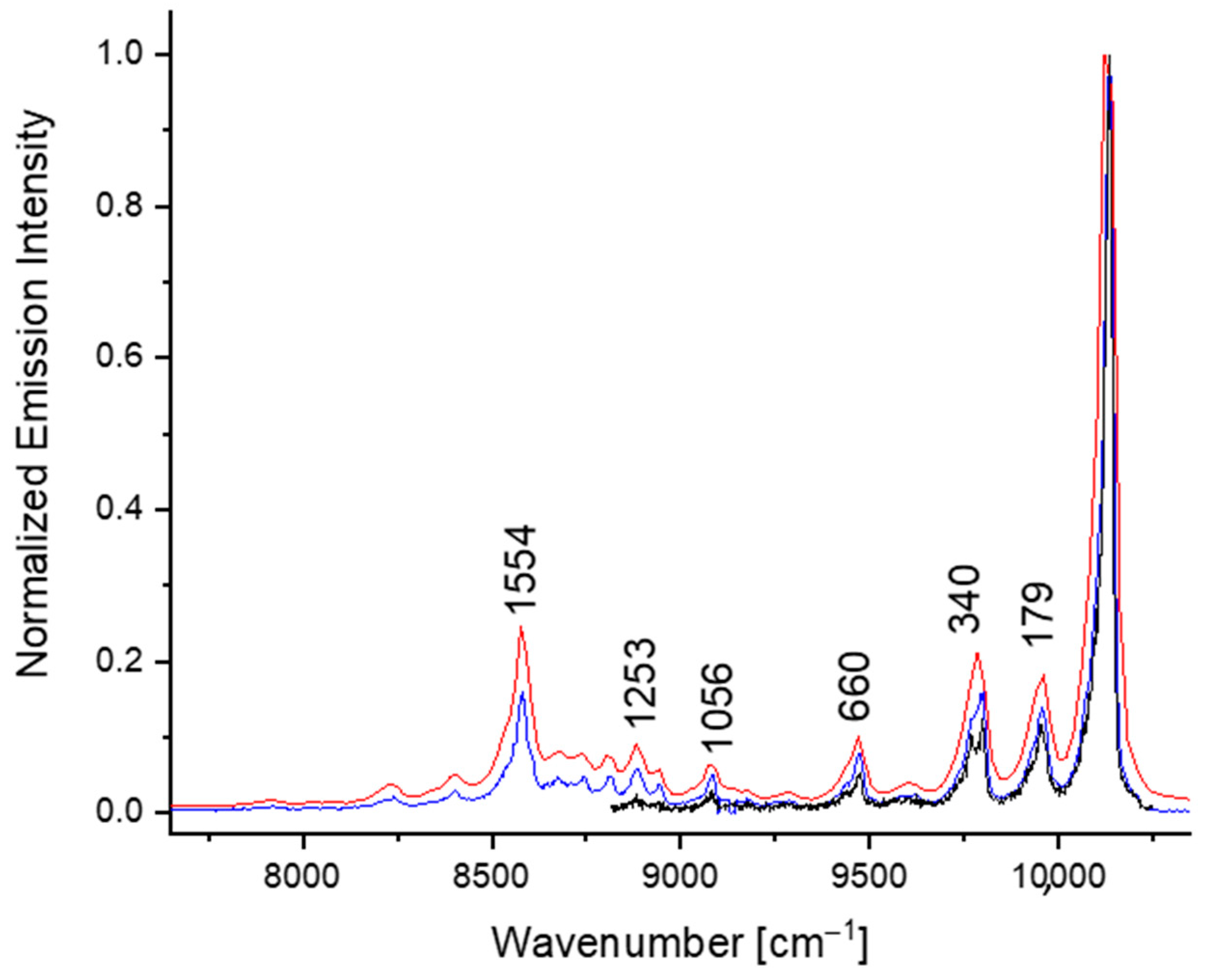
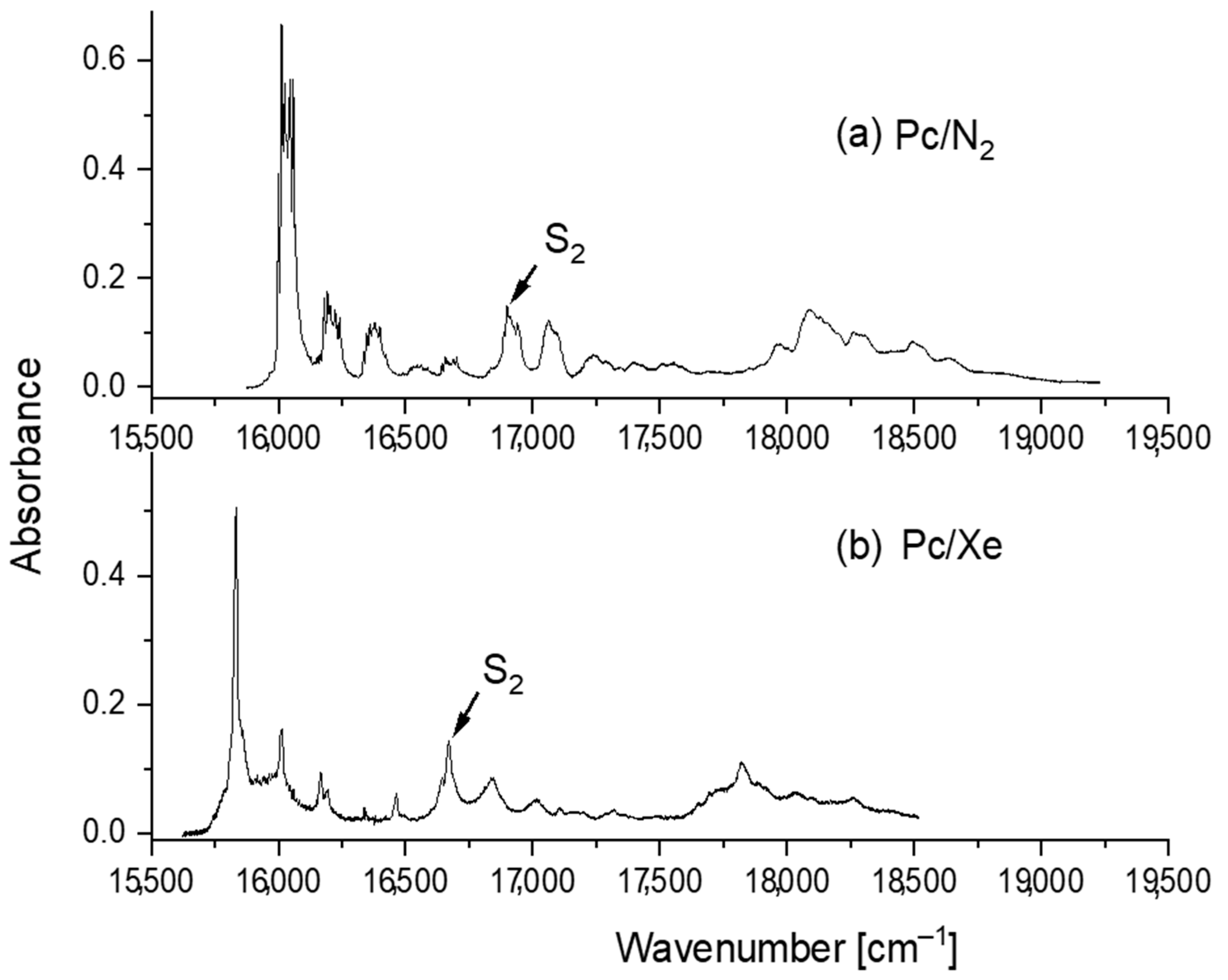
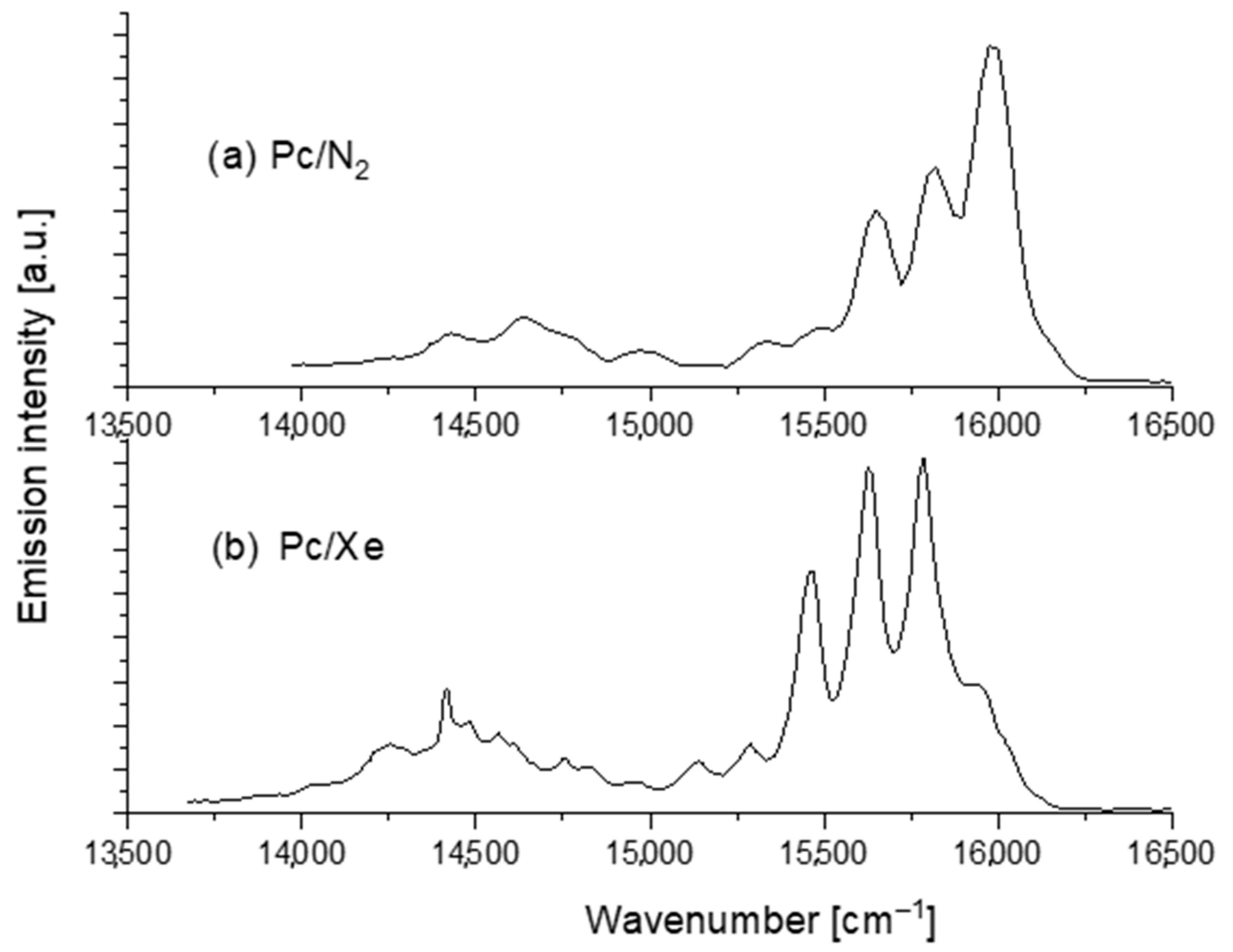
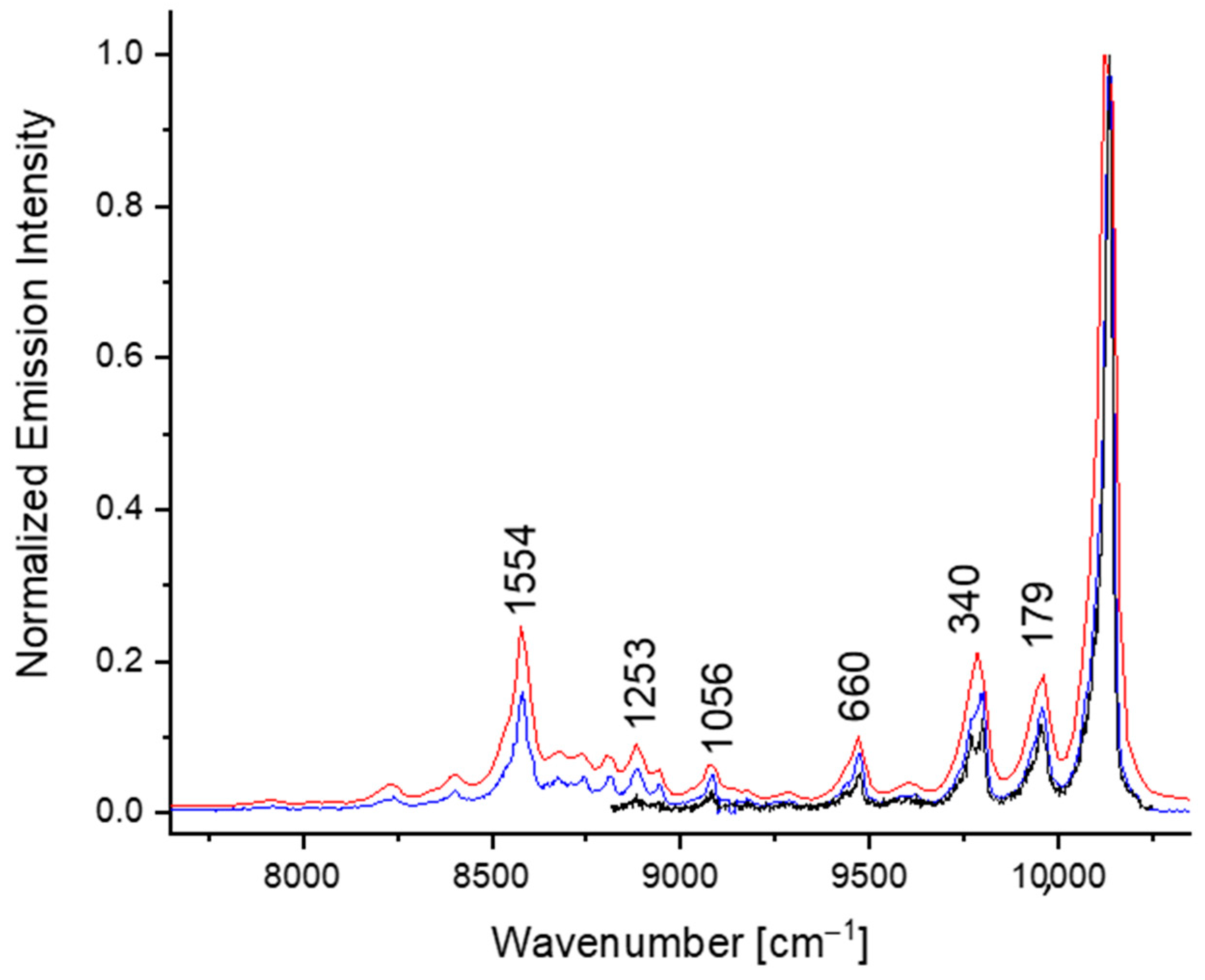
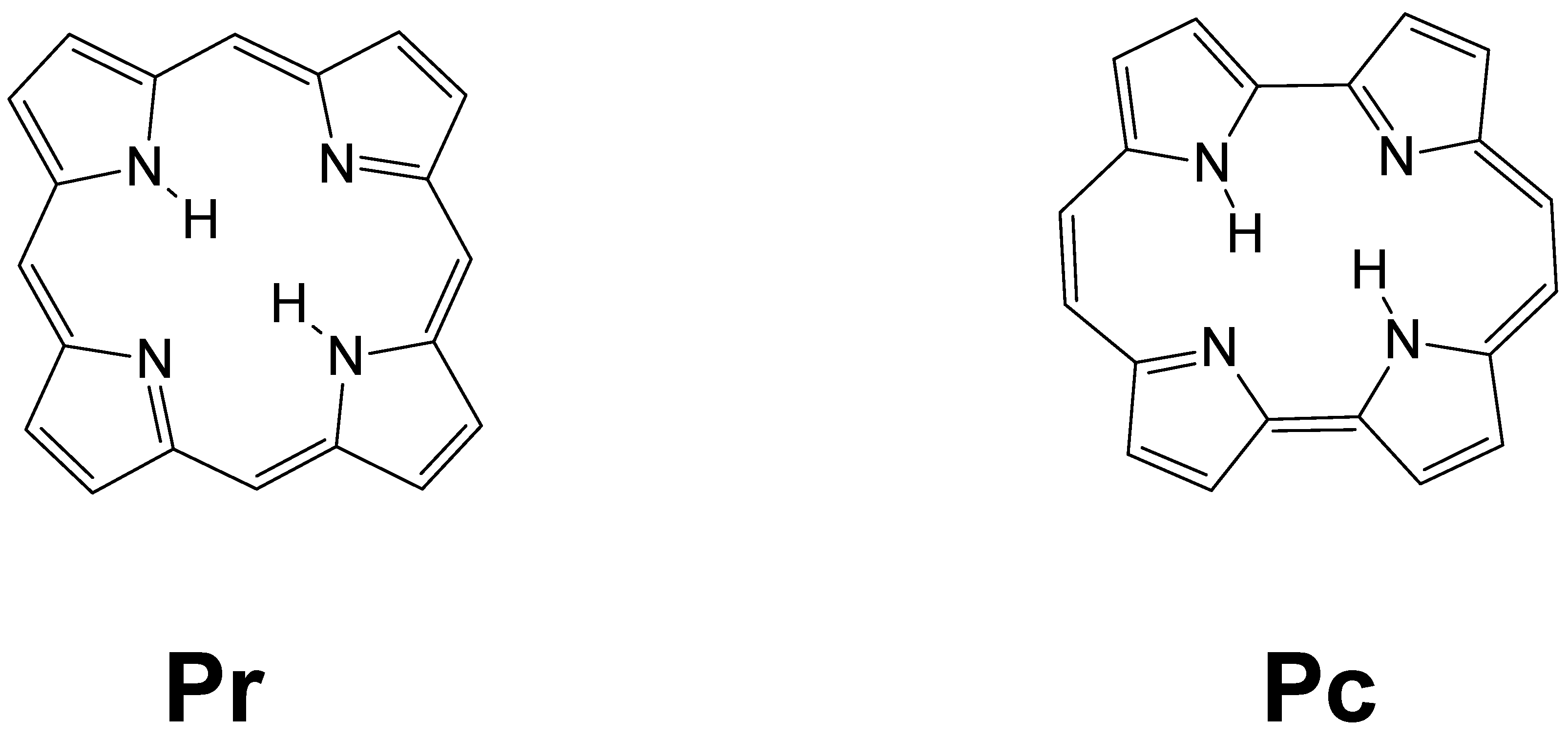
3. Results and Discussion
4. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; Montalti, M., Credi, A., Prodi, L., Gandolfi, M.T., Eds.; Taylor & Francis Group: Abingdon, UK, 2006. [Google Scholar]
- Battersby, A.R. Tetrapyrroles: The Pigments of Life. Nat. Prod. Rep. 2000, 17, 507–526. [Google Scholar] [CrossRef] [PubMed]
- Radziszewski, J.; Waluk, J.; Michl, J. FT Visible Absorption Spectroscopy of Porphine in Noble Gas Matrices. J. Mol. Spectrosc. 1990, 140, 373–389. [Google Scholar] [CrossRef]
- Radziszewski, J.G.; Waluk, J.; Nepraš, M.; Michl, J. Fourier Transform Fluorescence and Phosphorescence of Porphine in Rare Gas Matrices. J. Phys. Chem. 1991, 95, 1963–1969. [Google Scholar] [CrossRef]
- Waluk, J. Spectroscopy and Tautomerization Studies of Porphycenes. Chem. Rev. 2017, 117, 2447–2480. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C.; Cañete, M.; Juarranz, A.; Villanueva, A.; Horobin, R.W.; Borrell, J.; Teixidó, J.; Nonell, S. Porphycenes: Facts and Prospects in Photodynamic Therapy of Cancer. Curr. Med. Chem. 2007, 14, 997–1026. [Google Scholar] [CrossRef]
- Polo, L.; Segalla, A.; Bertoloni, G.; Jori, G.; Schaffner, K.; Reddi, E. Polylysine-Porphycene Conjugates as Efficient Photosensitizers for the Inactivation of Microbial Pathogens. J. Photochem. Photobiol. B Biol. 2000, 59, 152–158. [Google Scholar] [CrossRef]
- Planas, O.; Gallavardin, T.; Nonell, S. Unusual Properties of Asymmetric Porphycenes. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2016; Volume 41, pp. 299–349. [Google Scholar]
- Gil, M.; Dobkowski, J.; Wiosna-Sałyga, G.; Urbańska, N.; Fita, P.; Radzewicz, C.; Pietraszkiewicz, M.; Borowicz, P.; Marks, D.; Glasbeek, M.; et al. Unusual, Solvent Viscosity-Controlled Tautomerism and Photophysics: Meso-Alkylated Porphycenes. J. Am. Chem. Soc. 2010, 132, 13472–13485. [Google Scholar] [CrossRef]
- Aramendia, P.F.; Redmond, R.W.; Nonell, S.; Schuster, W.; Braslavsky, S.E.; Schaffner, K.; Vogel, E. The Photophysical Properties of Porphycenes: Potential Photodynamic Therapy Agents. Photochem. Photobiol. 1986, 44, 555–559. [Google Scholar] [CrossRef]
- Levanon, H.; Toporowicz, M.; Ofir, H.; Fessenden, R.W.; Das, P.K.; Vogel, E.; Köcher, M.; Pramod, K. Triplet-State Formation of Porphycenes—Intersystem Crossing Versus Sensitization Mechanisms. J. Phys. Chem. 1988, 92, 2429–2433. [Google Scholar] [CrossRef]
- Redmond, R.W.; Valduga, G.; Nonell, S.; Braslavsky, S.E.; Schaffner, K.; Vogel, E.; Pramod, K.; Köcher, M. The Photophysical Properties of Porphycene Incorporated in Small Unilamellar Lipid Vesicles. J. Photochem. Photobiol. B Biol. 1989, 3, 193–207. [Google Scholar] [CrossRef]
- Braslavsky, S.E.; Müller, M.; Mártire, D.O.; Pörting, S.; Bertolotti, S.G.; Chakravorti, S.; Koç-Weier, G.; Knipp, B.; Schaffner, K. Photophysical Properties of Porphycene Derivatives (18 π Porphyrinoids). J. Photochem. Photobiol. B Biol. 1997, 40, 191–198. [Google Scholar] [CrossRef]
- Nonell, S.; Aramendía, P.F.; Heihoff, K.; Negri, R.M.; Braslavsky, S.E. Laser-Induced Optoacoustics Combined with near-Infrared Emission. An Alternative Approach for the Determination of Intersystem Crossing Quantum Yields Applied to Porphycenes. J. Phys. Chem. 1990, 94, 5879–5883. [Google Scholar] [CrossRef]
- Urbańska, N.; Pietraszkiewicz, M.; Waluk, J. Efficient Synthesis of Porphycene. J. Porphyr. Phthalocyanines 2007, 11, 596–600. [Google Scholar] [CrossRef]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822–8824, Erratum: Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 34, 7406–7406. [Google Scholar] [CrossRef]
- Starukhin, A.; Vogel, E.; Waluk, J. Electronic Spectra in Porphycenes in Rare Gas and Nitrogen Matrices. J. Phys. Chem. A 1998, 102, 9999–10006. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Waluk, J. Molecular Dynamics Simulations of Matrix Deposition. Iii. Site Structure Analysis for Porphycene in Argon and Xenon. J. Chem. Phys. 2005, 123, 064706. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Gawinkowski, S.; Urbańska, N.; Pietraszkiewicz, M.; Waluk, J. Matrix Isolation Spectroscopy and Molecular Dynamics Simulations for 2,7,12,17-Tetra-Tert-Butylporphycene in Argon and Xenon. J. Chem. Phys. 2007, 127, 134501. [Google Scholar] [CrossRef]
- Gil, M.; Gorski, A.; Starukhin, A.; Waluk, J. Fluorescence Studies of Porphycene in Various Cryogenic Environments. Low Temp. Phys. 2019, 45, 656–662. [Google Scholar] [CrossRef]
- Kijak, M.; Nawara, K.; Listkowski, A.; Masiera, N.; Buczyńska, J.; Urbańska, N.; Orzanowska, G.; Pietraszkiewicz, M.; Waluk, J. 2+2 Can Make Nearly a Thousand! Comparison of Di- and Tetra-Meso-Alkyl-Substituted Porphycenes. J. Phys. Chem. A 2020, 124, 4594–4604. [Google Scholar] [CrossRef] [PubMed]
- Gawinkowski, S.; Walewski, Ł.; Vdovin, A.; Slenczka, A.; Rols, S.; Johnson, M.R.; Lesyng, B.; Waluk, J. Vibrations and Hydrogen Bonding in Porphycene. Phys. Chem. Chem. Phys. 2012, 14, 5489–5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penfold, T.J.; Gindensperger, E.; Daniel, C.; Marian, C.M. Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev. 2018, 118, 6975–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dorp, W.G.; Soma, M.; Kooter, J.A.; van der Waals, J.H. Electron Spin Resonance in the Photo-Excited Triplet State of Free Base Porphin in a Single Crystal of n-Octane. Mol. Phys. 1974, 28, 1551–1568. [Google Scholar] [CrossRef]
- Weiss, C.; Kobayashi, H.; Gouterman, M. Spectra of Porphyrins. Part III. Self-Consistent Molecular Orbital Calculations of Porphyrin and Related Ring Systems. J. Mol. Spectrosc. 1965, 16, 415–450. [Google Scholar] [CrossRef]
- Knop, J.V.; Knop, A. Quantenchemische Und Spektroskopische Untersuchungen an Porphyrinen. I. Freie Base Porphin Und Metallo-Porphin. Z. Für Naturforsch. 1970, 25, 1720–1725. [Google Scholar] [CrossRef]
- Sundbom, M. Semi-Empirical Molecular Orbital Studies of Neutral Porphin, PH2, the Dianion P2- and the Dication PH42+. Acta Chem. Scand. 1968, 22, 1317–1326. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| τF, ns | τT, μs | ΦT | |
|---|---|---|---|
| N2 | 21 ± 2 | 2000 ± 40 | 0.36 ± 0.04 |
| Xe | 0.35 ± 0.1 | 45 ± 0.5 | 1 ± 0.05 |
| Phosphorescence | Fluorescence | Assignment a |
|---|---|---|
| 179 | 184 | 2 Ag |
| 340 | 336 | 3 Ag |
| 368 | 358 | 4 Ag |
| 511 | 510 | 2 Ag + 3 Ag |
| 541 | 538 | 2 Ag + 4 Ag |
| 621 | 7 Bg | |
| 660 | 665 | 7 Ag |
| 686 | 2 × 3 Ag | |
| 696 | 9 Bg | |
| 703 | 3 Ag + 4 Ag | |
| 723 | 2 × 4 Ag | |
| 847 | 1 Ag + 7 Bg | |
| 863 | 9 Ag | |
| 881 | 14 Bg | |
| 959 | 961 | 10 Ag |
| 1010 | 3 × 3 Ag | |
| 1056 | 14 Ag | |
| 1079 | 3 × 4 Ag | |
| 1192 | 1203 | 17 Ag |
| 1253 | 1265 | 19 Ag |
| 1322 | 20 Ag | |
| 1354 | 21 Ag | |
| 1392 | 1395 | 23 Ag |
| 1426 | 25 Ag | |
| 1458 | 26 Ag | |
| 1486 | 1497 | 27 Ag |
| 1554 | 1562 | 29 Ag |
| 1585 | ||
| 1614 | 30 Ag | |
| 1730 | 2 Ag + 29 Ag | |
| 1891 | 3 Ag + 29 Ag | |
| 1918 | 4 Ag + 29 Ag | |
| 2215 | 7 Ag + 29 Ag |
| S1 | S2 | T1 | T2 | |
|---|---|---|---|---|
| Pc | 17,191 a | 18,226 | 11,898 | 12,449 |
| (0.0927) b | (0.1549) | |||
| 54.4.% sH-L c | 56.3% H-L | H-L | sH-L | |
| 26% H-L | 25.0% sH-L | |||
| Pr | 17,505 (0.0007) | 18,563 (0.0004) | 14,087 | 14,870 |
| 61.5% H-sL | 57.3% H-L | |||
| 37.5% sH-L | 41.4% sH-sL | H-L | H-sL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golec, B.; Gorski, A.; Waluk, J. Phosphorescence and Photophysical Parameters of Porphycene in Cryogenic Matrices. Photochem 2022, 2, 217-224. https://doi.org/10.3390/photochem2010016
Golec B, Gorski A, Waluk J. Phosphorescence and Photophysical Parameters of Porphycene in Cryogenic Matrices. Photochem. 2022; 2(1):217-224. https://doi.org/10.3390/photochem2010016
Chicago/Turabian StyleGolec, Barbara, Aleksander Gorski, and Jacek Waluk. 2022. "Phosphorescence and Photophysical Parameters of Porphycene in Cryogenic Matrices" Photochem 2, no. 1: 217-224. https://doi.org/10.3390/photochem2010016
APA StyleGolec, B., Gorski, A., & Waluk, J. (2022). Phosphorescence and Photophysical Parameters of Porphycene in Cryogenic Matrices. Photochem, 2(1), 217-224. https://doi.org/10.3390/photochem2010016